
ABSTRACT
Pin1 is a peptidyl-prolyl isomerase that induces the cis-trans conversion of specific Ser/Thr-Pro peptide bonds in phosphorylated proteins, leading to conformational changes through which Pin1 regulates protein stability and activity. Since down-regulation of Pin1 has been described in several neurodegenerative disorders, including Alzheimer's Disease (AD), Parkinson's Disease (PD) and Huntington's Disease (HD), we investigated its potential role in prion diseases. Animals generated on wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) background for Pin1 were experimentally infected with RML prions. The study indicates that, neither the total depletion nor reduced levels of Pin1 significantly altered the clinical and neuropathological features of the disease.
KEYWORDS:
Introduction
Prion diseases, or Transmissible Spongiform Encephalopathies (TSEs), are a group of neurological disorders that affects different animal species and humans. They can have sporadic, genetic or infectious origins [Citation1,Citation2]. All of them are characterized by the conformational conversion of the normal prion protein (PrPC) to a pathological isoform named PrPSc which propagates in the brain by recruiting and converting new PrPC molecules [Citation2]. Compared to PrPC, PrPSc is rich in β-sheet structures, is partially resistant to digestion with proteinase K (PK) and is prevalently insoluble in detergent. In contrast, PrPC is predominantly composed of α-helix structures, is soluble in detergent and is completely digested by PK [Citation3,Citation4]. Both proteins have two N-linked glycosylation sites that enable the generation of unglycosylated, monoglycosylated and diglycosylated isoforms. The relative abundance of each PrP species represents the glycoform ratio. Once misfolded, PrPSc can acquire different abnormal conformations [Citation5] also referred to as prion strains. Each conformer is associated with peculiar clinical, neuropathological and biochemical alterations [Citation6]. A prion strain can be identified either neuropathologically considering (i) the incubation and survival time [Citation7], (ii) the clinical signs, (iii) the pattern of PrPSc deposition [Citation8] and the severity of spongiform changes [Citation9]; or biochemically after PK digestion according to: (i) the electrophoretic mobility of the unglycosylated PrP band, (ii) the glycoform ratio [Citation10], (iii) the resistance to PK digestion [Citation11] and (iv) the conformational stability using chaotropic agents (e.g. Guanidine-hydrochloride) [Citation12]. The mechanism of conversion is not fully understood, but several factors, like glycosaminoglycans, lipids, nucleic acids, metals and phosphorylations are supposed to play an important role in this process [Citation13–20]. An important technique named Protein Misfolding Cyclic Amplification (PMCA) has been developed in 2001 with the aim of mimicking the process of prion conversion in vitro in an accelerated manner [Citation21]. PMCA consists of cycles of sonication and incubation of a sample containing minute amount of PrPSc and an excess of PrPC [Citation22]. During the incubation phase PrPSc recruits and converts PrPC to form polymers that are fragmented during the sonication phase whereby new PrPSc species are created and can induce further conversion of PrPC. PMCA enables exponential amplification of PrPSc and has been extensively used to study the potential role of cellular cofactors in PrPC to PrPSc conversion [Citation17,Citation23–28]. These studies highlighted the fact that different anionic conditions (anionic detergents, RNA and synthetic polyanions) were found to favor the formation of PrPSc [Citation26–29]. Phosphorylation of PrPC indeed provides anionic conditions that could severely affect its conformation [Citation20]. Neuronal Cdk5 is a kinase able to phosphorylate PrPC at Ser-43 position which induces the protein to change conformation and determines its conversion to PrPSc as showed either in vitro or in vivo [Citation20]. Phosphorylated proteins can be recognized by different proteins, usually polypeptides containing either WW domains or SH2 domains such as scaffold proteins or enzymes. Among others, Pin1, a member of to the peptidyl-prolyl cis/trans isomerase (PPIase) family. Pin1 is a 18 kDa protein that catalyzes phospho-Serine/Threonine-Proline (pSer/Thr-Pro) isomerization from cis to trans conformation thus modifying the molecular conformation of phosphorylated proteins [Citation30,Citation31]. The N-terminal binding domain of the protein (WW) allows Pin1 to interact with its substrates and the C-terminal domain is responsible for their cis/trans isomerization [Citation32]. This conformational modification represents a fundamental controlled signaling mechanism regulating the activity of different proteins, such as c-Jun, NF-κB, p53, p27, β-catenin, c-Myc, IRAK1, gephyrin, myeloid cell leukemia sequence-1 (MCL-1), cyclin D1, cyclin E, Notch1, Notch4, tau, APP, and others [Citation33–35]. Pin1 is ubiquitously expressed, especially in mitotic cells and in neurons during developmental phases, suggesting a possible involvement in cellular proliferation and neural differentiation [Citation36]. At subcellular level Pin1 can be detected in the nucleus, cytoplasm or mitochondria [Citation34]. Dysregulation of Pin1 activity has been implicated in different disorders, including cancers and neurodegenerative disorders [Citation37,Citation38]. The existence of epidemiological studies showing an inverse association between Alzheimer's disease and cancer might support the idea that the fate of a cell to degenerate or proliferate might be directed by a common mechanism which could involve the activity of Pin1 [Citation39–41]. Indeed, Pin1 up-regulation has been observed in several types of cancer, including prostate, lung, breast cancers [Citation42] and esophageal squamous cell carcinomas [Citation39,Citation43]. Pin1 deregulation has been reported in Alzheimer's Disease (AD), Parkinson's Disease (PD) and Huntington's Disease (HD) [Citation35,Citation44–51]. It has been described that brain regions of AD patients with low levels of Pin1 are more vulnerable and prone to neurofibrillary degeneration than areas where Pin1 is normally expressed [Citation35]. Supporting the important role of Pin1 in the field of neurodegenerative diseases is the fact that Pin1 knock-out mice (Pin1−/−) are described to develop age-dependent neuropathy (at around 9–14 months of age), including motor/behavioral deficits and accumulation of hyperphosphorylated tau [Citation35].
In this work we have investigated whether, other than AD, PD and HD, Pin1 might have a role in prion diseases by modulating the process of PrPC/PrPSc conversion. To this aim, we have performed RML inoculation in animals generated on wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) background for Pin1. Since the lack of Pin1 is associated with tau hyperphosphorylation, tau filaments formation and neuronal degeneration, we have then assessed whether the inoculation of RML could have exacerbated these conditions in mice sacrificed at terminal stage of the disease (at around 6–7 months of age, therefore younger than 9–14 months). In addition, we exploited the ability of PMCA to mimic the process of prion conversion in vitro to verify whether reduced levels of Pin1 or its depletion could modulate the efficiency of RML replication. These results might help at identifying any interplay between Pin1 and prions. Establishing whether Pin1 has any role in PrPSc formation would lay the basis for developing innovative therapies for these devastating disorders.
Results
Wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) mice for Pin1 express similar levels of PrPC
Since differences in PrPC levels can significantly alter the efficiency of prion propagation, we have initially evaluated the expression level of PrPC in wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) mice for Pin1. Western blot analysis of 10% (weight/volume) brain homogenates collected from three mice per group were performed. Samples were immunoblotted with the 6D11 antibody and resulting signal underwent to densitometric analysis normalized against α-tubulin. As shown in , the PrPC signal was similar in all mice, regardless of their Pin1 genetic background. Densitometric evaluations revealed the presence of small differences that were not statistically significant.
Figure 1. Western blot and PMCA analysis. (A) Ten % brain homogenates (BH) collected from Pin1+/+, Pin1+/− and Pin1−/− mice were analyzed by means of Western blot and showed similar levels of PrPC expression. Densitometric analysis confirmed the lack of statistically significant differences (t-test). Graphs represent the mean and bars indicate Standard Error of the Mean (SEM). (B) RML prion strain was spiked (from 10−5 to 10−9) in Pin1+/+, Pin1+/− or Pin1−/− BH and subjected to PMCA analysis. The amplification efficiency of RML was similar in all groups of mice and enable detection of all dilutions after one round of PMCA. Samples were immunoblotted with the 6D11 antibody after PK digestion. PrPC refers to normal mouse brain homogenate not digested with PK. Numbers in the left of the Western blot indicate molecular weight markers.
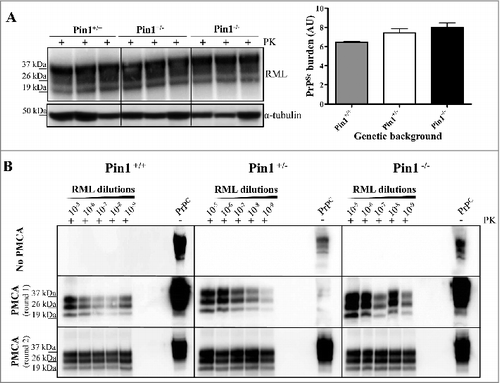
Brain homogenates of Pin1+/+, Pin1+/− or Pin1−/− mice were used as PMCA substrates and efficiently amplified RML
Considering that the brains of Pin1+/+, Pin1+/− and Pin1−/− mice contained similar level of PrPC, we homogenized those brains (as reported in materials and methods) and performed PMCA experiments. In particular, RML brain homogenate was serially diluted (from 10−5 to 10−9 volume/volume) in Pin1+/+, Pin1+/− or Pin1−/− substrates and subjected to 2 rounds of amplification. After one round of amplification, all RML-PrPSc dilutions were efficiently amplified, regardless of the substrate. Thus, using our experimental conditions, the lack of Pin1 did not significantly influence the amplification efficiency of RML by means of PMCA (). Furthermore, after PK digestion, we observed that the biochemical profiles of all the amplified PrPres were identical to that of the RML used as inoculum and were characterized by the predominance of the monoglycosylated PrPres species over the other. Taken together these results suggest that PMCA faithfully propagated the RML prion strain despite the use of three different substrates.
Incubation and survival times of RML injected animals were not statistically different among groups
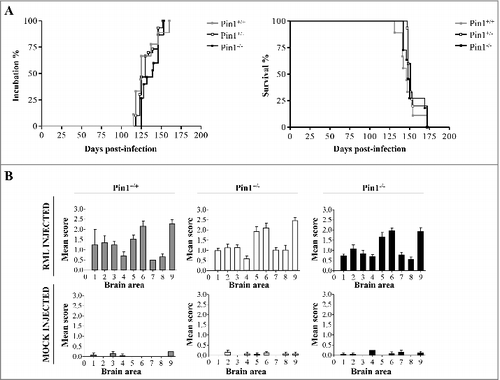
To assess whether different expression levels of Pin1 might have exerted any influence on prion propagation in vivo, we challenged intracerebrally Pin1+/+, Pin1+/− and Pin1−/− animals with RML prion strain. Regardless of their genetic background, all RML injected mice succumbed to prion disease with an incubation time of 129.78 ± 5, 130.8 ± 2.98 and 134.33 ± 2.9 days and a survival time of 147.44 ± 3.84, 154.27 ± 2.43 and 152.82 ± 3.64 days (mean ± Standard Error of the Mean, SEM) for Pin1+/+, Pin1+/− and Pin1−/− mice, respectively ( and ). Statistical analysis confirmed that neither the incubation time (p = 0.9881, Logrank test) nor the survival time (p = 0.4023, Logrank test) reached statistical significance among groups. None of the animals injected with mock inocula developed prion disease and their brains were used as control.
Table 1. Summary of the inoculation and main clinical findings in Pin1+/+, Pin1+/− and Pin1−/− mice. This table summarizes the experimental groups of animals challenged with RML or Mock. Attack rate, incubation and survival times (Mean ± SEM) of injected mice are reported.
Figure 2. Effect of different Pin1 expression levels on incubation time, survival time and spongiform changes of RML injected mice. (A) Incubation time (period between prion inoculation and the appearance of clinical signs) and survival time (period between inoculation and the day of sacrifice) were not statistically different (Logrank test) between groups of RML injected animals. (B) The lesion profiles were determined on H&E stained sections, by scoring the vacuolar changes in nine standard brain regions: 1 = dorsal medulla, 2 = cerebellar cortex, 3 = superior colliculus, 4 = hypotalamus, 5 = thalamus, 6 = hippocampus, 7 = septum, 8 = retrosplenial and adjacent motor cortex, 9 = cingulated and adjacent motor cortex. Double-tailed unpaired t-test (Mann-Whitney test) confirmed that these differences did not reach statistical significance.
Prion depositions, spongiform changes and glial activation were similar between groups of RML injected animals
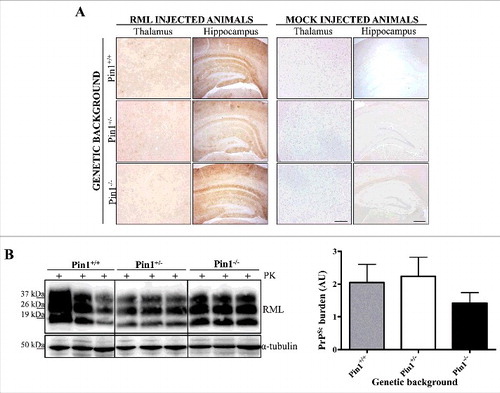
Severe spongiform changes were found in all groups of RML injected mice (Pin1+/+, Pin1+/− and Pin1−/−), especially confined to the thalamus, hippocampus and cingulated and adjacent motor cortex (). Few vacuoles were detected in the remaining areas of the brain, including cerebellum and medulla. Widespread and synaptic pattern of PK-resistant PrP deposits were observed in all infected animals, prevalently affecting the hippocampus, thalamus, cerebellar cortex and retrosplenial cortex (), with occasional accumulations in septum and striatum. Prion accumulation was accompanied by severe astroglial activation, mainly found in retrosplenial cortex, hippocampus and thalamus (Supplementary Figure 1A). Strong microglial activation was also observed in the hippocampus, but was very faint in retrosplenial cortex and thalamus (Supplementary Figure 1B). These alterations were similar in all RML injected mice, regardless of their genetic background for Pin1.
Figure 3. Neuropathological and biochemical analysis. (A) Immunohistochemical analysis showed no differences in PK-resistant PrP deposition throughout the brain of RML injected animals. Representative pictures of hippocampus and thalamus belonging to RML or mock injected mice are shown. As expected, no PK-resistant PrP deposits were found in the brain of mock injected animals. Sections were immunostained with the 6H4 antibody after PK digestion. Scale bar related to all hippocampal pictures: 60 µm. Scale bar related to all thalamic pictures: 40 µm. (B) Biochemical analysis showed similar amount of PrPres accumulation in the brain of all RML injected mice. Samples were immunoblotted with the 6D11 antibody after PK digestion. Densitometric analysis confirmed the lack of statistically significant differences (t-test). Graphs indicate the mean while bars represent SEM.
Prion inoculations did not exacerbate tau pathology in knock-out and hemizygous mice for Pin1
Considering the fact that Pin1 seems to protect against neurodegeneration and that its depletion increases neuron vulnerability and causes tau pathologies in knock-out animals at around 9–14 months of age [Citation35], we assessed whether RML inoculation might exacerbate tau pathology in Pin1−/− or Pin1+/− animals at terminal stage of prion disease (4–5 months after the inoculation). Immunohistochemical analysis revealed the complete lack of hyperphosphorylated tau deposits in the brain of Pin1−/−, Pin1+/− and Pin1+/+ animals sacrificed at around 6–7 months of age. Similarly, tau deposits were not detected in the brain of all mock-inoculated mice. Brain samples collected from a transgenic mouse model of tauopathy were used as positive controls for immunohistochemical analysis and confirmed the presence of abundant hyperphosphorylated tau (Supplementary Figure 2).
Biochemical analysis revealed no differences in PK-resistant PrP among groups of RML injected animals
Biochemical analysis of RML injected mice showed the presence of PK-resistant PrP typically characterized by a predominance of the monoglycosylated band. Densitometric analysis confirmed the lack of statistical differences in PrPres amount between Pin1+/+, Pin1+/− and Pin1−/− animals (). To assess whether reduced levels of Pin1 or its depletion might have affected the biochemical features of the RML prion strain, we have performed PK-resistant assays by treating samples with increasing concentration of PK (from 50 μg/mL to 400 μg/mL) and subsequently verified the remaining amount of PK-resistant PrP by Western blotting. Each animal accumulated PrP whose PK resistance was totally comparable to that of the other animal groups (Supplementary Figure 3), thus indicating that either the biochemical or the biophysical properties of the RML were retained, regardless of the genetic background of the animals for Pin1.
Prion infection did not alter Pin1 expression levels in the brain of mice sacrificed at terminal stage of the disease
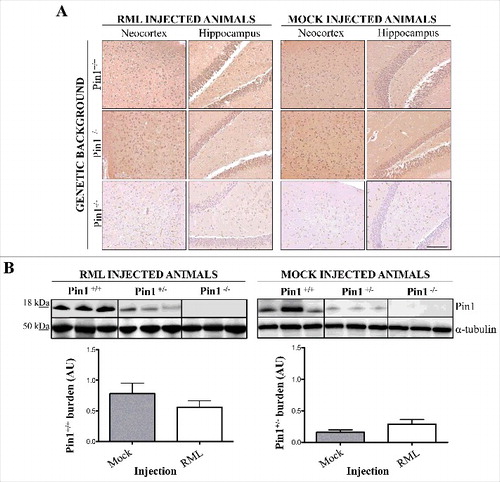
Brains of Pin1+/+, Pin1+/− and Pin1−/− animals injected with RML prions (or mock inoculum) were collected at terminal stage of the disease. Immunohistochemical and biochemical analysis were performed to verify whether prion infection could have modified the expression level of Pin1. In particular, immunohistochemical evaluations revealed the presence of synaptic and widespread Pin1 signal throughout the whole brain of Pin1+/+ and Pin1+/− animals. As expected, the signal was completely missing in Pin1−/− mice. shows representative pictures of neocortex and hippocampus of Pin1+/+, Pin1+/− and Pin1−/− animals injected with RML or mock inocula, and shows that Pin1 immunoreaction is similar in all groups of animals, regardless of the inoculum. Biochemical and densitometric analysis performed on the same samples confirmed that Pin1 expression levels were similar in all RML or mock injected mice, thus highlighting the fact that prion infection did not modify the expression level of Pin1 in a statistically significant manner ().
Figure 4. Neuropathological and biochemical analysis of Pin1 expression. (A) Immunohistochemical analysis showed similar Pin1 immunoreactivity in the brain of Pin1+/+ and Pin1+/− animals injected with RML or mock. Representative pictures of neocortex and hippocampus are shown. No signal was detected in the brain of Pin1−/− animals. Scale bar, 10 µm. (B) Biochemical and densitometric analysis confirmed the lack of statistically significant differences in the total expression level of Pin1 in RML or mock injected (Pin1+/+ and Pin1+/−) mice (t-test). Graphs indicate the mean while bars represent SEM.
Discussion
Neurodegenerative diseases share a common pathological hallmark represented by the accumulation of misfolded proteins in the brain. Indeed, extracellular deposits of amyloid β (Aβ) and intraneuronal accumulation of hyperphosphorylated tau forming neurofibrillary tangles (NFT) are found in AD, α-synuclein inclusions are typical of PD while prion aggregates are found in prion disorders. The exact mechanisms leading to the formation of these aggregates is still unknown, but recent evidence suggests that these insoluble species might have a neuroprotective function by sequestering oligomeric and toxic intermediates [Citation52].
Different causative mechanisms have been proposed to explain the phenomenon of protein aggregation, including abnormal nitration, ubiquitination and phosphorylation of physiological protein. Abnormal phosphorylation of physiological proteins might impair their functions and induce the formation of post-translationally modified proteins more prone to misfolding and aggregation. For examples, tau, neurofilaments (NF) and amyloid precursor protein (APP) are proteins constantly subjected to physiological phosphorylation. Once phosphorylated these proteins interacts with other proteins and might undergo to Pin1 mediated cis-trans isomerization. Dysregulation of both phosphorylation and Pin1 activity might alter the equilibrium between cis and trans protein conformations leading to a predominant production of the pathogenic cis isoforms. This impairment has been associated with NFTs and amyloid plaques formation in AD. Recent studies have suggested that dysregulation of Pin1 activity and abnormal phosphorylation of α-synuclein and p53 are involved in Parkinson's disease and Huntington's disease, respectively [Citation49,Citation51,Citation53].
In this work, we have analysed whether Pin1 has any pathological role also in prion diseases. Indeed, it has been reported that the phosphorylation of the residue Ser-43 at the N-terminal of PrPC might favour its misfolding into the abnormal conformation (PrPSc) [Citation20]. Therefore, we wondered whether Pin1 might take part in this process thus sustaining the phenomenon of prion formation and propagation in the brain. To analyse whether Pin1 and PrPC interact in prion diseases, we have performed in vitro and in vivo experiments aimed at verifying whether reduced levels of Pin1 or even its depletion could affect the clinical and neuropathological course of prion diseases in mice on wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) background for Pin1. Initially, we evaluated whether all genetically modified animals (Pin1+/+, Pin1+/−, Pin1−/−) expressed similar levels of PrPC, the substrate for prion propagation. Our results show that regardless of their genetic background the brains of all the animals expressed similar level of PrPC. Based on these premises, we performed preliminary PMCA experiments to assess whether different levels of Pin1 could affect the amplification efficiency of the RML prion strain in vitro. We have spiked RML (from 10−5 to 10−9) in brain homogenates of Pin1+/+, Pin1+/− and Pin1−/− mice. One round of amplification was able to efficiently amplify all these dilutions thus suggesting that, under these experimental conditions, different expression levels of Pin1 did not affect the RML prions replication properties. Although these results failed to provide any evidence that Pin1 could take part in the process of PrPC misfolding, we decided to perform in vivo studies because PMCA is an artificial system for prion amplification and there are no physiological processes (e.g. protein-to-protein interaction, presence of chaperones or foldases which are known to assist the folding of PrPC, presence of phosphatases, kinases, etc.) that can finely take part and regulate the intricate process of PrPC misfolding and aggregation. Therefore, we performed intracerebral inoculations of RML prions or mock inocula in the striatum of Pin1+/+, Pin1+/− and Pin1−/− mice. As already observed in the in vitro experiment, regardless of their genetic background, all RML inoculated animals succumbed to prion disease (with 100% attack rate) with incubation and survival times comparable and typical of the RML prion strain. After sacrifice, the brains of these animals were collected and analyzed biochemically and immunohistochemically to verify whether the genetic background for Pin1 might have modified the neuropathological changes typically associated to RML. In particular, immunohistochemical analysis revealed synaptic PK-resistant PrP deposition throughout the brain accompanied by severe astroglial and microglial activation. These alterations were comparable among the different groups of mice. Similarly, spongiform changes severely affected the thalamus, hippocampus and the cingulated and adjacent motor cortex of all groups of animals. Biochemically all the animals accumulated high levels of PK-resistant PrP characterized by the typical glycoform profile of the RML prion strain (predominance of the monoglycosylated band of PrP). Compelling evidences suggest that prions can change their properties in response to changes in the environment of propagation [Citation54], in our case represented by different expression levels (or even its depletion) of Pin1. This phenomenon is not surprising and has been already observed after pharmacological treatment of cell cultures [Citation55] or infected animals [Citation56,Citation57]. After PK-digestion we found that the biochemical properties of the RML were retained in all animal groups further sustaining that, under our experimental conditions, different levels of Pin1 did not alter the process of prion propagation in vivo.
We also verified whether the injection of RML might have affected the expression of Pin1 in animals sacrificed at terminal stage of the disease (especially in Pin1+/+ and Pin1+/− mice). Neither biochemical nor immunohistochemical results showed differences in Pin1 expression levels in the brain of RML or mock inoculated animals. Densitometric analysis showed a slight reduction of Pin1 in the brain of RML injected Pin1+/+ animals compared to mock inoculated mice but this difference did not reach statistical significance. Similarly, no differences in Pin1 expression levels were found in the brains of Pin1+/− animals.
Taken together these data suggest that Pin1 does not significantly interact with PrPC in prion diseases and other pathological mechanisms should be considered. Since PrPC is rich in prolines, and considering that two proline substitutions (P102L and P105L) cause a genetic form of prion disease known as GSS [Citation58,Citation59], other than Pin1, different peptidylprolyl cis-trans isomerases (PPIases) may play a role in PrPC misfolding. Little is known about the mechanisms that induce PrPC to PrPSc conversion but many enzymatically driven processes might take part in this process. Finally we assessed whether the lack of Pin1 might have exacerbated tau pathology (which spontaneously occurs in these animals at around 9–14 months of age) in RML injected mice. Specific immunohistochemical analysis revealed that RML inoculated Pin1+/− or Pin−/− mice did not accumulated hyperphosphorylated tau in the brain. Thus, although lacking Pin1 (which is known to have neuroprotective function), tau accumulation was not exacerbated by prion inoculation.
This work provides the first evidence that, in animals experimentally infected with the RML prion strain, the impairment of Pin1 activity or its total depletion did not clearly and significantly alter the clinical and neuropathological changes associated to this disease. It would be worthy to extend the analysis to human prions collected from patients with different forms of the disease. Indeed, considering the phenotypic variability of human prion pathologies, it might still be possible that Pin1 can be involved only in some of them. If this were the case, Pin1 could be considered as disease-specific therapeutic target for such devastating diseases.
Materials and methods
Ethics statement
B6.129P2-Pin1tm1Tuc mice were obtained from The Jackson Laboratory, Bar Harbor, ME. Mice were housed in groups of 2–5 animals in individually ventilated cages, daily fed and water provided ad libitum. Lighting was on an automatic 12 h basis. Regular veterinary care was daily performed for assessment of animal health. Animal facility is licensed and inspected by the Italian Ministry of Health. Current animal husbandry and housing practices comply with the Council of Europe Convention ETS123 (European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes; Strasbourg, 18.03.1986), Italian Legislative Decree 116/92 (Gazzetta Ufficiale della Repubblica Italiana, 18th February 1992) and with the 86/609/EEC (Council Directive of 24 November 1986 on the approximation of laws, regulations and administrative provisions of the Member States regarding the protection of animals used for experimental and other scientific purposes). The study, including its Ethics aspects, was approved by the Italian Ministry of Health.
Genetic analysis
Genetic analyses were performed to identify animals with wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) background for Pin1. To this aim, genomic DNA was extracted from ear biopsies using the NucleoSpin Tissue kit (Macherey-Nagel) and analyzed by PCR. Two pairs of primers were used: 1.2 L (5′-AGCACCCGATCCTGTTCTGCAA-3′) / WILD 1.2A (5′-AAGGGATTAGAAGCAAGATTCG-3′) that specifically amplify the wild-type allele, and 1.2 L / START2 (5′-CAGAGGCCACTTGTGTA-3′), specific for the Pin1 knock-out allele [Citation60]. An initial denaturation at 95°C for 5 minutes was followed by 35 cycles of amplification (95°C for 1 min, 60°C for 1 min, 72°C for 1 min) and 10 minutes extension at 72°C. PCR products were resolved by agarose gel electrophoresis.
Animal groups and intracerebral inoculations
Eight-week old animals were anesthetized with 2,2,2-tribromoethanol i.p-administered (100 µL/10 g) and stereotaxically inoculated in the striatum using these coordinates: −0.5 caudal, + 2 lateral, + 3.5 depth. All surgical procedures were performed under sterile conditions. Inoculations were performed in 3 groups of animals generated on wild-type (Pin1+/+), hemizygous (Pin1+/−) or knock-out (Pin1−/−) background for Pin1. In particular, 20 µl of 10% (weight/volume) RML brain homogenate was prepared from a pool of terminally RML-sick CD1 mouse brain and injected in Pin1+/+ (n = 9), Pin1+/− (n = 15) and Pin1−/− (n = 11) mice. Similarly, 20 µl of 10% (weight/volume) mock homogenate was obtained from a pool of healthy CD1 mouse brains and was inoculated in Pin1+/+ (n = 6), Pin1+/− (n = 10) and Pin1−/− (n = 6) mice.
Behavioral testing
Incubation time was calculated as the period between the day of prion intracerebral inoculation and the day of appearance of at least two of the following clinical signs: piloerection and kyphosis, decreased locomotor activity and curiosity, uncoordinated movements, awkwardness of movement on a metal grid, inability to hang on inverted grid. Terminally sick animals, unable to ambulate and to eat, were sacrificed and survival time was calculated as the period between RML intracerebral inoculation and the day of euthanasia. Mock injected animals were sacrificed along with RML injected animals and used as controls.
Euthanasia and brains collection
RML-injected mice were sacrificed at the agonic stage of disease along with age-matched mock-injected animals that were used as negative controls. All mice were euthanized by 2,2,2-tribromoethanol i.p.-injected and cervical dislocations were finally performed. Brains were harvested and half used for biochemical studies and the other half processed for histological analysis.
Western blot analysis
Ten percent (weight/volume) brain homogenates from frozen tissues were prepared in lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.5% Sodium Deoxycholate, 10 mM Tris-HCl, pH 7.4). Samples were centrifuged at 4°C, 2000 x g, 1 minute in order to remove cellular debris. Supernatant was then collected and stored at −80°C for further use. Twenty μL of supernatant was heated at 100°C for 10 minutes and loaded into 12% Bolt Bis-Tris Plus gels. Proteins were fractionated by means of SDS-PAGE under reducing conditions and transferred onto Polyvinylidene difluoride (PVDF) membrane and incubated with 5% (wt/vol) dry nonfat milk in TBST, 0.05%-Tween20 (prepared in Tris-HCl) for 1 h at room temperature with shaking. Membranes were finally incubated with antibodies against PrP (Clone 6D11, Covance), Pin1 (Clone G-8, Santa Cruz) and α-Tubulin (Sigma). Blots were developed using the ECL Prime detection system (Amersham) and visualized using a G:BOX Chemi Syngene system.
PK digestion
Before PrP analysis, samples were digested with proteinase K (PK) (Invitrogen) [100 μg/mL] for 1 hour at 37°C. Digestion was stopped by the addition of LDS-PAGE loading buffer (Invitrogen) and subjected to Western blot analysis with 6D11 (0.2 µg/mL, Covance).
PK resistance assay
Five aliquots (20 μL) of brain homogenates were digested with increasing concentration of PK: 50, 100, 200, 300, 400 µg/mL) and immunoblotting was performed using the 6D11 antibody. The PK-resistant signal was quantified and normalized using ImageJ software (1.48v).
Neuropathological analysis
Brains were fixed in Alcolin (Diapath), dehydrated and embedded in paraplast. Seven-micrometer thick serial sections were stained with hematoxylin-eosin (H&E) or immunostained with monoclonal antibodies to PrP (6H4, Prionics), Pin1 (Pin1, Santa Cruz Biotechnology), hyperphosphorylated tau (AT8, Thermo Scientific Pierce Antibodies), polyclonal antibodies to astrocyte activation (GFAP, Dako) and microglial activation (Iba-1, Abcam). Before PrP immunostaining the sections were sequentially treated with proteinase K (VWR) (10 μg/mL, 5 min) and guanidine isothiocyanate (Carlo Erba) (3 M, 20 min), and non-specific binding was prevented using ARK kit (Dako). Immunoreactions were visualized using 3–3′-diaminobenzidine (DAB, Dako) as chromogen. Spongiform profiles were determined on H&E-stained sections, by scoring the vacuolar changes in nine standard gray matter areas (1): dorsal medulla (2), cerebellar cortex (3), superior culliculus (4), hypothalamus (5), thalamus (6), hippocampus (7), septum (8), retrosplenial and adjacent motor cortex and (9) cingulated and adjacent motor cortex, as previously described [Citation9]. Brains collected from genetically modified mice to express the human tau with P301L mutation (JPNL3) were used as positive control for tau immunostaining [Citation61].
PMCA procedures
PMCA was performed as previously described [Citation62]. Briefly, brain samples obtained from mice on wild-type (Pin1+/+), hemizygous (Pin1+/−) and knock-out (Pin1−/−) background for Pin1 were used as substrates. The animals were intracardially perfused with PBS (containing 0.5 mM EDTA, Sigma) and brains harvested and homogenized at 10% (weight/volume) in conversion buffer (PBS containing 150 mM sodium chloride and 1% Triton X-100) with the addition of protease inhibitors (Roche). To evaluate the effect of Pin1 on RML amplification, RML (Rocky Mountain Laboratories) prion strain was spiked at different dilutions (from 10−5 to 10−9) in each substrate (Pin1+/+, Pin1+/−, Pin1−/−). Samples were then transferred in 0.2 mL tubes, positioned on an adaptor placed on the plate holder of a microsonicator (Misonix, Model S3000) and subjected to 96 cycles of PMCA. Each cycle (referred to as PMCA round) consisted of 29 min and 40 sec of incubation at 37–40°C followed by 20 sec of sonication set at potency of 260–270 Watt. After one round of PMCA, an aliquot of the amplified material was diluted 10-folds into fresh substrate and a further PMCA was performed following the same procedure. To increase PMCA efficiency, teflon beads (n = 3) were added to the samples before each round of amplification. To avoid samples cross-contamination between each round, thorough decontamination of instruments and equipment was performed using 2N sodium hydroxide (NaOH) or 4M guanidine hydrochloride (Gdn-HCl).
Statistical analysis
Densitometric analysis were carried out using ImageJ. Statistical analysis was performed using the GraphPad-Prism 5.0 software. Kaplan-Meier survival curves were plotted and differences in incubation or survival times between groups were compared using the Logrank test. Densitometric analysis of immunoblot data were performed using a double-tailed unpaired t-test (Mann-Whitney test).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
suppl_mat.zip
Download Zip (3.2 MB)Acknowledgments
We are grateful to prof. Giannino Del Sal (University of Trieste) for providing a colony of transgenic mice with different genetic background for Pin1.
Additional information
Funding
Related Research Data
References
- Prusiner SB. Molecular structure, biology, and genetics of prions. Adv Virus Res. 1988;35:83–136. doi:10.1016/S0065-3527(08)60709-5
- Prusiner SB. Molecular biology of prion diseases. Science. 1991;252(5012):1515–22. doi:10.1126/science.1675487
- Prusiner SB. Molecular biology and genetics of prion diseases. Philos Trans R Soc Lond B Biol Sci. 1994;343(1306):447–63. doi:10.1098/rstb.1994.0043
- Prusiner SB. Shattuck lecture-neurodegenerative diseases and prions. N Engl J Med. 2001;344(20):1516–26. doi:10.1056/NEJM200105173442006
- Tanaka M, Chien P, Naber N, et al. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428(6980):323–8. doi:10.1038/nature02392
- Bartz JC. Prion strain diversity. Cold Spring Harb Perspect Med. 2016;6(12):1–14. doi:10.1101/cshperspect.a024349.
- Dickinson AG, Meikle VM. Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol Gen Genet. 1971;112(1):73–9. doi:10.1007/BF00266934
- Fraser H. Diversity in the neuropathology of scrapie-like diseases in animals. Br Med Bull. 1993;49(4):792–809. doi:10.1093/oxfordjournals.bmb.a072647
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78(3):301–11. doi:10.1016/0021-9975(68)90006-6
- Khalili-Shirazi A, Summers L, Linehan J, et al. PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J Gen Virol. 2005;86(Pt 9):2635–44. doi:10.1099/vir.0.80375-0
- McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35(1):57–62. doi:10.1016/0092-8674(83)90207-6
- Peretz D, Williamson RA, Legname G, et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron. 2002;34(6):921–32. doi:10.1016/S0896-6273(02)00726-2
- Wolf H, Grassmann A, Bester R, et al. Modulation of Glycosaminoglycans Affects PrPSc Metabolism but does not block PrPSc Uptake. J Virol. 2015;89(19):9853–64. doi:10.1128/JVI.01276-15
- Zhou Z, Xiao G. Conformational conversion of prion protein in prion diseases. Acta Biochim Biophys Sin (Shanghai). 2013;45(6):465–76. doi:10.1093/abbs/gmt027
- Wang F, Yang F, Hu Y, et al. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry. 2007;46(23):7045–53. doi:10.1021/bi700299h
- Silva JL, Cordeiro Y. The “Jekyll and Hyde” Actions of Nucleic Acids on the Prion-like Aggregation of Proteins. J Biol Chem. 2016;291(30):15482–90. doi:10.1074/jbc.R116.733428
- Deleault NR, Harris BT, Rees JR, et al. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104(23):9741–6. doi:10.1073/pnas.0702662104
- Supattapone S. Synthesis of high titer infectious prions with cofactor molecules. J Biol Chem. 2014;289(29):19850–4. doi:10.1074/jbc.R113.511329
- Kim NH, Choi JK, Jeong BH, et al. Effect of transition metals (Mn, Cu, Fe) and deoxycholic acid (DA) on the conversion of PrPC to PrPres. FASEB J. 2005;19(7):783–5. doi:10.1096/fj.04-2117fje
- Giannopoulos PN, Robertson C, Jodoin J, et al. Phosphorylation of prion protein at serine 43 induces prion protein conformational change. J Neurosci. 2009;29(27):8743–51. doi:10.1523/JNEUROSCI.2294-09.2009
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–3. doi:10.1038/35081095
- Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 2002;25(8):390–4. doi:10.1016/S0166-2236(02)02195-1
- Deleault NR, Walsh DJ, Piro JR, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A. 2012;109(28):E1938–46. doi:10.1073/pnas.1206999109
- Torrent J, Marchal S, Tortora P, et al. High pressure, an alternative approach to understand protein misfolding diseases. Cell Mol Biol (Noisy-le-grand). 2004;50(4):377–85.
- Xiong LW, Raymond LD, Hayes SF, et al. Conformational change, aggregation and fibril formation induced by detergent treatments of cellular prion protein. J Neurochem. 2001;79(3):669–78. doi:10.1046/j.1471-4159.2001.00606.x
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425(6959):717–20. doi:10.1038/nature01979
- Supattapone S. Prion protein conversion in vitro. J Mol Med (Berl). 2004;82(6):348–56. doi:10.1007/s00109-004-0534-3
- Deleault NR, Geoghegan JC, Nishina K, et al. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem. 2005;280(29):26873–9. doi:10.1074/jbc.M503973200
- Geoghegan JC, Valdes PA, Orem NR, et al. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem. 2007;282(50):36341–53. doi:10.1074/jbc.M704447200
- Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380(6574):544–7. doi:10.1038/380544a0
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8(11):904–16. doi:10.1038/nrm2261
- Jager M, Nguyen H, Dendle M, et al. Influence of hPin1 WW N-terminal domain boundaries on function, protein stability, and folding. Protein Sci. 2007;16(7):1495–501. doi:10.1110/ps.072775507
- Liou YC, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci. 2011;36(10):501–14. doi:10.1016/j.tibs.2011.07.001
- Lin CH, Li HY, Lee YC, et al. Landscape of Pin1 in the cell cycle. Exp Biol Med (Maywood). 2015;240(3):403–8. doi:10.1177/1535370215570829
- Liou YC, Sun A, Ryo A, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424(6948):556–61. doi:10.1038/nature01832
- Nakamura K, Greenwood A, Binder L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. 2012;149(1):232–44. doi:10.1016/j.cell.2012.02.016
- Driver JA, Lu KP. Pin1: a new genetic link between Alzheimer's disease, cancer and aging. Curr Aging Sci. 2010;3(3):158–65. doi:10.2174/1874609811003030158
- Lee TH, Pastorino L, Lu KP. Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev Mol Med. 2011;13:e21. doi:10.1017/S1462399411001906
- Angelucci F, Hort J. Prolyl isomerase Pin1 and neurotrophins: a loop that may determine the fate of cells in cancer and neurodegeneration. Ther Adv Med Oncol. 2017;9(1):59–62. doi:10.1177/1758834016665776
- Ma LL, Yu JT, Wang HF, et al. Association between cancer and Alzheimer's disease: systematic review and meta-analysis. J Alzheimers Dis. 2014;42(2):565–73.
- Musicco M, Adorni F, Di Santo S, et al. Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology. 2013;81(4):322–8. doi:10.1212/WNL.0b013e31829c5ec1
- Ayala G, Wang D, Wulf G, et al. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63(19):6244–51.
- Lin FC, Lee YC, Goan YG, et al. Pin1 positively affects tumorigenesis of esophageal squamous cell carcinoma and correlates with poor survival of patients. J Biomed Sci. 2014;21:75. doi:10.1186/s12929-014-0075-1
- Rudrabhatla P. Regulation of neuronal cytoskeletal protein phosphorylation in neurodegenerative diseases. J Alzheimers Dis. 2014;41(3):671–84.
- Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci. 2014;7:42.
- Pastorino L, Sun A, Lu PJ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440(7083):528–34. doi:10.1038/nature04543
- Ghosh A, Saminathan H, Kanthasamy A, et al. The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J Biol Chem. 2013;288(30):21955–71. doi:10.1074/jbc.M112.444224
- Martin LJ, Pan Y, Price AC, et al. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26(1):41–50. doi:10.1523/JNEUROSCI.4308-05.2006
- Ryo A, Togo T, Nakai T, et al. Prolyl-isomerase Pin1 accumulates in lewy bodies of parkinson disease and facilitates formation of alpha-synuclein inclusions. J Biol Chem. 2006;281(7):4117–25. doi:10.1074/jbc.M507026200
- Sorrentino G, Comel A, Mantovani F, et al. Regulation of mitochondrial apoptosis by Pin1 in cancer and neurodegeneration. Mitochondrion. 2014;19(Pt A):88–96. doi:10.1016/j.mito.2014.08.003
- Grison A, Mantovani F, Comel A, et al. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc Natl Acad Sci U S A. 2011;108(44):17979–84. doi:10.1073/pnas.1106198108
- Soto C, Estrada LD. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65(2):184–9. doi:10.1001/archneurol.2007.56
- Anderson JP, Walker DE, Goldstein JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739–52. doi:10.1074/jbc.M600933200
- Weissmann C, Li J, Mahal SP, et al. Prions on the move. EMBO Rep. 2011;12(11):1109–17. doi:10.1038/embor.2011.192
- Li J, Browning S, Mahal SP, et al. Darwinian evolution of prions in cell culture. Science. 2010;327(5967):869–72. doi:10.1126/science.1183218
- Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5(11):e1000673. doi:10.1371/journal.ppat.1000673
- Bian J, Kang HE, Telling GC. Quinacrine promotes replication and conformational mutation of chronic wasting disease prions. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(16):6028–33. doi:10.1073/pnas.1322377111
- Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 1989;338(6213):342–5. doi:10.1038/338342a0
- Yamazaki M, Oyanagi K, Mori O, et al. Variant Gerstmann-Straussler syndrome with the P105L prion gene mutation: an unusual case with nigral degeneration and widespread neurofibrillary tangles. Acta Neuropathol. 1999;98(5):506–11. doi:10.1007/s004010051116
- Fujimori F, Takahashi K, Uchida C, et al. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochem Biophys Res Commun. 1999;265(3):658–63. doi:10.1006/bbrc.1999.1736
- Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging. 2009;30(1):1–13. doi:10.1016/j.neurobiolaging.2007.05.010
- Morales R, Duran-Aniotz C, Diaz-Espinoza R, et al. Protein misfolding cyclic amplification of infectious prions. Nat Protoc. 2012;7(7):1397–409. doi:10.1038/nprot.2012.067