
ABSTRACT
The yeast Saccharomyces cerevisiae has proven to be a useful model system to investigate the mechanism of prion generation and inheritance, to which studies in Sup35 made a great contribution. Recent studies demonstrated that ‘protein misfolding and aggregation’ (i.e. amyloidogenesis) is a common principle underlying the pathogenesis of neurodegenerative diseases including prion, amyotrophic lateral sclerosis (ALS), Perkinson’s (PD), Alzheimer’s (AD) diseases and polyglutamine (polyQ) diseases such as spinocerebellar ataxia (SCA) and Hantington’s disease (HD). By these findings, the yeast has again been drawing increased attention as a useful system for studying neurodegenerative proteinopathies. So far, it has been reported that proteolytic cleavage of causative amyloidogenic proteins might affect the pathogenesis of the respective neurodegenerative diseases. Although those reports provide a clear phenomenological description, in the majority of cases, it has remained elusive if proteolysis is directly involved in the pathogenesis of the diseases. Recently, we have demonstrated in yeast that proteolysis suppresses prion generation. The yeast-based strategy might make a breakthrough to the unsolved issues.
Introduction
During the process of amyloidogenesis, proteins are converted from a soluble form into an insoluble aggregated form and assembled into highly ordered fibers with antipararel β-sheet structure. The abnormal aggregates act as self-propagating infectious agents. Such transitions can give rise to pathological conditions ranging from neurodegenerative disorders to systemic amyloidoses. One of the representative examples, the polyQ disease, is a group of neurodegenerative disorders caused by expanded cytosine-adenine-guanine (CAG) repeats encoding an abnormally long polyQ tract in the respective proteins. PolyQ diseases include HD, which is a fatal neurodegenerative disorder characterized by motor dysfunction, cognitive deficits and psychosis. HD results from the expansion of polyQ repeats in the Huntintin (HTT) protein due to mutations in HTT gene. Second example, ALS, is one of the common neurodegenerative disease and causes muscle weakness, spasticity, paralysis and atrophy due to the degeneration of both upper and lower motor neurons. Trans-activation response element (TAR) DNA-binding protein of 43 kDa (TDP-43) has been reported to be the amyloidogenic component of intracellular inclusions in the affected regions of the spinal cord and brain in patients with ALS [Citation1]. Third example, systemic amyloidoses include AD and Creutzfeldt-Jakob disease (CJD). Amyloidosis due to the accumulation of amyloid-beta (Aβ) released from the amyloid precursor protein (APP) was initially regarded as a feature of neurodegenerative alterations seen in the brain of AD patients. Extracellular senile plaques, pathophysiological feature of AD, are composed of a dense core of amyloid fibrils associated with degenerating neurites, astrocytes and astrocytic processes. In humans, prion-related protein (PrP) is responsible for CJD, Fatal Familial Insomnia (FFI), Gertsmann-Strӓussler-Scheinker syndrome (GSS), and Kuru [Citation2]. These Prion diseases are a group of neurodegenerative diseases caused by the abnormal conversion of the normal prion protein to pathogenic isoform, which stands for proteinaceous infectious particle (PRION) [Citation2]. Remarkably, proteins that can form infectious amyloid conformations are widespread in lower eukaryotes [Citation3]. Yeast (Saccharomyces cerevisiae) prions, depending on genetics and the environment, act as an epigenetic element of inheritance associated with a genetic phenotype that may be harmful or beneficial [Citation3]. While several yeast prions have been identified, the yeast [PSI+] prion, which results from the misfolded and amyloid form of the translation termination factor (Sup35), is the best-studied prion and provides a useful model system for studying protein aggregation diseases.
Amyloids formed by these amyloidogenic proteins APP, HTT, PrP, TDP-43, Sup35 are characterized in common by the cross-beta-sheet structure, congo red birefringence, and formation of insoluble fibrils. Although protease resistance is a hallmark of the amyloids, there are many reports showing that amyloid formation is affected positively and negatively by the proteolysis. In this EXTRA VIEWS, we will focus on the role of proteolytic cleavage of amyloidogenic proteins (HTT, APP, PrP, Sup35, TDP-43) for the generation of abnormal amyloids and the development of amyloidoses.
Proteolysis Promotes amyloidogenesis of HTT, TDP-43 and APP
The representative examples that proteolysis promotes amyloidogenesis are HTT and TDP-43. HD is caused by an abnormal polyQ expansion at the extreme N-terminus of the protein HTT. The cleavage of HTT is a crucial step in HD pathogenesis and gives rise to release of smaller N-terminal fragments which contain the polyQ stretch and are toxic to neurons (). Released polyQ-containing fragments from HTT have been detected in mouse and human HD tissues. Several proteases including caspase, calpain, cathepsin, metalloproteinase, are reported to cleave HTT and inhibition of HTT cleavage reduces toxicity and aggregation [Citation4,Citation5]. However, proteases that mediate the cleavage in vivo is still enigmatic. In addition, the role of HTT cleavage in the pathogenesis of HD in vivo remains to be elucidated [Citation6].
TDP-43 is aggregated into neuronal cytoplasmic inclusions in vast majority of ALS [Citation1]. TDP-43 is a heterogeneous nuclear ribonucleoprotein (hnRNP) with two highly conserved RNA-recognition motifs (RRM) and a C-terminal glycine-rich domain with glutamine/asparagine (Q/N)-rich content (). Overexpression of TDP-43 in cultured cells and animals causes fragmentation of the protein, generating N-terminally truncated C-terminal fragments (CTFs) [Citation7,Citation8]. Several proteases including caspase and calpain are suggested to mediate C-terminal cleavage of TDP43. CTFs mislocalize from the nucleus to the cytoplasm due to removal of NLS and have a strong tendency to self-aggregate owing to C-terminal-Q/N-rich region [Citation9]. It is suggested that CTFs further serves as a seed to facilitate aggregation of full-length TDP-43 [Citation10]. However, it remains to be determined whether the TDP-43 aggregates induced by the proteolytic cleavage is the primary cause of ALS [Citation11].
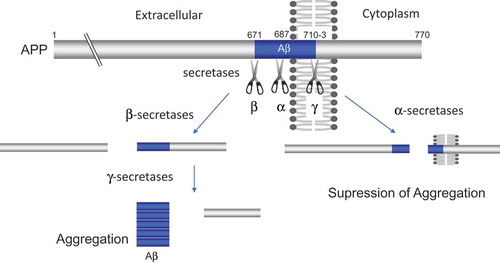
APP is one of a large number of membrane proteins that are proteolytically converted to their soluble counterparts. The Aβ, an amyloidogenic cleavage product of APP, is a critical initiator that triggers the progression of AD via misfolding and aggregation. In the amyloidogenic pathway, Aβ production results from the sequential proteolytic cleavage of APP by β- and γ-secretases (Figure 3). In mammals, beta-site APP cleaving enzyme 1 and 2 (BACE1 and BACE2) and transmembrane aspartate-type proteases are the β-secretase [Citation12], and γ-secretase is a complex composed of presenilins (PSEN1 or PSEN2), Nicastrin, Presenilin enhancer 2 (PEN2), and anterior pharynx defective 1 (APH-1) [Citation13]. Studies with knockout mice confirmed that β- and γ-secretase activities are responsible for the pathogenesis of AD.
Proteolysis suppresses amyloidogenesis of APP and PRP
In sharp contrast to the above described β- and γ-secretases, proteolytic cleavage of APP by α-secretase suppresses amyloidogenesis. Several members of the ADAM (a Disintegirin and metalloptroteinase) family proteins including ADAM9, ADAM10 and ADAM17 possess α-secretase activity and are able to cleave APP at the α-secretase site [Citation14] (). In primary neurons, ADAM10 is the major physiological α-secretase of APP. Alpha-secretase cleaves within the Aβ sequence of APP, thereby suppressing Aβ generation. In addition, α-secretase cleavage generates non-toxic N-terminal fragment of APP (APPsα), which has been reported to have neurotrophic and neuroprotective properties [Citation15], whereas the slightly shorter form (APPsβ) generated by β-secretase has a proapoptotic function [Citation16]. A moderate neuronal overexpression of ADAM10 in mice increased the secretion of APPsα, reduced the formation of Aβ peptides, and prevented their deposition in plaques [Citation17]. In addition, a reduced level of ADAM10 is observed in platelets and cerebrospinal fluid obtained from AD patients as compared to age-matched controls [Citation18]. Furthermore, a decrease in α-secretase (81% of normal) and a large increase in β-secretase (185%) are observed in sporadic Alzheimer’s disease temporal cortex [Citation19]. Thus, α-secretase cleavage of APP is protective against Alzheimer’s disease. However, the pathogenic processes of AD that is regulated by α-secretase cleavage are mostly veiled. Further studies are required to clarify whether decrease in the α-secretase activity is directly involved in the pathogenesis of AD.
Figure 1. Proteolysis of HTT promotes aggregation of N-terminal fragments of HTT. The blue, yellow, and red columns indicate polyglutamine repeats (polyQ), HEAT repeats, and nuclear export signal (NES), respectively. The scissors indicate proteases that contribute proteolysis.
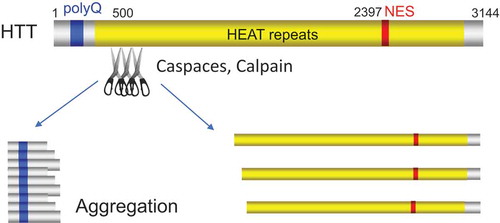
Figure 2. Proteolysis of TDP-43 promotes aggregation of C-terminal fragments (CTFs) of TDP-43. The red, yellow and blue columns indicate RNA recognition motif (RRM), nuclear localization signal (NLS) and glutamine/asparagine (Q/N)-rich, respectively.
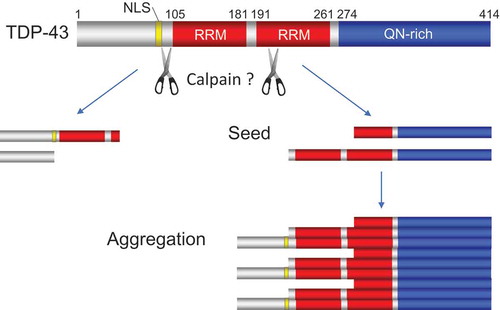
Figure 3. β- and β-secretase cleavages of APP promote aggregation of Aβ, whereas α-secretase cleavage suppresses it. The blue columns indicate amyloid-beta (Aβ).
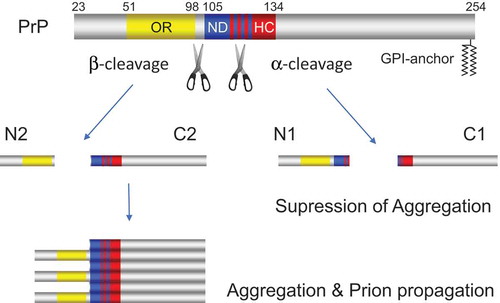
Similar observations have been reported for PrP. PrP is a membrane-anchored protein, in which the N-terminal is flexible and contains an octameric repeats region (OR), a neurotoxic domain (ND), and a hydrophobic core (HC), while the C-terminal is more structured and contains the glycosylphosphatidylinositol (GPI)-anchor for attachment to outer leaflet of membrane (). A non-pathogenic isoform of PrP (PrPC) is converted into the pathogenic isoform (PrPSc; Sc for scrapie, a prion disease of sheep) giving rise to prion diseases or transmissible spongiform encephalopathies (TSE). Prion is a fatal neurodegenerative disease resulting from sporadic or genetic etiology or exposure to infectious prions [Citation2]. In normal tissues, PrP is proteolytically cleaved mainly at positions 110/111 or 111/112 peptide bond (termed α-cleavage), yielding the N-terminally truncated membrane-attached fragment (termed C1 fragment) and releasing the soluble N-terminal fragment (termed N1 fragment) [Citation20]. In the brains of Creutzfeldt-Jakob disease patients and prion-infected cells, the cleavage of PrP takes place upstream of α-cleavage site around the end of the octapeptide repeat (OR) region (termed β-cleavage) to generate the so-called C2 and N2 fragments, indicating a pathophysiological relevance between β-cleavage and prion disease [Citation20,Citation21]. The ADAM (a Disintegirin and metalloptroteinase) family of proteases, plasmin/plasminogen and Protein kinase C are thought to contribute to α-cleavage, while the reactive oxygen species, Calpain and Cathepsin are implicated contribution to β-cleavage of PrP. The proteases responsible for the α-cleavage are still controversial [Citation22]. Interestingly, α-cleavage occurs within the neurotoxic domain (ND) of PrP. ND is a structurally essential region when PrPC changes its conformation to PrPSc and is thought to be mainly related to the property of PrPSc amyloid formation. Therefore, α-cleavage destroys the region essential for the conformational change from PrPC to PrPSc [Citation21]. In addition, α-cleavage removes the polybasic region of PrP, which is important for the first physical interaction between PrPC and PrPSc in the process of misfolding. Moreover, N1 fragment released by α-cleavage provides neuroprotective action both in vitro and in vivo and blocks a neurotoxic effect caused by Aβ (β-amyloid) related to Alzheimer’s disease [Citation23]. Furthermore, N1 fragment could more generally interferes with the toxicity produced by the β sheet structure rich oligomer, which is not limited to Aβ found in several neurodegenerative diseases. C1 fragment released by α-cleavage is unable to misfold into the pathogenic isoform PrPSc due to the loss of the neurotoxic domain and might be a dominant negative inhibitor of the conversion process [Citation20]. These data suggest that the α-cleavage of PrPc might be a defense mechanism against human prion propagation. However, it remains unknown whether the proteolysis actually suppresses prion generation in vivo and the prion disease is caused by a defect in α-cleavage [Citation22].
Figure 4. α-cleavage of PrP suppresses aggregation and prion propagation of PrP in marked contrast to β-cleavage which promotes that of PrP. The blue, yellow, and red columns indicate octameric repeats region (OR), neurotoxic domain (ND), and hydrophobic core (HC), respectively. The blue and red striped pattern shows the ND region that overlap with HC region.
Proteolysis Suppresses Prion Generation in Yeast
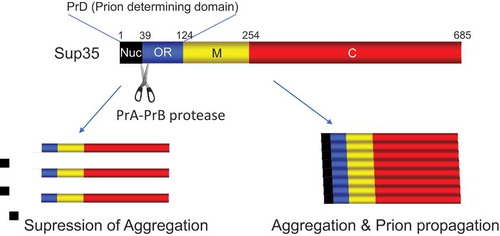
The yeast prion [PSI+] is a self-propagating amyloidogenic form of Sup353. Similar to other prions, Sup35 carries a prion-determining domain (PrD), which is required for the formation of self-perpetuating protein aggregates. PrD consists of a Q/N-rich nucleation domain and adjacent oligopeptide repeat (OR) region. Although both regions are essential for transmission of the [PSI+] prion, the QN-rich stretch mediates the initial formation of aggregates, while OR region is required for the replication and stable inheritance of these aggregates [Citation24]. Although the process of spontaneous de novo prion formation remains elusive, the process of prion induction by Sup35 overproduction is associated with the actin cytoskeletal networks and components of the endocytic/vacuolar pathway [Citation25]. Overexpression of Sup35 or its PrD gives rise to peripheral ring-like aggregates along the cell membrane. The peripheral ring then collapses and is transported to surround the vacuole [Citation26]. The cells with the ring-like aggregates generate [PSI+] prion. Assembly of the endocytic vesicles and their movement towards the endosomal/vacuolar system involves actin polymerization/depolymerization. The cytoskeleton-assisted generation of aggregates of prion-forming proteins resembles formation of aggresome, which accumulates misfolded proteins in mammalian cells. We have recently shown that proteolytic cleavage of Sup35 suppresses spontaneous de novo formation of [PSI+] prion [Citation27]. This cleavage occurs even after deletion of the SLA1 or END3 gene encoding proteins involved in actin cytoskeletal organization/endocytosis and therefore may occur before proteins are transported to the vacuole via the actin/cytoskeletal pathway. Interestingly, the PrB protease, whose conformationally altered form is found to act as another prion [β] by Wickner’s group [Citation28], mediates Sup35 cleavage at site Ala39 which is located exactly between the Q/N-rich stretch and oligopeptide repeat region, resulting in the removal of the Q/N-rich nucleation domain, critical for prion formation, from the N terminus of Sup35 (). This leads to the inhibition of spontaneous de novo formation of [PSI+]. Whereas the amyloid conformers of Sup35 in [PSI+] strains were not cleaved and preexisting [PSI+] was not eliminated by the activation of the proteases. These results indicate that the protease-mediated anti-prion system prevents prion generation but cannot cure pre-existing prions. Our results indicate that proteolysis acts as a defense against prion generation in yeast.
Figure 5. PrA-PrB-mediated cleavage of Sup35 suppresses aggregation as well as prion propagation of Sup35. The black, blue, yellow, and red columns indicate QN-rich nucleation domain (Nuc), oligopeptide repeats (OR) region, middle (M) domain, and C-terminal translation termination (C) domain, respectively.
During the course of investigating the proteolytic cleavage of Sup35, we have unexpectedly found that cleavage of Sup35 occurs only when the yeast is grown in a medium containing glucose as a carbon source (). It is interesting that the cleavage is not observed with other carbon sources such as galactose or glycerol. Yeast cells grow faster in medium containing glucose than in medium containing other carbon sources. The growth rate is tightly correlated with cytosolic pH, which is sensitive to the quality and quantity of the available carbon sources [Citation29]. In sharp contrast to the growth in glucose media, growth on alternative carbon sources results in decrease of cytosolic pH. Since proteolytic activity of PrB is optimal at the neutral pH [Citation27], it may therefore be reasonable that cleavage of Sup35 occurs only when cells are grown in glucose media. The higher growth rate in glucose media is accompanied by an increased rate of protein synthesis. This might increase the risk of misfolding of Sup35 during translation termination, for which pH-dependent cleavage of Sup35 might be prepared as a defense mechanism. Alternatively, it could also be explained by the production of ROS. It is known that yeast cells grow faster in glucose media with concomitant increase in the production of ROS. This implies that proteins actively produced in cells grown in glucose media may be exposed to the oxidative stress and undergo misfolding. PrB-mediated cleavage of Sup35 may function to remove oxidatively damaged Sup35 before formation of abnormal aggregates. In this sense, similar observation has been made by Grant’s group. They reported that autophagy system removes the oxidatively damaged Sup35 and prevents prion formation [Citation30]. However, since PrB-mediated Sup35 cleavage is observed independently of autophagy [Citation27], these systems may function redundantly or co-ordinately to prevent prion formation. On the other hand, it was also reported that even fibrils formed by Sup35, as well as another prion forming protein Ure2, can be degraded by a proteasome in an in vitro experiment using proteasome degradation assays [Citation31,Citation32]. Therefore, proteolysis not only functions as a defense against prion disease by preventing prion formation but also may function to cure prion disease by clearing pre-existing abnormal prion aggregates.
Figure 6. Sup35 is specifically cleaved by PrA-PrB proteases when cells are grown in glucose media. A GT17 [psi− pin−] cells grown in the mid log-phase were starved in YPA (1% yeast extract, 2% peptone, 100 μg/ml adenine) medium for 12 h and cultured in YPA supplemented with vehicle control (lanes 1–3), 2% glucose (lanes 4–6), 2% glycerol (lanes 7–9) and 2% galactose (lanes 10–12) for the specified times. Cells were harvested and analyzed by Western blotting using anti-Sup35 antibody. Sup35s indicates cleaved product of Sup35.
![Figure 6. Sup35 is specifically cleaved by PrA-PrB proteases when cells are grown in glucose media. A GT17 [psi− pin−] cells grown in the mid log-phase were starved in YPA (1% yeast extract, 2% peptone, 100 μg/ml adenine) medium for 12 h and cultured in YPA supplemented with vehicle control (lanes 1–3), 2% glucose (lanes 4–6), 2% glycerol (lanes 7–9) and 2% galactose (lanes 10–12) for the specified times. Cells were harvested and analyzed by Western blotting using anti-Sup35 antibody. Sup35s indicates cleaved product of Sup35.](/cms/asset/34c8484d-b657-4aba-ae9d-dfeda10e1726/kprn_a_1521234_f0006_c.jpg)
Conclusion
Proteolytic cleavage is one of the common post-translational modifications and often affects aggregation properties of the target proteins. This review focused on the influence of proteolytic cleavage on the amyloidogenicity of HTT, TDP43, APP, PrP, Sup35 proteins. In the case of HTT, TDP43 and APP, proteolytic cleavage (e.g. APP by β- and γ-secretases) accelerates aggregation and toxicity of the proteins. While in the case of APP and PrP, proteolytic cleavage (e.g. APP by α-secretase and PrP by α-cleavage) suppresses aggregation and exerts neurotrophic and neuroprotective effects. The difference in these effects seems to depend on whether the sequence necessary for aggregation as well as replication is destroyed by proteolytic cleavage or not. However, in the majority of cases it has not been demonstrated whether the proteolytic cleavage is directly involved in the pathogenesis by the amyloidogenic proteins. We have shown that the cleavage of Sup35 by PrA-PrB proteases suppresses spontaneous prion generation in yeast. The yeast Saccharomyces cerevisiae is a simple and genetically tractable organism that has been successfully used as a model for numerous neurological diseases including prion, Parkinson’s disease, polyQ expansion disorders, Friedreich’s ataxia, and Batten disease etc [Citation33]. Recently, yeast systems that enable to evaluate aggregation and toxicity of human polyQ/amyloid diseases have been developed. For example, Chandramowlishwaran et al. have reported a de novo nucleation system that detects the ability of mammalian amyloidogenic protein (Aβ and PrP etc.) to promote prion nucleation by attaching the mammalian amyloidogenic protein to prion domain of Sup35 [Citation34]. Also, aggregation and toxicity of human polyQ diseases (HD etc.) can be reproduced in yeast [Citation35]. By using these systems, it would be possible to identify proteases that modify amyloidogenesis. Moreover, a yeast system that reconstitutes β-secretase has also been established [Citation36]. Apart from proteolysis, alternative methodology using extracellular vesicles to promote/prevent prion formation has been reported in yeast [Citation37,Citation38]. Yeast-based strategy will further contribute to clarifying the mechanism for the pathogenesis of the neurodegenerative disorders by amyloidogenic proteins, their temporal and spatial regulation by the proteolytic cleavage and developing new drugs affecting the proteolytic cleavage for the treatment of amyloidoses.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. PMID:17023659
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363–13383. PMID:9811807
- Halfmann R, Jarosz DF, Jones SK, et al. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature. 2012;482(7385):363–368. PMID:22337056
- Gafni J, Ellerby LM. Calpain activation in huntington’s disease. J Neurosci. 2002;22(12):4842–4849. PMID:12077181
- Gafni J, Hermel E, Young JE, et al. Inhibition of calpain cleavage of Huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279(19):20211–20220. PMID:14981075
- Lee CY, Cantle JP, Yang XW. Genetic manipulations of mutant huntingtin in mice: new insights into Huntington’s disease pathogenesis. FEBS J. 2013;280(18):4382–4394. PMID:23829302
- Zhang YJ, Xu YF, Dickey CA, et al. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27(39):10530–10534. PMID:17898224
- Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2010;107(8):3858–3863. PMID:20133711
- Ayala YM, Zago P, D’Ambrogio A, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121(Pt22):3778–3785. PMID:18957508
- Yang C, Tan W, Whittle C, et al. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One. 2010;5:e15878. PMID:21209826.
- Ishii T, Kawakami E, Endo K, et al. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS One. 2017;12(6):e0179375. PMID:28599005
- Vassar R, Kovacs DM, Yan R, et al. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neuro sci. 2009;29(41):12787–12794. PMID:19828790
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38(1):9–12. PMID:12691659
- Saftig P, Lichtenthaler SF. The α secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog Neurobiol. 2015;135:1–20. PMID:26522965.
- Furukawa K, Sopher BL, Rydel RE, et al. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neuro chem. 1996;67(5):1882–1896. PMID:8863493
- Nikolaev A, McLaughlin T, O’Leary DD, et al. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457(7232):981–989. PMID:19225519
- Postina R, Schroeder A, Dewachter I, et al. A disintegrin-metalloproteinaseprevents amyloid plaque formation and hippocampal defects in an Alzheimerdisease mouse model. J Clin Invest. 2004;113(10):1456–1464. PMID:15146243.
- Colciaghi F, Borroni B, Pastorino L, et al. [alpha]-secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med. 2002;8(2):67–74. PMID:12080182
- Tyler SJ, Dawbarn D, Wilcock GK, et al. alpha- and beta-secretase: profound changes in Alzheimer’s disease. Biochem Biophys Res Commun. 2002;299(3):373–376. PMID:12445809
- Jiménez-Huete A, Lievens PM, Vidal R, et al. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol. 1998;153:1561–1572. PMID:9811348.
- Chen SG, Teplow DB, Parchi P, et al. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270(32):19173–19180. PMID:7642585
- Liang J, Kong Q. α-Cleavage of cellular prion protein. Prion. 2012;6(5):453–460. PMID:23052041
- Guillot-Sestier MV, Sunyach C, Druon C, et al. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem. 2009;284(51):35973–35986. PMID:19850936.
- Liebman SW, Chernoff YO. Prions in yeast. Genetics. 2012;191(4):1041–1072. PMID:22879407
- Ganusova EE, Ozolins LN, Bhagat S, et al. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol Cell Biol. 2006;26(2):617–629. PMID:16382152
- Manogaran AL, Hong JY, Hufana J, et al. Prion formation and polyglutamine aggregation are controlled by two classes of genes. PLoS Genet. 2011;7(5):e1001386. PMID:21625618
- Okamoto A, Hosoda N, Tanaka A, et al. Proteolysis suppresses spontaneous prion generation in yeast. J Biol Chem. 2017;292(49):20113–20124. PMID:29038292
- Tibor Roberts T, Wickner RB. Heritable activity: a prion that propagates by covalent autoactivation. Genes Dev. 2003;17:2083–2087. PMID:12923060.
- Dechant R, Saad S, Ibanez AJ, et al. Cytosolic pH regulates cell growth through distinct GTPases, Arf1 and Gtr1, to promote Ras/PKA and TORC1 activity. Mol Cell. 2014;55:409–421. PMID:25002144.
- Speldewinde SH, Doronina VA, Grant CM. Autophagy protects against de novo formation of the [PSI+] prion in yeast. Molecular Biology of the Cell. 2015;26:4541–4551. PMID:26490118.
- Kabani M, Redeker V, Melki R. A role for the proteasome in the turnover of Sup35p and in [PSI+] prion propagation. Mol Microbiol. 2014;92:507–528. PMID:24589377.
- Wang K, Redeker V, Madiona K, et al. 26S proteasome degrades the soluble but not the fibrillar form of the yeast prion Ure2p in vitro. PLOS One. 2015;10:e0131789. PMID: 26115123.
- Khurana V, Lindquist S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker’s yeast? Nat Rev Neurosci. 2010;11(6):436–449. PMID:20424620
- Chandramowlishwaran P, Sun M, Casey K, et al. Mammalian amyloidogenic proteins promote prion nucleation in yeast. J Biol Chem. 2018;293(9):3436–3450. PMID:29330303
- Alexandrov AI, Serpionov GV, Kushnirov VV, et al. Wild type hungtingtin toxicity in yeast: implications for the role of amyloid cross-seeding in polyQ diseases. Prion. 2016;10:221–227. PMID:27220690.
- Futai E, Osawa S, Cai T, et al. Suppressor mutations for presenilin 1 familial Alzheimer diseas mutants modulate g-secretase activities. J Biol Chem. 2016;291:435–446. PMID:26559975.
- Kabani M, Melki R. Sup35p in its soluble and Prion States is packaged inside extracellular vesicles. MBio. 2015;6:e01017–15. PMID:26286691.
- Kabani M, Melki R. More than just trash bins? Potential roles for extracellular vesicles in the vertical and horizontal transmission of yeast prions. Curr Genet. 2016;62:265–270. PMID:26553335.