
ABSTRACT
The major neurological feature of prion diseases is a neuronal loss accomplished through either apoptosis or autophagy. In this review, I compared axonal alterations in prion diseases to those described 40 years earlier as a result of nerve ligation. I also demonstrated that autophagic vacuoles and autophagosomes are a major part of dystrophic neurites. Furthermore, I summarized the current status of the autophagy in prion diseases and hypothesize, that spongiform change may originate from the autophagic vacuoles. This conclusion should be supported by other methods, in particular laser confocal microscopy.
We observed neuronal autophagic vacuoles in different stages of formation, and our interpretation of the ‘maturity’ of their formation may or may not equate to actual developmental stages. Initially, a part of the neuronal cytoplasm was sequestrated within double or multiple membranes (phagophores) and often exhibited increased electron-density. The intracytoplasmic membranes formed labyrinth-like structures that suggest a multiplication of those membranes. The autophagic vacuoles then expand and eventually, a vast area of the cytoplasm was transformed into a merging mass of autophagic vacuoles. Margaret R. Matthews published a long treatise in the Philosophical Transactions of the Royal Society of London in which she had described in great detail the ultrastructure of postganglionic branches of the superior cervical ganglion in the rat following ligation of them. The earliest changes observed by Matthews between 6 h to 2 days in the proximal stump were distensions of proximal axons. Analogously, in our models, an increased number of ‘regular’ (round) and ‘irregular’ MVB and some autophagic vacuoles were observed collectively, both processes were similar.
Introduction
Prion diseases are a group of noninflammatory transmissible diseases of the central nervous system (CNS) of both humans and animals. In humans, they comprise kuru, Creutzfeldt-Jakob disease (CJD; sporadic, familial, iatrogenic and variant); Gerstmann-Sträussler-Scheinker (GSS) disease and fatal familial insomnia. In animals, they include scrapie, bovine spongiform encephalopathy (BSE), chronic wasting disease (CWD) in captive and wild cervids and transmissible mink encephalopathy (TME) in ranch-reared mink. Recently a prion disease in dromedary camel was described [Citation1]. They are caused by unusual infectious agent designated prion, from proteinaceous infectious particles, widely believed to be composed exclusively of an abnormal isoform (PrPSc) of a normal cellular protein (PrPc). How PrPc converts to PrPSc, whether a cofactor is needed and how the aggregate of PrPSc becomes infectious has not been well envisaged [Citation2].
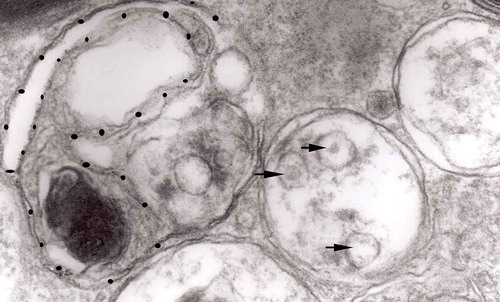
Figure 13. Three autophagic vacuoles in different stages of formation. The left one presents two adjacent membranes marked with black ovals; those membranes form a loop. The middle autophagic vacuole contains three electron-lucent vesicles (additional autophagic vacuoles ?); original magnification, x 33,000.
Prion diseases are neurodegenerative disorders – i.e. they are not inflammatory disorders [Citation3]. It has been recently suggested that other neurodegenerative disorders, i.e. Alzheimer disease, Parkinson’s disease, frontotemporal dementias and many more are in reality prion-like diseases caused by misfolding proteins [Citation4,Citation5]. It seems also that ‘infectious’ proteins may be transferred horizontally as ‘regular’ prions [Citation6,Citation7]. The neuropathological alterations of prion diseases consist of spongiform change of the neuropil or in neurons, the latter more characteristic for animal diseases, astrocytosis, a microglial response and, in some but not all diseases, amyloid plaques. The major neurological feature is a neuronal loss accomplished through either apoptosis or autophagy. In this review, we shall compare axonal alterations in prion diseases to those described some 40 years earlier as a result of nerve ligation. We are aware that electron microscopy is subject to bias; however, we compared structures that are easy to pick up against the background. Thus, we believe that we noticed every one of them in all model. We shall also demonstrate that autophagic vacuoles and autophagosomes are a major part of dystrophic neurites. Furthermore, we shall summarize the current status of the autophagy in prion diseases and hypothesize, that spongiform change may originate from the autophagic vacuoles. This conclusion should be supported by other methods, in particular laser confocal microscopy.
Neuronal cell death in TSEs
As in many neurodegenerative diseases caused by the accumulation of ‘toxic’ misfolded proteins [Citation8,Citation9] prion diseases die via programmed cell death of which only the apoptotic process is relatively well understood. According to the recommendations of the Nomenclature Committee on Cell Death [Citation10–Citation12], three major types of programmed cell death (PCD) can be discriminated.
The first type (apoptosis) is characterized by electron microscopy by specific alterations – cell shrinkage, condensation of chromatin and, eventually, the formation of so-called ‘apoptotic bodies’ that are actively phagocytosed by macrophages. Apoptosis is regulated by a highly conservative network of molecules consisting of Bcl-2 family, caspases, and many others, but some evidence suggests that apoptosis without caspases activation may also occur [Citation13].
The second type – involving macroautophagy (called ‘autophagy’ through this text) – is characterized by the presence of numerous autophagosomes that subsequently fuse with lysosomes to form autolysosomes. The molecular mechanism differs from that of apoptosis and consists of a complex interplay of numerous proteins including the mTor (mammalian target of rapamycin) kinase.
The third type is similar to the second type, except for the negligible or absent involvement of lysosomes. Electron microscopically, type 3 cell death is characterized by swelling of intracellular organelles resulting in the formation of large empty spaces within the cytoplasm. Of interest, TSEs are characterized by ‘spongiform vacuole’ formation within neuronal elements. While the latter have never been linked to the type 3 PCD, the ultrastructural resemblance of ‘large empty spaces’ to ‘spongiform vacuoles’ appears to be worthy of consideration (see below).
The Nomenclature Committee on Cell Death [Citation10] also recognizes ‘mitotic catastrophe’, ‘anoikis’, ‘cornification’, ‘Wallerian degeneration’ and ‘excitotoxicity’: these will not be discussed here. It should be stressed, however, that the major categories of cell death have been defined based mostly on in vitro observations, and the strict usage of such formulated criteria for tissues in vivo may not be entirely appropriate.
As an evolutionarily ancient cellular response to intra- and extracellular noxious stimuli, autophagy may precede or co-exist with apoptosis, and the process may be induced by apoptotic stimuli. Furthermore, the level of autophagy may define the sensitivity of a given neuronal population to apoptotic stimuli, which may underlie the phenomenon of ‘selective neuronal vulnerability’. Thus, autophagy and apoptosis are often interwoven.
Cellular autophagy is a physiological degradation process employed, like apoptosis, in embryonic growth and development, cellular remodelling and the biogenesis of some subcellular organelles ‒ viz. multilamellar bodies [Citation14,Citation15]. A portion of the cytoplasm is engulfed by a semi-circular membrane which closes to create a double membrane vesicle – an autophagosome [Citation16]. Autophagosomes fuse with lysosomes and deliver its cargo to them to form autolysosomes and, as in apoptosis, only excessive or misdirected autophagy causes a pathological process basically regarded as protective against aggregation of misfolded proteins, including PrPSc. Autophagy is highly enhanced in many brain amyloidoses or conformational disorders, Alzheimer’s disease, Parkinson’s disease [Citation17] and Huntington’s disease, in which the signal for autophagy is Huntington [Citation18]. Autophagy is largely regarded as a protective mechanism helping in the removal by organelles of misfolded proteins [Citation19,Citation20].
Neuronal autophagy in prion diseases: ultrastructural observations
Data on autophagy in prion diseases and in yeast prions are meagre [Citation21–Citation26]. Our initial strategy using the hamster-adapted 263K or 22C-H strains of scrapie [Citation26–Citation30] was subsequently broadened by exploration of human brain biopsies from patients with sporadic CJD, variant CJD, and FFI [Citation31,Citation32]. Experimentally infected animal prion disease models are widely used because of their relatively short incubation periods that, for mice, range from 16 to 18 weeks, and for hamsters from 9 to 10 weeks for the 263K scrapie strain and 24–26 weeks for the 22C-H scrapie strain.
For human TSE strains, mice were inoculated either with the Fujisaki (Fukuoka 1) strain of GSS [Citation33,Citation34], and hamsters were inoculated with either the Echigo-1 strain of CJD or the 263K strain of scrapie [Citation28,Citation35]. The Echigo-1 strain of CJD was isolated by Mori and colleagues [Citation35] from a case of 33-year-old female with a panencephalopathic type of GSS [Citation36]. The inoculum was originally prepared from brain tissue and was passaged through guinea pigs with incubation periods (IP) of 728 days at primary and approximately 400 days at subsequent passages. From an animal that exhibited hyperactivity and an excessive response to external stimuli, a substrain was isolated with substantially reduced IP (254 days). This strain was re-isolated in hamsters with IP of 141 days at the third passage. Control animals were sham-inoculated with saline. The Fujisaki strain of GSS was isolated from a GSS case by Tateishi et al. [Citation37].
Formation of autophagic vacuoles in prion-affected brains
Autophagic vacuoles developed not only in neuronal perikaryal but also in neuronal processes eventually replacing the whole cross-section of affected neuritis. In a few specimens, we found round electron-dense structures that we identified as aggresomes [Citation38]. In general, there was little qualitative difference between hamsters infected with either the 263K strain of scrapie or the 22L-H strain, although hamsters inoculated with the 263K strain showed a more robust pathology.
Autophagic vacuoles are areas of the cytoplasm sequestrated within double or multiple membranes (phagophores) of unknown origin; the endoplasmic reticulum may be a possible source. Sequestrated piece of the cytoplasm within membranes contains ribosomes, small secondary vacuoles, and occasional mitochondria. Some vacuoles present a homogeneously dense appearance.
We observed neuronal autophagic vacuoles in different stages of formation in the same specimens, and our interpretation of the ‘maturity’ of their formation may or may not equate to actual developmental stages. Initially, a part of the neuronal cytoplasm was sequestrated within double or multiple membranes (phagophores) and often exhibited increased electron-density. The intracytoplasmic membranes formed labyrinth-like structures that suggest a multiplication of those membranes. The autophagic vacuoles then expand and eventually, a vast area of the cytoplasm was transformed into a merging mass of autophagic vacuoles.
Autophagy and neuritic degeneration
Our interest in neuritic changes in prion disease is a long-lasting one [Citation39–Citation41]. The interpretation of neuritic changes in prion diseases has changed, however. In a seminal Lampert [Citation42] papers, definitions of ‘dystrophic’, ‘regenerative’ and ‘degenerative’ neurites were established based on transmission electron microscopy and accumulations of altered mitochondria and heterogeneous dense bodies were regarded as an ultrastructural correlate dystrophic neurites. With the passage of time, it appeared that distinction between those three classes of neurites are not that clear cut [Citation43] and we used the term ‘dystrophic neurites’ as an ‘umbrella term’ to cover all classes of altered neuronal branches. Furthermore, in the quantitative study, we observed that alterations in the number of dystrophic neurites during the incubation period involved only those neurones containing more than one dense body [Citation41]. Recently a change in the interpretation of the nature of those accumulations has occurred as following a renewed interest in autophagy. Nixon et al. [Citation44,Citation45] interpreted many of those dense-bodies as small autophagic vacuoles.
Of interest, in 1973 Margaret R. Matthews published a long treatise in the Philosophical Transactions of the Royal Society of London [Citation46] in which she had described in great detail, the ultrastructure of postganglionic branches of the superior cervical ganglion in the rat following ligation of them. This study was based on enormous number, in terms of electron microscopy, of 48 rats that underwent either cutting or constriction of external carotid nerve/or internal carotid nerve. Those animals were left to survive for until 143 days and electron microscopic alterations were observed and categorized.
In the following paragraph, I decided to re-arrange our electron microscopic data on neuroaxonal dystrophy in prion diseases as to follow the findings in the Matthews review and to compare those two interpretations.
A putative sequence of changes in neurites in hamster brains infected with the Echigo-1 and the 263K strains of CJD and scrapie, respectively, as deduced against alterations published for sympathetic nerves constrictions [Citation46].
The earliest changes observed by Matthews between 6 h to 2 days in the proximal stump were distensions of proximal axons. Those distensions were relatively large encompassing up to 0.4 μm of the axons and were already filled with packed organelles comprising mitochondria, vesicles and short tubular profiles (,)). By the 12th and 13th hours in the Matthews experiment, mitochondria appeared clustered and these changes were also readily observed in our models (). Multivesicular bodies (MVB), dense cytoplasmic bodies (DCB) and clumps of electron-dense vesicles were also seen. Analogously, in our models, an increased number of ‘regular’ (round) and ‘irregular’ MVB and some autophagic vacuoles were observed ().
Figure 1. (a, b) An axon in an early phase of degeneration. There are numerous slender mitochondria, a multivesicular body (short arrow) and a small autophagic vacuole (arrowhead). Numerous lucent vesicles and dense-core vesicles (bent arrows) are observed. Original magnification, x 8300.
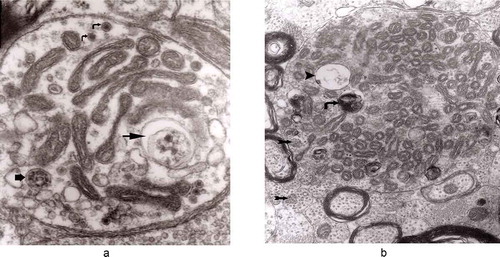
Figure 2. Degenerating axon accumulating some elongated mitochondria, ‘regular’ and ‘irregular’ MVB (arrows) and early autophagic vacuole (arrowhead pointing toward the autophagosome membrane); original magnification, x 8300.
From 12 and 13 hours onward, the clusters of mitochondria were seen in Matthews experiment (Matthews, –; mine, –) followed by increased number of DCB, MVB and autophagic vacuoles. By 38 h following constriction, Matthews observed typical dystrophic neurites filled with ‘masses of DCB’ and a large number of autophagic vacuoles. Our data closely followed the Matthews description (–). Complex ‘irregular’ MVB taking part in the formation of autophagic vacuoles were visible (–). In the latter situation, the MVB form a ‘cap’ sitting on a membrane enveloping electron-lucent vacuole. Numerous autophagic vacuoles also exhibit, as Matthews described, different stages of darkening and degradation (–).
Figure 7. A fragment of a dystrophic axon containing numerous dense bodies and ‘forming and formed’ autophagic vacuoles; original magnification, 6600. This figure corresponds to Matthews Fig. 22. The details are shown in and .
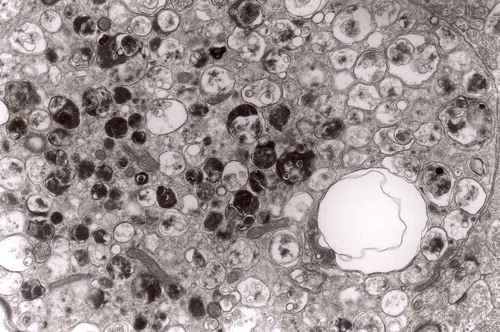
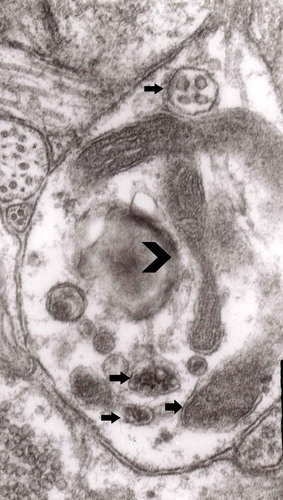
Figure 3. Dilated myelinated axons filled with a large number of MVB (arrows). This electron-micrograph is analogous to . of Matthews [Citation46]. Note that some MVB take part in a formation of autophagic vacuoles and , original magnification, x 33 000.
![Figure 3. Dilated myelinated axons filled with a large number of MVB (arrows). This electron-micrograph is analogous to Figure 16. of Matthews [Citation46]. Note that some MVB take part in a formation of autophagic vacuoles and Figure 5, original magnification, x 33 000.](/cms/asset/7a6e0060-5173-4f14-91b5-9da6037eec88/kprn_a_1595315_f0003_oc.jpg)
Figure 4. Higher magnification of part of dystrophic neurite depicted in . Note that some ‘irregular’ MVB take part in autophagic vacuole formation.
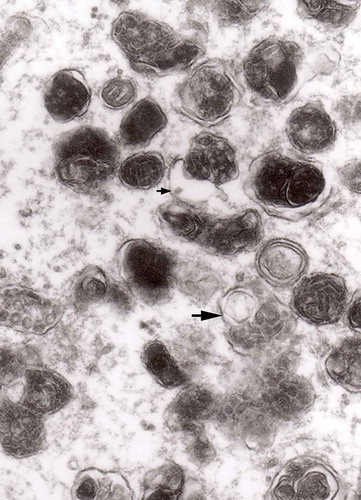
Figure 5. A large magnification of numerous ‘irregular’ MVB (arrows). This electron micrograph corresponds to . of Matthews. Note that MVB marked with double arrows presents a dark part, probably an autophagic vacuole. Original magnification, x 20 000.
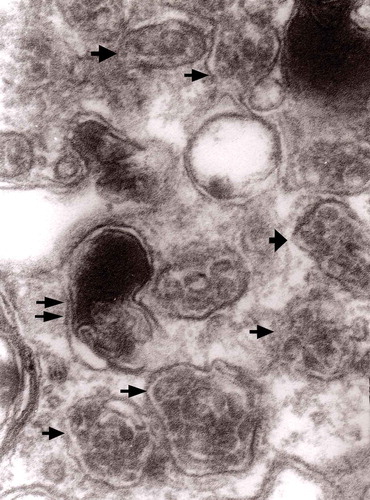
Figure 6. A fragment of a dystrophic axon containing three autophagic vacuoles with content (arrows) of ‘different stages of darkening and degradation’, original magnification, x 33,000. This figure corresponds to Matthews .
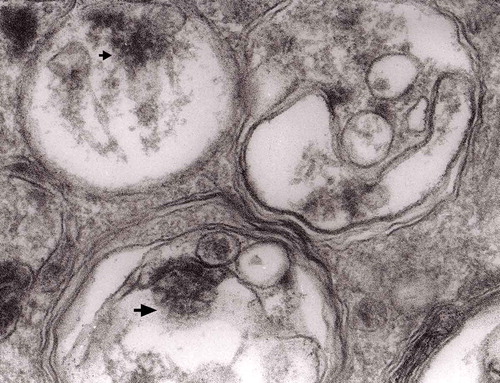
Figure 8. A fragment of . Note autophagic vacuoles (arrows) with partially electron-dense content; original magnification, x 6600.
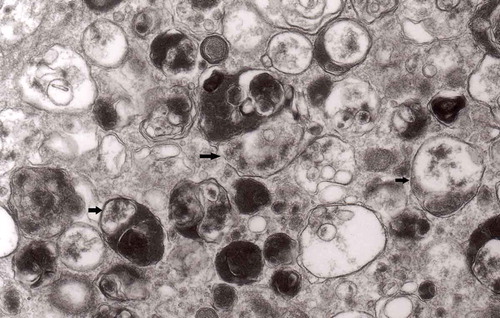
Figure 9. Another fragment of . Note two larger autophagic vacuoles (arrows) and a complex dense body. Note also, that larger vacuole of those two contains electron-dense fragment, this corresponds to partially digested content.
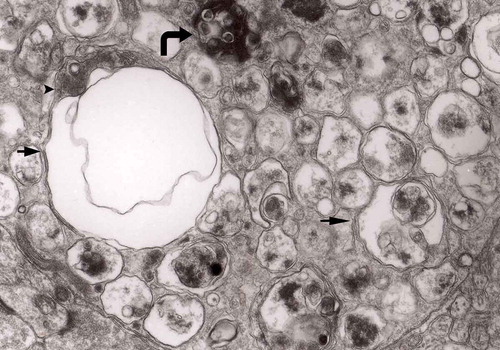
Figure 16. A typical round ‘regular’ MVB in unmyelinated nerve process; original magnification, x 16,000.
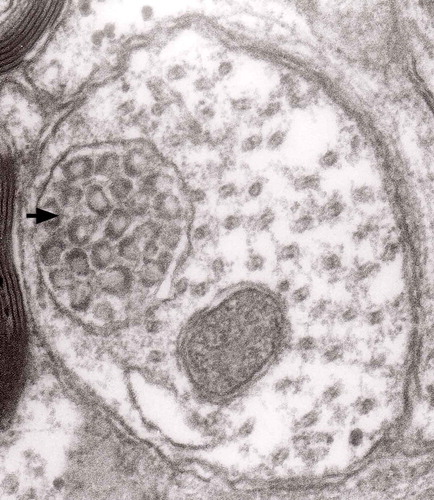
Figure 19. Accumulation of numerous MVBs of irregular sizes and partially flattened; original magnification, x 33,000.
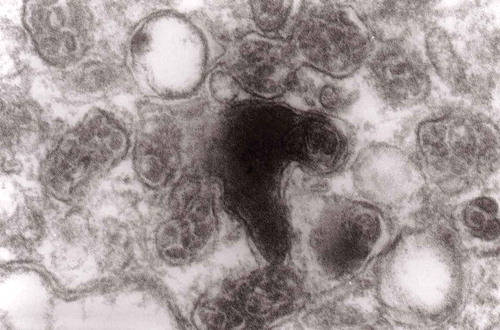
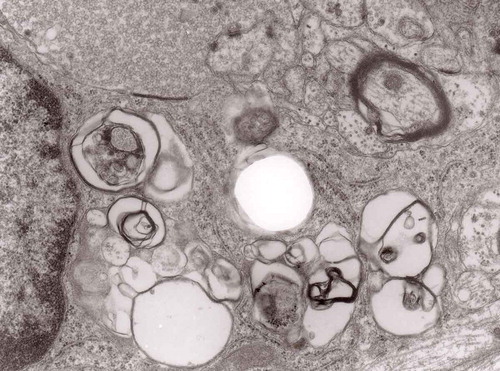
Figure 21. Another fragment of cytoplasm of the macrophage filled with numerous lysosomes and autophagic vacuoles, original magnification, x 16 000.
The formation of autophagic vacuoles may be followed in our material and closely corresponds to those depicted by Matthews. It must be stressed, however, that we compared chronic experimental prion diseases with a model of acute nerve ligation. On the basis of our findings, we suggest that the changes observed on electron microscopy in chronic experimental prion diseases represent numerous episodes of the acute changes observed following nerve ligation, seen in cross section. Autophagic vacuoles are enclosed by paired membranes (; corresponding to Matthews –22). These membranes proliferate, form loops or ‘flattened sacs’ and encircle the fragment of the cytoplasm to form a complex autophagic vacuole composed of several chambers (). Such loops in the process of penetrating the cytoplasm are occasionally discernible (–); of note, such a loop is somehow reminiscent of a loop of inner mesaxon during myelin formation. Cytoplasmic dense bodies are frequent and their similarity to autophagic vacuoles was stressed by Matthews ().
Figure 10. An enlarged fragment of dystrophic neurite showing an autophagic vacuole lined with double membrane. Original magnification, x 33,000.
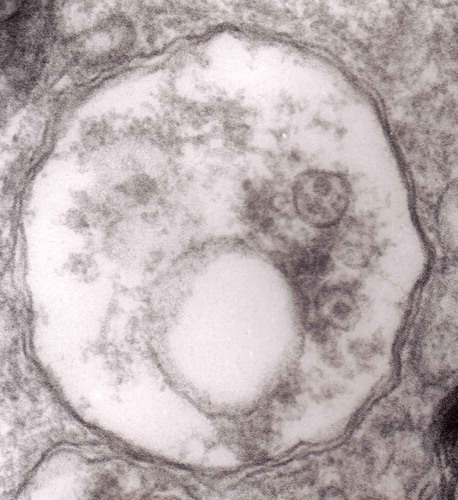
Figure 11. An enlarged fragment of dystrophic neurite showing a complex autophagic vacuole with a darkened content. Original magnification, x 33,000.
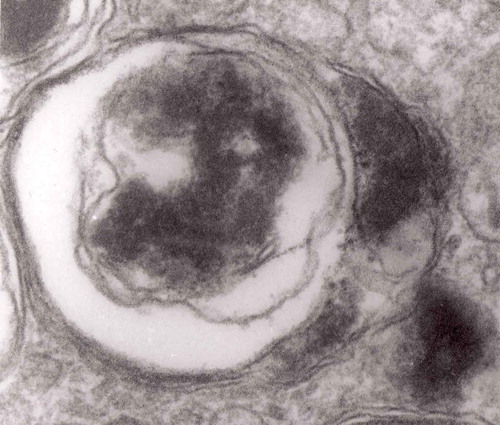
Figure 12. An enlarged fragment of dystrophic neurite showing a complex autophagic vacuole with the formed loop (two arrows) penetrating the adjacent cytoplasm. Original magnification, x 33,000.
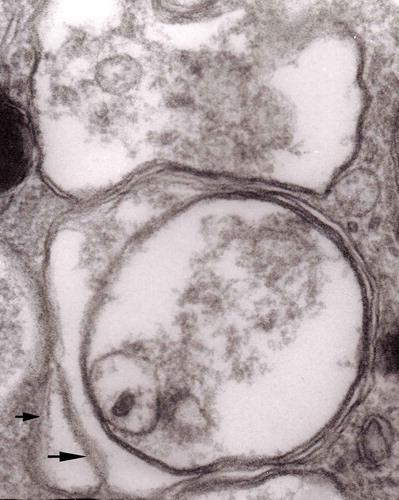
Figure 14. Several autophagic vacuoles. Note the one marked with arrows – it demonstrates elongated protrusions in a process of encircling another vacuole (bent arrow). A complex autophagic vacuole is visible in the vicinity (semicircular arrow); original magnification, x 33,000.
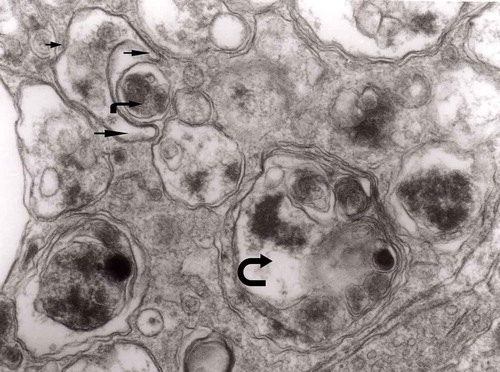
Figure 15. Accumulation of dense bodies – the similarities or even identity with autophagic vacuoles is noticeable; original magnification, x 20,000.
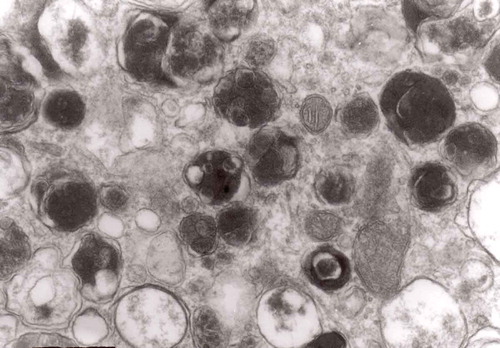
Multivesicular bodies are frequent in both the Matthews’ experiments [Citation46] and in experimental prion diseases described here [Citation28]. In control material they were not very frequent and were of a rather regular form, filled with vesicles with one semicircular end – a ‘cap’ presenting increased electron-density (). In prion diseases, MVB accumulated either as round structures (–) or as more elongated forms () [Citation28]. MVBs tend to demonstrate increased density both of inside vesicles and the intravesicular matrix (). MVB that accumulate both in constricted sympathetic nerves and in TSEs tend to be numerous, more complex, irregular in contour and somehow flattened ().
Figure 17. A dystrophic axon with numerous complex MVB (arrows). Those MVB marked with arrows contained vesicles of increased electron density while others (semicircular arrow) contain electron-lucent vesicles; original magnification, x 16,000.
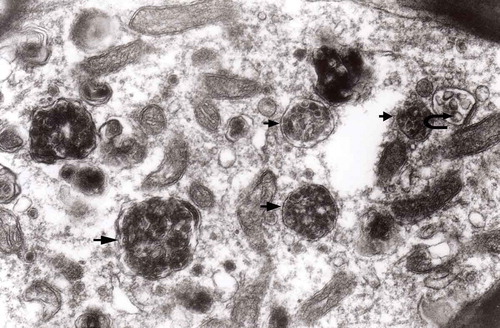
Figure 18. Enlarged fragment of the previous electron-micrograph. Note MVBs – one of normal electron density (semicircular arrow) and one with increased density (arrow); original magnification, x 16,000.
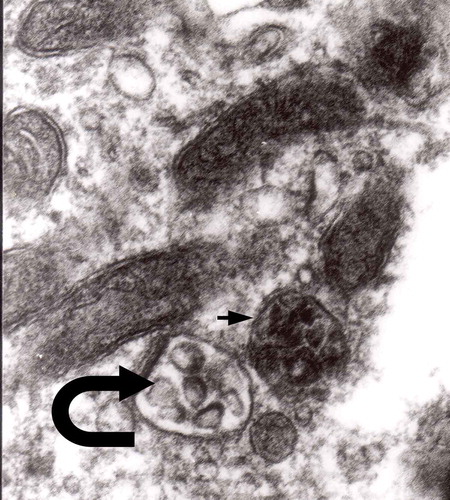
In a group of animals 2 to 7 days following ligature [Citation46], late phagosomes were observed in Schwann cells. From obvious reasons, there are no Schwann cells in our TSE model, and Schwann cells function in a similar manner to brain macrophages (–).
Figure 20. Cytoplasm of the macrophage filled with numerous lysosomes and autophagic vacuoles, original magnification, x 16 000.
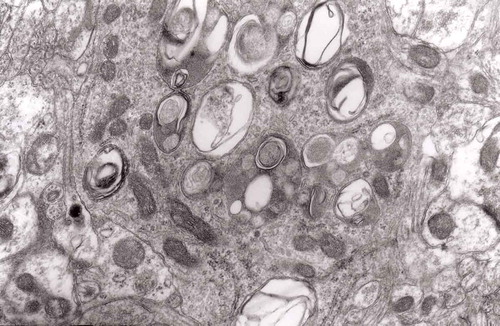
With the time between constriction of the nerve and the electron microscopic observation extended to 143 days, the distended axons became less frequent [Citation46].
In this reconstruction, we artificially grouped electron microscopic images according to the framework provided by Matthews in her study of ligatured sympathetic nerves [Citation46]. In prion diseases, those changes were observed simultaneously, but we believe that our isolated images represent a ‘frozen’ processes that affect multiple neurons and their processes at the same time. We could follow distension of axons, accumulations of cellular dense bodies, MVB and autophagic vacuoles as reported by us [Citation28] and by others [Citation47] in experimental scrapie and CJD.
Neurodegenerative disorders – dystrophic neurites are the major neuronal alterations
The dystrophic neurites in prion diseases are caused by the impairment of axonal transport as was suggested many years ago by Gajdusek [Citation48] and we have shown their presence ultrastructurally in experimental scrapie [Citation39–Citation41], BSE [Citation33], CJD [Citation32,Citation49,Citation50] and GSS [Citation28,Citation51,Citation52] and, finally in chronic wasting disease [Citation53]. Collectively dystrophic neurites form a constant alteration in the prion-affected brains. However, the mechanism(s) of their formation may only be speculated upon. I believe that constriction of neurites, and slowing of the axoplasmic flow leads to their formation.
Using a novel method to study prion diseases, ex-vivo cultures organotypic cerebellar slices, we recapitulated all the scrapie (RML strain) neuropathology at the electron microscopy level [Citation54–Citation56]. The scrapie changes started 5 weeks following inoculation and numerous dystrophic neuritis were readily observed.
One of the most studied prionoid diseases is Alzheimer disease where NAD forms the major part of pathology [Citation7,Citation57,Citation58]. The first description of DN in Alzheimer disease was provided by Lampert [Citation59] – and this definition became the basis of further research ever since. As new transgenic models of AD became available, neuroaxonal dystrophy may be studied in those experimental conditions [Citation60–Citation63]. DN are very plastic, long living and change the size and shape over time; this finding may suggest that what we observe in prion diseases are just different stages of neuritic degeneration. DN are removed by microglial and astrocytic cells [Citation64]. However, the presence of DN within reactive astrocytes were not observed by us in any natural and experimental situations. The exact role of DN in the pathogenesis of Alzheimer disease is unclear; however, they may contribute to the loss of components of the synaptic transmission [Citation62].
Taken together, DN in Alzheimer disease are very similar to those seen in prion diseases. What is their exact role in pathogenesis, remains a mystery.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Babelhadj B, Di Bari MA, Pirisinu L, et al. Prion disease in dromedary camels, Algeria. Emerg Infect Dis. 2018;24(6):1029‒1036.
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601‒623.
- Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nat Rev Genet. 2018;19(7):405‒418.
- Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112(38):E5308‒17.
- Woerman AL, Stöhr J, Aoyagi A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. 2015;112(35):E4949‒58.
- Jaunmuktane Z, Quaegebeur A, Taipa R, et al. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018;135(5):671‒679.
- Jaunmuktane Z, Mead S, Ellis M, et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015;525(7568):247‒250.
- Walker LC. Proteopathic strains and the heterogeneity of neurodegenerative diseases. Annu Rev Genet. 2016;50:329‒346.
- Das AS, Zou WQ. Prions: beyond a single protein. Clin Microbiol Rev. 2016;29(3):633‒658.
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19(1):107‒120.
- Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009;16(1):3‒11.
- Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016;23(4):209‒213.
- Kiraz Y, Adan A, Kartal Yandim M, et al. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016;37(7):8471‒8486.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1‒222.
- Fernández ÁF, Sebti S, Wei Y, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558(7708):136‒140.
- Coutts AS, La Thangue NB. Regulation of actin nucleation and autophagosome formation. Cell Mol Life Sci. 2016;73(17):3249‒3263.
- Nilsson P, Saido TC. Dual roles for autophagy: degradation and secretion of Alzheimer’s disease Aβ peptide. Bioessays. 2014;36(6):570‒578.
- Martin DD, Ladha S, Ehrnhoefer DE, et al. Autophagy in Huntington disease and Huntingtin in autophagy. Trends Neurosci. 2015;38(1):26‒35.
- Redmann M, Darley-Usmar V, Zhang J. The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: implications for neurodegenerative diseases. Aging Dis. 2016;7(2):150‒162.
- Saá P, Harris DA, Cervenakova L. Mechanisms of prion-induced neurodegeneration. Expert Rev Mol Med. 2016;18:e5.
- Boellaard JW, Kao M, Schlote W, et al. Neuronal autophagy in experimental scrapie. Acta Neuropathol. 1991;82(3):225‒228.
- Kovacs GG, Budka H. Molecular pathology of human prion diseases. Int J Mol Sci. 2009;10(3):976‒999.
- Moon JH, Lee JH, Nazim UM, et al. Human prion protein-induced autophagy flux governs neuron cell damage in primary neuron cells. Oncotarget. 2016;7(21):29989‒30002.
- Cortes CJ1, Qin K, Cook J, et al. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Sträussler-Scheinker disease. J Neurosci. 2012;32(36):12396‒12405.
- Speldewinde SH, Doronina VA, Grant CM. Autophagy protects against de novo formation of the [PSI+] prion in yeast. Mol Biol Cell. 2015;26(25):4541‒4551.
- Liberski PP, Gajos A, Bogucki A. Robust autophagy in optic nerves of experimental Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease. Folia Neuropathol. 2017;55(4):289‒294.
- Joshi-Barr S, Bett C, Chiang WC, et al. De novo prion aggregates trigger autophagy in skeletal muscle. J Virol. 2014 Feb;88(4):2071‒2082.
- Liberski PP, Sikorska B, Gibson P, et al. Autophagy contributes to widespread neuronal degeneration in hamsters infected with the Echigo-1 strain of Creutzfeldt-Jakob disease and mice infected with the Fujisaki strain of Gerstmann-Sträussler-Scheinker (GSS) syndrome. Ultrastruct Pathol. 2011;35(1):31‒36.
- Liberski PP, Brown DR, Sikorska B, et al. Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol. 2008;46(1):1‒25.
- Sikorska B. Mechanisms of neuronal death in transmissible spongiform encephalopathies. Folia Neuropathol. 2004;42(Suppl B):89‒95.
- Sikorska B, Liberski PP, Giraud P, et al. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: a brain biopsy study. Int J Biochem Cell Biol. 2004;36(12):2563‒2573.
- Liberski PP, Sikorska B, Hauw JJ, et al. Ultrastructural characteristics (or evaluation) of Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies or prion diseases. Ultrastruct Pathol. 2010 Dec;34(6):351‒361.
- Liberski PP, Yanagihara R, Wells GA, et al. Ultrastructural pathology of axons and myelin in experimental scrapie in hamsters and bovine spongiform encephalopathy in cattle and a comparison with the panencephalopathic type of Creutzfeldt-Jakob disease. J Comp Pathol. 1992 May;106(4):383‒398.
- Liberski PP, Yanagihara R, Gibbs CJ Jr, et al. Experimental Creutzfeldt-Jakob disease: light microscopic, immunohistochemical and ultrastructural studies of the Fujisaki strain of Creutzfeldt-Jakob disease virus in NIH Swiss mice. Neuropatol Pol. 1991;29(1–2):1‒17.
- Mori S, Hamada C, Kumanishi T, et al. A Creutzfeldt-Jakob disease agent (Echigo-1 strain) recovered from brain tissue showing the ‘panencephalopathic type’ disease. Neurology. 1989;39(10):1337‒1342.
- Liberski PP, Mori S. The Echigo-1: a panencephalopathic strain of Creutzfeldt-Jakob disease: a passage to hamsters and ultrastructural studies. Folia Neuropathol. 1997;35(4):250‒254.
- Tateishi J, Kitamoto T, Doh-Ura K, et al. Immunochemical, molecular genetic, and transmission studies on a case of Gerstmann-Sträussler-Scheinker syndrome. Neurology. 1990;40(10):1578‒1581.
- Sikorska B, Liberski PP, Brown P. Neuronal autophagy and aggresomes constitute a consistent part of neurodegeneration in experimental scrapie. Folia Neuropathol. 2007;45(4):170‒178.
- Liberski PP. Electron microscopic observations on dystrophic neurites in hamster brains infected with the 263K strain of scrapie. J Comp Pathol. 1987 Jan;97(1):35‒39.
- Gibson PH, Liberski PP. An electron and light microscopic study of the numbers of dystrophic neurites and vacuoles in the hippocampus of mice infected intracerebrally with scrapie. Acta Neuropathol. 1987;73(4):379‒382.
- Liberski PP, Yanagihara R, Gibbs CJ Jr, et al. Scrapie as a model for neuroaxonal dystrophy: ultrastructural studies. Exp Neurol. 1989 Nov;106(2):133‒141.
- Lampert PW. A comparative electron microscopic study of reactive, degenerating, regenerating, and dystrophic axons. J Neuropathol Exp Neurol. 1967 Jul;26(3):345‒368.
- Liberski PP, Yanagihara R, Budka H, et al. Neuroaxonal dystrophy in unconventional slow virus diseases. In: Liberski PP editor. Light and electron microscopic neuropathology of slow virus disorders. Boca Raton: CRC Press; 1992. p. 251‒268.
- Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113‒122.
- Bordi M, Berg MJ, Mohan PS, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12(12):2467‒2483.
- Matthews MR. An ultrastructural study of axonal changes following constriction of postganglionic branches of the superior cervical ganglion in the rat. Philos Trans R Soc Lond B Biol Sci. 1973;264(866):479‒505.
- Yim YI, Park BC, Yadavalli R, et al. The multivesicular body is the major internal site of prion conversion. J Cell Sci. 2015;128(7):1434‒1443.
- Gajdusek DC. Hypothesis: interference with axonal transport of neurofilament as a common pathogenetic mechanism in certain diseases of the central nervous system. N Engl J Med. 1985;312(11):714‒719.
- Sikorska B, Hainfellner JA, Mori S, et al. Echigo-1: a panencephalopathic strain of Creutzfeldt-Jakob disease. II. Ultrastructural studies in hamsters. Folia Neuropathol. 2004;42(Suppl B):167‒175.
- Liberski PP, Budka H, Yanagihara R, et al. Neuroaxonal dystrophy in experimental Creutzfeldt-Jakob disease: electron microscopical and immunohistochemical demonstration of neurofilament accumulations within affected neurites. J Comp Pathol. 1995;112(3):243‒255.
- Liberski PP, Yanagihara R, Asher DM, et al. Reevaluation of the ultrastructural pathology of experimental Creutzfeldt-Jakob disease. Serial studies of the Fujisaki strain of Creutzfeldt-Jakob disease virus in mice. Brain. 1990;113(Pt1):121‒137.
- Liberski PP, Budka H. Ultrastructural pathology of Gerstmann-Sträussler-Scheinker disease. Ultrastruct Pathol. 1995;19(1):23‒36.
- Guiroy DC, Williams ES, Liberski PP, et al. Ultrastructural neuropathology of chronic wasting disease in captive mule deer. Acta Neuropathol. 1993;85(4):437‒444.
- Falsig J, Julius C, Margalith I, et al. A versatile prion replication assay in organotypic brain slices. Nat Neurosci. 2008 Jan;11(1):109‒117.
- Sigurdson CJ, Nilsson KP, Hornemann S, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A. 2009;106(1):304‒309.
- Falsig J, Sonati T, Herrmann US, et al. Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog. 2012;8(11):e1002985.
- Hall GF, Patuto BA. Is tau ready for admission to the prion club? Prion. 2012;6(3):223‒233.
- Kovacs GG, Lutz MI, Ricken G, et al. Dura mater is a potential source of Aβ seeds. Acta Neuropathol. 2016;131(6):911‒923.
- Lampert P. Fine structural changes of neurites in Alzheimer’s disease. Acta Neuropathol. 1971;5(Suppl 5):49‒53.
- Blazquez-Llorca L, Valero-Freitag S, Rodrigues EF, et al. High plasticity of axonal pathology in Alzheimer’s disease mouse models. Acta Neuropathol Commun. 2017;5(1):14.
- Christensen DZ, Huettenrauch M, Mitkovski M, et al. Axonal degeneration in an Alzheimer mouse model is PS1 gene dose dependent and linked to intraneuronal Aβ accumulation. Front Aging Neurosci. 2014;6:139.
- Adalbert R, Nogradi A, Babetto E, et al. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132(Pt 2):402‒416.
- Gowrishankar S, Yuan P, Wu Y, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112(28):E3699‒708.
- Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia. 2018;66(3):637‒653.