
ABSTRACT
Transformation of astrocytes into reactive states is considered one of the major pathological hallmarks of prion and other neurodegenerative diseases. Recent years witnessed a growing appreciation of the view that reactive astrocytes are intimately involved in chronic neurodegeneration; however, little is known about their role in disease pathogenesis. The current article reviews the progress of the last few years and critically discusses controversial questions of whether reactive astrocytes associated with prion diseases are neurotoxic or neuroprotective and whether bidirectional A1–A2 model is applicable for describing polarization of astrocytes. Moreover, other important topics, including reversibility of a transition to a reactive state, along with the role of microglia and other stimuli in triggering astrocyte activation are reviewed. Defining the role of reactive astrocytes in pathogenesis of neurodegenerative diseases will open unrealized opportunities for developing new therapeutic approaches against prion and other neurodegenerative diseases.
Unsettled questions of astrocyte biology
Astrocytes are responsible for a number of homoeostatic functions required for proper functioning of CNS [Citation1–3]. Under chronic neurodegeneration associated with prion and other neurodegenerative diseases, astrocytes undergo significant transcriptional, morphological and functional transformation resulting in reactive phenotypes [Citation4,Citation5]. Recent years witnessed a growing appreciation of the view that reactive astrocytes are intimately involved in chronic neurodegeneration [Citation6–9]. However, the precise role of reactive astrocytes in disease pathogenesis remains highly controversial (reviewed in [Citation6,Citation10]). A number of important questions remain unsettled. The extent to which normal homoeostatic functions are altered in the reactive states is unknown. It is not clear whether polarization into reactive state produces the net neurotoxic or neuroprotective outcome [Citation6–8]. Another controversial topic is whether microglia trigger astrocyte activation and dictate their reactive phenotype. However, among the most important questions is whether the polarization of astrocytes into reactive states represents a downstream response to altered brain homoeostasis or, on the contrary, drives prion pathogenesis. Finally, it is also unclear whether the transformation of astrocytes into reactive states is fully reversible.
Is A1–A2 polarization model applicable to prion diseases?
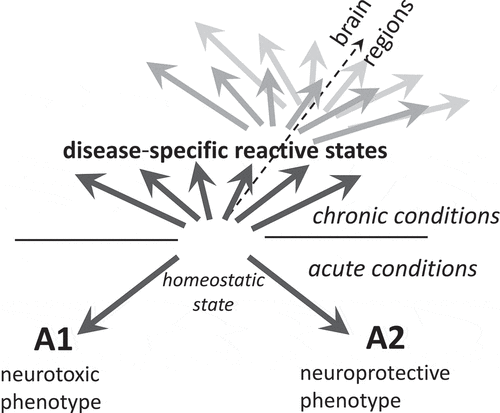
According to the hypothesis introduced by Barres and co-authors, astrocytes can polarize into well-defined neurotoxic (A1) or neuroprotective (A2) reactive states, which exhibit distinct transcriptional profile and opposing effects on neuronal survival [Citation11,Citation12]. The hypothesis proposing alternative A1 and A2 reactive states was developed using animals treated with LPS or subjected to ischaemic stroke [Citation11], conditions that do not induce long-term chronic effects (). The question whether bidirectional polarization model is applicable to chronic neurodegenerative illnesses is highly controversial [Citation10]. Moreover, assessing polarization phenotypes based on animal models is tricky, because most neurodegenerative diseases rely on genetically modified animals that might not faithfully reproduce all aspects of chronic neuroinflammation or neurodegeneration of human diseases [Citation13–15]. Wild type on inbreed animals infected with prions of natural or synthetic origin develops bona fide prion disease [Citation16–20]. As judged from several independent transcriptome studies, in prion-infected animals, astrocytes do not follow the A1–A2 polarization model [Citation21–26]. Instead, upregulation of a mixture of A1-, A2- and pan-reactive markers was observed [Citation21–26]. At present, it is not clear, whether the mixed A1/A2 profile, which was observed in bulk tissues, arise as a result of the actual mixture of A1 and A2 astrocytes, existence of multiple activation states, co-expression A1- and A2-specific markers within individual cells, or all of the above. Application of single cell transcriptome approach should answer the questions regarding diversity of the reactive phenotypes associated with prion diseases along with identifying molecular sub-types not distinguishable from the analyses of bulk tissues. Up to date, very similar profiles of A1-, A2- and pan-reactive markers were found regardless of a brain region or prion strain [Citation21,Citation26], suggesting that in prion diseases, astrocyte adopt relatively uniform reactive state, which might be different from the reactive states in other chronic illnesses. Moreover, the reactive phenotype of astrocytes associated with prion disease was universal across strains with different cell tropism, that is, regardless of whether prion strains prefer to propagate in neurons or astrocytes [Citation26]. Notably, echoing findings in the prion field, a panel of experts in astrocyte biology, recommended moving beyond the A1–A2 labels in describing phenotypes of astrocytes in acute injuries and chronic illnesses of CNS [Citation10].
Figure 1. The diagram illustrating that in neurodegenerative diseases, instead of polarization according to the bidirectional A1-A2 or neurotoxic-neuroprotective model, reactive astrocytes adopt multiple, disease-specific states dictated in part by a nature of an insult. Within individual diseases, reactive states might vary across brain regions at any given time point of the disease (represented by dark and light grey arrows)
Is reactive phenotype associated with prion disease neurotoxic or neuroprotective?
Do reactive astrocytes contribute to diseases pathogenesis? Considering that a mixture of A1- and A2-reactive markers were upregulated in prion diseases, while neither markers being liked to homoeostatic or toxic functions of astrocytes, the bidirectional A1-A2 concept does not provide a cue whether reactive astrocytes are expected to produce neuroprotective or neurotoxic effects. PrPSc strains that target astrocytes often have shorter incubation times to the disease in comparison with the strains that colocalize with neurons [Citation22,Citation27]. Moreover, transmissible PrP states that are characterized by very mild reactive astrogliosis were shown to propagate in CNS silently, without causing clinical signs of the diseases despite substantial synaptic immunoreactivity [Citation28]. These results brings up a possibility that a causative link between astrocyte response to transmissible PrP states and disease pathogenesis exists.
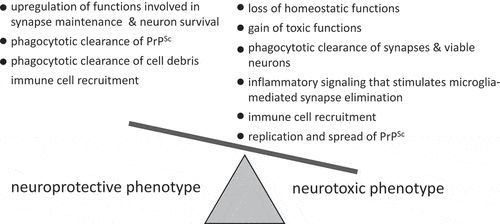
Several possible mechanisms, including upregulation of functions involved in synapse maintenance and neuronal survival, phagocytic clearance of PrPSc and cell debris or, in opposite, spread of PrPSc, elimination of synapses along with viable neurons, loss of homoeostatic functions, gain of toxic functions, inflammatory signalling that stimulates microglia-mediated synapse elimination, recruitment of immune cells and others mechanisms, should be examined to determine the net impact of reactive astrocytes ().
Figure 2. Schematic diagram illustrating that dysregulation of multiple neuroprotective and neurotoxic mechanisms might contribute to defining a net outcome of reactive astrocyte phenotype
Recent studies suggested that reactive astrocytes associated with prion diseases are neurotoxic [Citation29,Citation30]. Using primary astrocyte cultures isolated from prion-infected animals, our recent work revealed that in the reactive states, astrocytes exbibit profound neurotoxic effects mediated via astrocyte-conditioned media and also manifested directly in co-cultures with primary neurons [Citation29]. As tested in co-cultures with neurons, homoeostatic functions responsible for neuronal growth, spine development and synapse maturation were impaired in reactive astrocytes isolated from the prion-infected animals [Citation29]. The media conditioned by the reactive astrocytes too had deleterious effects on primary neurons, including reduction in density and size of the dendritic spines, disintegration of synapses, reduced expression of pre- and post-synaptic proteins along with a decrease in viability of neurons [Citation29]. Genes involved in formation, maturation and stability of synapses and dendritic spine were downregulated in astrocytes derived from prion-infected animals [Citation29]. These results argue that in the reactive state associated with prion disease, the net effect of astrocyte is neurotoxic, which manifest itself as loss of homoeostatic functions responsible for synapse maintenance. In other study, selective astrocyte-specific targeting of unfolded protein response, which is exuberated in reactive astrocytes, was found to extend the incubation time to the terminal disease in mice [Citation30]. This also suggests that reactive astrocytes are neurotoxic [Citation30].
Among possible neuroprotective effects of the reactive state is phagocytosis of PrPSc. Indeed, primary astrocyte cultures and astrocyte cell line were found capable of phagocytic uptake or internalization of PrPSc in vitro [Citation31,Citation32]. Moreover, PrPSc aggregates were found in reactive astrocytes in animals infected with prions [Citation22,Citation27,Citation33]. However, whether phagocytic clearance is up- or downregulated in reactive state versus homoeostatic astrocytes has never been tested. It is also not clear whether reactive astrocytes can selectively upregulate phagocytosis of PrPSc but not synapses or viable neurons.
The net neurotoxic phenotype does not exclude upregulation of neuroprotective functions in parallel with the loss of homoeostatic functions and/or to activation of neurotoxic mechanisms (). Moreover, considering significant region-specific heterogeneity in astrocyte homoeostatic phenotypes [Citation4,Citation9,Citation34–36], asynchronous progression of the disease in different brain regions along with selective tropism of prion strains to different regions [Citation37,Citation38], it cannot be excluded that distinct reactive states with opposite net effects on neuronal survival are present in different brain regions during diseases time course.
Recent studies on transcriptome analysis of astrocyte-specific genes revealed that the manifestation of the reactive states associated with prion diseases was not in dysfunction of any specific pathway, but a global transformation of the physiological state of astrocytes, characterized by disturbance in multiple functions [Citation26]. While both sets of genes, those involved in neuroprotection and neurotoxic functions, were disturbed, the net result of disturbances produced a neurotoxic phenotype [Citation26]. Remarkably, the degree of astrocyte activation along with disturbance in functional pathways showed strong reverse correlation with the incubation time to disease [Citation26]. The most rapid disease progression was found in the animal groups with the most severe astrocyte response. Analysis of astrocyte-specific genes raised the possibility that the degree of astrocyte activation contribute to the faster progression of the disease and perhaps even drive prion pathogenesis [Citation26].
Does microglia trigger astrocyte activation?
Because microglia and astrocytes often become reactive in parallel and both contribute to neuroinflammation, it is difficult to pinpoint the contribution of each cell type. According to the Barres hypothesis that gained popularity in neurodegeneration field, reactive microglia drives the polarization of astrocytes into neurotoxic A1 state via the upregulation of three secreted factors TNF-α, IL-1α and C1qa [Citation39]. In conflict with this hypothesis, the ablation of these three factors in triple TNF−/-/IL1a−/-/C1qa−/- knockout mice was found to accelerate the progression of prion diseases resulting in only modest suppression in A1-specific markers in prion-infected animals [Citation23]. Moreover, contrary to this hypothesis, the partial ablation of microglia by PLX5622 exacerbated the reactive phenotype of astrocyte and, again, accelerated disease progression [Citation40]. These studies argued that in prion diseases, instead of driving neurotoxic phenotype in astrocytes, reactive microglia seem to attenuate it. Moreover, it appears that the reactive phenotype of astrocyte is not dictated entirely by reactive microglia, and independent mechanisms of activation exist. Indeed, the analysis of transcriptome revealed that astrocytes respond to prion infection prior to clinical signs and, perhaps, even prior to microglia [Citation21]. Astrocytes are sensitive to neuronal activity and respond to abnormalities in synaptic transmission [Citation41]. Furthermore, studies that employed cell cultures showed that astrocytes can detect PrPSc and, in response, upregulate chemokine gene expression releasing signals that trigger microglia migration [Citation42]. Reactive astrocytes isolated from prion-infected mice upregulate the expression of pro-inflammatory genes (IL6, IL12b, IL33, Ccl4) along with the secretion of IL-6, suggesting that the reactive phenotype is relatively stable and can be maintained in vitro in the absence of pro-inflammatory CNS environment [Citation29]. IL-33 secreted by astrocytes is known to drive microglia-dependent synapse engulfment and elimination [Citation43], while elevated levels of IL-6 triggers pathways are linked to neurodegeneration [Citation44]. In mouse models of Alzheimer’s and Huntington’s disease, suppressing astrocyte reactivity by inhibiting activation of transcription factor STAT3 selectively in astrocytes reduced neuroinflammation and activation of microglia [Citation45]. To summarize, the reactivity phenotypes of microglia and astrocytes appear to be closely coordinated and rely on multiple feedback loops. Both cell types are capable of serving as the first responders to CNS insults and drive neuroinflammation.
Is reactive state of astrocyte reversable?
Is presence of a stimulus that triggers reactive states important for maintaining the reactive phenotype? Studies of optic nerve subjected to mild injury introduced by a brief ocular pressure showed that astrocyte reactivity could be fully resolved if the insult is removed [Citation46]. In more severe insults such as spinal cord injury that lead to the formation of glial scares consisting of reactive astrocytes, phenotypic changes have long been considered irreversible. However, recent studies showed that reactive astrocytes isolated from injured spinal cord reverted their phenotype upon transplantation into a naïve spinal cord and vice versa [Citation47], suggesting that preservation of reactive phenotypes relies on the persistent stimulus or the presence of environmental factors. In our study on isolation of astrocytes from prion-infected animals, the reactive phenotypes were preserved, at least in part, for three weeks post-isolation [Citation29].
Is it possible to reverse reactive states in the presence of persistent stimulus? Activation of STAT3 transcription factor was identified as universal feature of astrocyte reactivity in neurodegenerative diseases shared between different species, brain regions and different types of illnesses (reviewed in [Citation6,Citation48]. Selective inhibition of STAT3 pathway in astrocytes was found to suppress astrocyte activation or reverse their reactive phenotype, and improved the disease outcomes in several animal models of neurodegenerative diseases [Citation45,Citation49,Citation50]. Activation of STAT3 was observed in animals infected with prions [Citation51]; however, its role in driving astrocyte reactive states associated with prion diseases has not yet been examined.
What stimuli trigger astrocyte activation?
In prion diseases, astrocytes respond to prion infection prior to clinical symptoms or neuronal damage [Citation21]. In mice infected with prions, the onset of GFAP upregulation appeared to be triggered by the accumulation of PrPSc over a certain threshold, while the kinetics in GFAP overexpression followed very closely the kinetics of PrPSc accumulation [Citation52]. Phagocytosis of PrPSc in vitro by cultured astrocytes [Citation31,Citation32] along with intracellular localization of PrPSc aggregates in reactive astrocytes in animals [Citation22,Citation27,Citation53] suggests that astrocytes have the ability to recognize PrPSc directly. Indeed, in response to scrapie brain homogenate, cultured astrocytes upregulated the expression of cytokine genes [Citation42]. It is not clear what receptors are responsible for recognition of PrPSc in astrocytes and neurons share the expression of lipoprotein receptor-related protein 1 (LR1P), which in neurons was found to be involved in endocytosis of PrPSc [Citation54]. Astrocytes and microglia share a wide range or receptors that might participate in PrPSc phagocytosis, including toll-like receptors (TLRs), which are involved in the recognition of pathogens and extracellular protein aggregates; Axl receptor activates JAK/STAT pathway; a heterogeneous family of scavenger receptors is capable of recognition of danger- and pathogen-associated molecular patterns along with extracellular protein aggregates [Citation55]; and MEGF10, an important receptor, is involved in phagocytosis on synapses and apoptotic cells [Citation56]. Cultured microglia react to purified PrPSc by upregulating proinflammatory signalling [Citation57]. Notably, the degree of response in microglia was found to be dictated by the sialylation status of N-linked glycans on the surface of PrPSc [Citation57], with a stronger response caused by a PrPSc with desialylated glycans [Citation57]. Like normal form of the prion protein or PrPC, PrPSc is sialylated [Citation58]; however, the level and density of sialylation of PrPSc particles are variable among prion strains and dictated by a strain identity [Citation59–62]. It is not known whether the same signalling pathways that respond in microglia to asialoglycans are also active in astrocytes. Remarkably, significant reduction in PrPSc sialylation levels that accompanied cross-species adaptation of a strain to a new host produced a new strain characterized by a very profound neuroinflammation and the shortest incubation time to the diseases [Citation22]. These results suggested that a causative link between sialylation status of PrPSc, the degree of glia activation and the rate of disease progression exists [Citation22].
Concluding remarks
With the development of new tools and gaining more knowledge regarding the role of reactive astrocyte in neurodegenerative disease, new questions have to be answered. To what extent the heterogeneity in homoeostatic state dictates reactive state of astrocytes? Do reactive phenotypes drive diseases pathogenesis? Is it possible to fully and selectively reverse reactive states of astrocytes in the presence of persistent proinflammatory stimulus? Does reversing of the reactive states represent an effective therapeutic approach? Answering these question brings a new opportunity for developing unexplored therapeutic approaches.
Acknowledgments
We thank Kara Molesworth for editing this manuscript. This work was supported by NIH grants NS045585 and AI128925.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the author(s).
Abbreviations
PrPC, normal cellular isoform of the prion protein; PrPSc, abnormal, disease-associated isoform of the prion protein
Additional information
Funding
References
- Dallérac G, Zapata J, Rouach N. Versatile control of synaptic circuits by astrocytes: where, when and how? Nat Rev Neurosci. 2018 2018/12/01;19(12):729–743.
- Santello M, Toni N, Volterra A. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci. 2019 Feb 1;22(2):154–166.
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010 Jan 1;119(1):7–35.
- Makarava N, Chang JC-Y, Kushwaha R, et al. Region-specific response of astrocytes to prion infection [original research]. Front Neurosci. 2019 Oct 9;13(1):e1048.
- Bradford BM, Wijaya CAW, Mabbott NA. Discrimination of prion strain targeting in the central nervous system via reactive astrocyte heterogeneity in CD44 expression [original research]. Front Cell Neurosci. 2019 Sept 10;13(411). DOI:https://doi.org/10.3389/fncel.2019.00411
- Ben Haim L, M-a C-DS, Ceyzériat K, et al. Elusive roles for reactive astrocytes in neurodegenerative diseases [Review]. Front Cell Neurosci. 2015 Aug 3;9(278): e278.
- Oksanen M, Lehtonen S, Jaronen M, et al. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol Life Sci. 2019 July 1;76(14):2739–2760.
- Acioglu C, Li L, Elkabes S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021 Jan;28:147291.
- Habib N, McCabe C, Medina S, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020 Jun;23(6):701–706.
- Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021 Mar;24(3):312–325.
- Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391–6410.
- Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–967.
- Morrissette DA, Parachikova A, Green KN, et al. Relevance of transgenic mouse models to human Alzheimer disease. J Biol Chem. 2009 Mar 6;284(10):6033–6037.
- Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci. 2018 Oct 1;21(10):1370–1379.
- Friedman BA, Srinivasan K, Ayalon G, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018;22(3):832–847.
- Watts JC, Prusiner SB. Mouse models for studying the formation and propagation of prions. J Biol Chem. 2014 Jul 18;289(29):19841–19849.
- Wang F, Wang X, Yuan CG, et al. Generating a prion bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132–1135.
- Makarava N, Kovacs GG, Savtchenko R, et al. A new mechanism for transmissible Prion diseases. J Neurosci. 2012;32(21):7345–7355.
- Makarava N, Kovacs GG, Savtchenko R, et al. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathogen. 2011;7(12):e1002419.
- Deleault NR, Walsh DJ, Piro JR, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. ProcAcadNatlSciUSA. 2012;109(28):E1938–E1946.
- Makarava N, Chang JC-Y, Molesworth K, et al. Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol Dis. 2020 Apr 1;137(1):e104783.
- Makarava N, Chang JC-Y, Molesworth K, et al. Posttranslational modifications define course of prion strain adaptation and disease phenotype. J Clin Invest. 2020 Aug 3;130(8):4382–4395.
- Hartmann K, Sepulveda-Falla D, Rose IVL, et al. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia [journal article]. Acta Neuropathol Commun. 2019 May 22;7(1):83.
- Scheckel C, Imeri M, Schwarz P, et al. Ribosomal profiling during prion disease uncovers progressive translational derangement in glia but not in neurons. Elife. 2020 Sep 22;9. DOI:https://doi.org/10.7554/eLife.62911
- Carroll JA, Race B, Williams K, et al. RNA-seq and network analysis reveal unique glial gene expression signatures during prion infection. Mol Brain. 2020 May 7;13(1):71.
- Makarava N, Mychko O, Chang JC-Y, et al. The degree of astrocyte activation is predictive of the incubation time to prion disease. Acta Neuropathol Commun. 2021 May 12;9(1):87.
- Carroll JA, Striebel JF, Rangel A, et al. Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: comparison of three prion strains. PLoS Pathog. 2016;12(4):e1005551.
- Kovacs GG, Makarava N, Savtchenko R, et al. Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am J Pathol. 2013;183(5):1539–1547.
- Kushwaha R, Sinha A, Makarava N, et al. Non-cell autonomous astrocyte-mediated neuronal toxicity in prion diseases. Acta Neuropathol Commun. 2021 Feb 5;9(1):22.
- Smith HL, Freeman OJ, Butcher AJ, et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron. 2020 Mar 4;105(5):855–866.e5.
- Choi YP, Head MW, Ironside JW, et al. Uptake and degradation of protease-sensitive and -resistant forms of abnormal human prion protein aggregates by human astrocytes. Am J Pathol. 2014;184(12):3299–3307.
- Hollister JR, Lee KS, Dorward DW, et al. Efficient uptake and dissemination of scrapie prion protein by astrocytes and fibroblasts from adult hamster brain. PLOS ONE. 2015;10(1):e0115351.
- Jeffrey M, McGovern G, Makarava N, et al. Pathology of SSLOW, a transmissible and fatal synthetic prion protein disorder, and comparison with naturally occurring classical transmissible spongoform encephalopathies. NeuropathApplNeurobiol. 2014;40(3):296–310.
- Zeisel A, Hochgerner H, Lonnerberg P, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174(4):999–1014.
- Bayraktar OA, Bartels T, Holmqvist S, et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci. 2020 Apr;23(4):500–509.
- Batiuk MY, Martirosyan A, Wahis J, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun. 2020 Mar 5;11(1):1220.
- Makarava N, Chang JC-Y, Baskakov IV. Region-specific sialylation pattern of prion strains provides novel insight into prion neurotropism. Int J Mol Sci. 2020;21(3):828.
- Karapetyan YE, Saa P, Mahal SP, et al. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One. 2009;4(5):e5730.
- Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487.
- Carroll JA, Race B, Williams K, et al. Microglia are critical in host defense against prion disease. J Virol. 2018;92(15):e00549–18.
- Blanco-Suárez E, Caldwell AL, Allen NJ. Role of astrocyte-synapse interactions in CNS disorders. J Physiol. 2017 Mar 15;595(6):1903–1916.
- Marella M, Chabry J. Neurons and astrocytes respond to prion infection by inducing microglia recruitment. J Neurosci. 2004 Jan 21;24(3):620–627.
- Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018 Mar 16;359(6381):1269–1273.
- Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta, Mol Cell Res. 2016 Jun 1;1863(6, Part A):1218–1227.
- Ben Haim L, Ceyzériat K, Carrillo-de Sauvage MA, et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J Neurosci. 2015 Feb 11;35(6):2817–2829.
- Sun D, Qu J, Jakobs TC. Reversible reactivity by optic nerve astrocytes. Glia. 2013 Aug;61(8):1218–1235.
- Hara M, Kobayakawa K, Ohkawa Y, et al. Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin–N-cadherin pathway after spinal cord injury. Nat Med. 2017 Jul 1;23(7):818–828.
- Yan Z, Gibson SA, Buckley JA, et al. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol. 2018 Apr;189:4–13.
- Ceyzériat K, Ben Haim L, Denizot A, et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol Commun. 2018 Oct 16;6(1):104.
- Reichenbach N, Delekate A, Plescher M, et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol Med. 2019;11(2):Feb.
- Na Y-J, Jin J-K, Kim J-I, et al. JAK-STAT signaling pathway mediates astrogliosis in brains of scrapie-infected mice. J Neurochem. 2007;103(2):637–649.
- Lasmézas CI, Deslys JP, Demaimay R, et al. Strain specific and common pathogenic events in murine models of scrapie and bovine spongiform encephalopathy. J Gen Virol. 1996 Jul;77(Pt 7):1601–1609.
- Diedrich JF, Bendheim PE, Kim YS, et al. Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. ProcNatlAcadSciUSA. 1991;88:375–379.
- Jen A, Parkyn CJ, Mootoosamy RC, et al. Neuronal low density lipoprotein receptor-related protein 1 binds and endocytoses infectious fibrils via receptor cluster 4. J Cell Sci. 2010;123(2):246–255.
- Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013 Sep;13(9):621–634.
- Chung WS, Clarke LE, Wang GX, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013 Dec 19;504(7480):394–400.
- Srivastava S, Katorcha E, Makarava N, et al. Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci Rep. 2018;8(1):e11326.
- Katorcha E, Makarava N, Savtchenko R, et al. Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLOS Pathog. 2014;10(9):e1004366.
- Baskakov IV, Katorcha E. Multifaceted role of sialylation in prion diseases. Front Neurosci. 2016;10(1):e358.
- Katorcha E, Makarava N, Savtchenko R, et al. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep. 2015;5(1):16912.
- Srivastava S, Makarava N, Katorcha E, et al. Post-conversion sialylation of prions in lymphoid tissues. Proc Acad Natl Sci USA. 2015;112(48):E6654–6662.
- Baskakov IV, Katorcha E. Prion Strain-specific structure and pathology: a view from the perspective of glycobiology. Viruses. 2018;10(12):723.