
ABSTRACT
We report a case of genetic Creutzfeldt-Jakob disease (gCJD), which has a clinical phenotype that is highly similar to Fatal Family Insomnia (FFI) and has a triad of Wernicke-Korsakoff syndrome (WKs) at the developmental stage of the disease. The 51-year-old male complained of sleep disorder and imbalance who had visited five different hospitals before diagnosed. A neurological examination revealed a triad of symptoms characteristic for WKs such as gaze paresis, ataxia of limbs and trunk, and memory disturbances. The disturbances increased during the course of the disease, which led to the death of the patient 18 months after the appearance of the signs. Although the patient show negative in brain magnetic resonance imaging (MRI) and 14-3-3 protein of cerebrospinal fluid (CSF), he was finally diagnosed with gCJD disease by the human prion protein (PRNP) gene mutations.
1. Introduction
It is well known that Prion diseases, a fatal neurodegenerative disorder, differ from the viral or bacterial infectious diseases, because prion-protein (PrP) has no DNA or RNA [Citation1]. Although most patients have no definable cause, about 10% to 15% of patients are caused by point mutations or insertions of octapeptide repeats in PRNP, and they are dominantly inherited. Up to date, at least 30 pathogenic mutations of the PRNP worldwide have led to familial prion diseases [Citation2]. Based on clinical and pathological features, genetic prion diseases are classified into 3 phenotypes: Genetic Creutzfeldt-Jakob disease (gCJD), Gerstmann-Sträussler-Scheinker disease (GSS) and Fatal familial insomnia (FFI) are different subtypes of human genetic prion-protein (PrP) diseases [Citation3]. In this study, We are presenting a case of 51-year-old man with a long history of alcoholism who was admitted to the hospital with intractable insomnia, dizziness and progressive ataxia. The patient was also suffered from noted impaired memory. The rare finding of the case is a gCJD patient shows FFI phenotype.
2. Case presentation
2.1. History
Since the sleep disorder was initial discovered 12 months ago, the patient has been hospitalized in five different hospitals. He attributed his symptoms to busy government tasks, so he did not go to the doctor. Five months after initial symptoms, episodes of dizziness occurred. The patient was no longer able to drive that had previously been easy for him. During his first hospitalization, neurologists found no positive sign, and his Magnetic Resonance Imaging(MRI) was also negative. He was diagnosed with Generalized anxiety disorder(GAD) and discharged from hospital with Paroxetine. After taking the medicine for one month, the patient’s symptoms did not improve and went to another hospital in Beijing. His Mini-Mental State Examination score was 25 and Montreal Cognitive Assessment was 16 on a scale of 0 to 30. Lumbar puncture was performed, and tests for paraneoplastic antibodies and autoimmune encephalitis antibodies in the blood and cerebrospinal fluid(CSF) were negative. MRI of the head exhibits no observable changes from those obtained one month earlier. The electroencephalogram (EEG) and electromyogram(EMG) showed normal waves. After excluding other possible diseases, the patient was still diagnosed with GAD and discharged. One month after the second discharge, the patient was hospitalized in a psychiatric hospital, and he developed leg muscle atrophy this time. Although he could walk independently, he was afraid of falling and reduced his activity. There is no change in the diagnosis and treatment in this hospitalization. Two months after his third discharge, he was hospitalized again because couldn’t stand and keep balance alone. The results of EEG, EMG and brain MRI did not change significantly compared with previous examination results in Beijing. The Montreal Cognitive Assessment(MOCA) score was 14 and the Mini–Mental State Examination (MMSE) score was 22. One month after the fourth discharge, the patient presented dreaminess and sleepwalking, and was hospitalized in the psychiatric hospital again. His sleepwalking was stopped when psychiatrists replaced paroxetine with sertraline, trazodone, and oxazepam. One month after his fifth discharge (12 months after the initial symptoms), he was admitted to our hospital. He was unable to stand steadily even with help of his wife. He had trouble falling asleep and looked anxious. The patient had previously been healthy. He lived in an apartment in Hainan island with his wife and had no sick contacts. He drank alcohol intermittently for more than 10 years. After suffering from sleep disorder 12 months ago, he began to drink heavily, about 6 unit of beer per day.
2.2. Examinations on admission
On physical examination, the patient’s temperature was 36.7°C, heart rate 91 beats per minute, blood pressure 154/93 mmHg, respiratory rate 15 breaths per minute, and oxygen saturation 98% while he was breathing ambient air. He appeared to be fatigued and was temporal and spatial disoriented but able to converse at very low volume. His pupils were 3mm in diameter, round, and reactive to light. The funduscopic examination revealed no evidence of papilloedema. He had horizontal gaze nystagmus. His finger to finger contact was inaccurate, and failed to find knee with heel. His limbs strength decreased slightly, and lower limb tuning fork vibration sense decreased. His deep tendon reflexes were symmetric hyperactivity, Babinski sign was positive on both sides. His MoCA was 11 on a scale of 0 to 30, the loss points focused on visuospatial and executive functions(0/5), delayed recall(1/5), and orientation(2/6). The remainder of neurologic examination was normal.
3. Diagnoses
Localization: Dysfunction of the temporal limbic system can cause emotional disorders, especially sleep disorders, sleepwalking, and memory impairment. The patient was unable to drive because of impaired visual space and executive stress at an early stage, and presented comprehensive cognitive impairment when he was admited, which could not be explained simply by the damage of limbic system. We considered cortical damage caused this phenomenon. Dysfunction of the somatosensory and vestibular inputs into the cerebellum can cause ataxia, and sensory ataxia typically worsens with removal of visual fixation. The patient didn’t present this phenomenon, and had deficits in executive function, linguistic fluency. Another important function of the cerebellum is oculovestibular coordination. This patient had horizontal nystagmus. The patient had tendon hyperreflexia and positive bilateral Babinski signs.
The differential diagnosis of this patient is focused on the progressive disease which starts in limbic system and spreads to other parts of central nervous system. Our initial diagnosis was WKs because of the mental and cognitive dysfunction, oculomotor paralysis, ataxia triad typically observed in alkoholics. However, the patient’s serum thiamine was negative and did not respond to the daily 300 mg thiamine intramuscular. Autoimmune encephalitis may represent limbic lesions, encephalitis syndrome characterized by diffuse brain damage, and the patient’s EEG shows epileptiform discharges in the right frontal lobe. These were the reasons we consider autoimmune encephalitis after ruling out WKs. We tried hormone and immunoglobulin therapy, but the patient continued worsening. At this time, CSF/blood Guillain Barre Syndrome (GBS) related antibody, Neuromyelitis optica spectrum disorder(NMOSD) related antibody negative, cerebrospinal CSF/blood autoimmune encephalitis and paraneoplastic antibody were all(-). Family members began to feel dissatisfied.
Two weeks after admission, the patient’s sleep disorder aggravated further. He did not sleep all night, wandered around in the ward, accompanied by throat wheezing, increased blood pressure(160/96 mmHg) and increased heart rate(103 bpm). We highly suspected prion disease after excluding WKs and autoimmune encephalitis. Except no family history, the patient currently had all three major symptom groups of Fatal Familial insomnia (FFI): sleep related symptoms, neuropsychiatric symptoms, and progressive sympathetic symptoms. However, early memory impairment is rare in FFI.The patient’s family members refused brain biopsy. The CSF 14-3-3 protein was negative, tau/p-tau was significantly increased. At this time, genetic testing is very important for distinguishing FFI and other genetic prion diseases. Finally, the patient’s genetic examination found that the human prion protein gene(RPNP) 129 amino acid polymorphism was M/M type, 219 amino acid polymorphism was E/E type, E200K gene mutation were found. gCJD was diagnosed.
4. Discussion
Alcoholics often suffer from vitamin B12 deficiency and malnutrition. The main manifestations were mental and emotional disorders, cognitive dysfunction, ataxia and eye movement disorder. Less than one-third of the patients who had the above-mentioned triad were finally diagnosed as WKs, it is the main reason we initially considered WKs and decided on thiamine treatment. However, the patient’s MRI did not find WKs typical lesions [Citation4] of the papillary body, thalamus, periaqueduct of the midbrain, or atypical cortical, cerebellar, brainstem lesions (). Considering that the sensitivity of these typical positive MRI findings was only 50% [Citation5], we also performed brain enhanced MRI scan at the same time to reduce false negative [Citation6], but still got negative results (). Stone et al [Citation7].reported a patient with rapidly progressive cognitive impairment who was treated with thiamine 100 mg once per day because of triad. He performed pathological biopsy to confirm CJD because his treatment was not effective. The final pathological diagnosis was WKs, and his patient’s condition was improved with 300 mg thiamine daily. The difference between our case and Stone’s report was the persistent negative MRI and the initial administration of 300 mg thiamine daily [Citation8]. After high-dose thiamine treatment, the patient’s symptoms did not improve. We began to doubt the diagnosis of WKs. The clinical features of autoimmune encephalitis are limbic lobe lesions, such as rapid progression of cognitive dysfunction, sleep disorders and emotional disorders. This patient showed multiple lesions in the cortex and subcortical, cerebellum, and the EEG showed epileptiform discharges in the right frontal lobe, which were consistent with the manifestations of autoimmune encephalitis. Evidences of nonsupporting autoimmune encephalitis were the patient’s emotional disorder without typical behaviour abnormality, had never present epileptic seizures, and negative autoimmune related antibody and MRI examination. Lucchinetti et al [Citation9]. believes that in the early stage of autoimmune encephalitis, the positive predictive value for detection rates vary from 33% for voltage-gated potassium channel complex(VGKC) antibodies to more than 80% for Type 1 antineuronal nuclear antibody(ANNA-1), so the possibility of false negative autoantibodies is high. Since a lesser delay to treatment is shown, immunotherapy should be started immediately once autoimmune dementia is suspected. According to Flanagan et al [Citation10]., more than 35% patients with suspected autoimmune dementia who were initially suspected rapidly progressive neurodegenerative lesions responded to immunotherapy. We gave methylprednisolone 1 g per day and immunoglobulin 0.4 g/kg/day after the failure of thiamine treatment. The biological markers of autoimmune encephalitis may not be found until the clinical symptoms and signs appear. Therefore, we took cerebrospinal fluid and blood samples for examination, including vgkc antibody, ANNA-1 and α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) propionate receptor, which are closely related to limbic encephalitis [Citation11,Citation12]. Finally, we got the negative results again, and the patient did not respond to immunotherapy. The possibility of autoimmune encephalitis decreased. CJD should be highly light when patients have rapidly progressive cognitive impairment, especially when other suspicious diagnoses have been ruled out. Various types of Prion disease account for more than 50% of rapidly progressive cognitive impairment disease [Citation13], and sCJD is the most common type of CJD [Citation14]. Myoclonus does not necessarily appear in early CJD patients. Most patients have typical periodic three-phase wave in the late stage while only atypical diffuse slow wave in the early stage. The sensitivity and specificity of 14-3-3 protein has been questioned, but the positive 14-3-3 protein is also one of the strong supporting evidences for CJD.
Figure 1. MRI DWI: no abnormal signal intensity in papillary body, thalamus, periaqueduct of the midbrain, cerebellar, brainstem
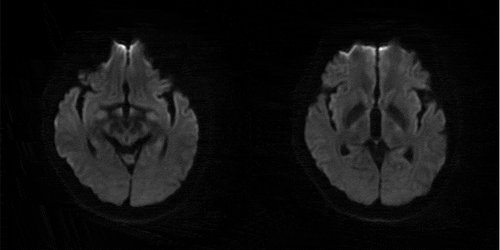
Figure 2. Enhanced MRI: no abnormal signal intensity in papillary body, thalamus, periaqueduct of the midbrain, cerebellar, brainstem
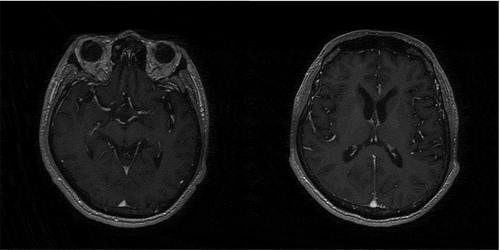
The main manifestations of this patient were progressive mental and emotional disorders, rapidly progressive cognitive impairment, cerebellar ataxia and positive bilateral Babinski signs. Compared the clinical manifestations of FFI [Citation15], this patient also had sleep related uncomfortable movement, laryngeal stridor, hypertension, tachycardia and hyperhidrosis. But there was no confirmed or suspected FFI patient in the first degree relatives, and cognitive dysfunction in the early stage was also uncommon in FFI patients [Citation16]. However, the clinical manifestations that are highly overlapped with FFI make it impossible for us to distinguish before genetic testing. The patient’s CSF 14-3-3 protein was negative, tau/p-tau was significantly increased. 129 amino acid polymorphism of PRNP gene was M/M type, 219 amino acid polymorphism was E/E type, E200K related mutation were found, and gCJD was diagnosed. The Creutzfeldt Jakob disease standard of the world health organization requires that the typical EEG (periodic spike wave complex) or the positive 14-3-3 in CSF, accompanied progressive dementia, leading to death within 2 years. In CSF biomarkers, t-tau shown the highest diagnostic accuracy (79.6%), but 14-3-3 protein might be detected in other acute neuronal atrophy diseases. The approximate sensitivity of 14-3-3 protein was 50% to 92%, and the approximate specificity was 80% [Citation17,Citation18], while the sensitivity and specificity of t-tau are more than 90% [Citation19,Citation20]. Forner et al [Citation21]. pointed out that DWI was more accurate (97%) than CSF biomarkers (14-3-3, t-tau). In this case, MRI and 14-3-3 protein negative are also rare. The final diagnosis of gCJD requires confirmed or clinically confirmed gCJD in first-degree relatives, and/or PrP gene mutations specific to the disease. No confirmed or suspected gCJD cases were found in this patient’s family. The diagnosis of our patient was based on E200K related mutations. Glu200lys (E200K) and asp178asn (D178N) were the most common PRNP mutations. Among more than 30 mutations of PRNP [Citation22], Kim reported a sCJD case with E200G mutation, different from E200K mutation the, patient with this variant had a longer course of disease [Citation23].
5. Diagnostic trap
5.1. Sleep disturbances, such as insomnia and sleep loss, are the common foremost symptoms of the Chinese FFI patients [Citation1,Citation24]. This is consistent with the initial performance of this patient. The clinical manifestations of the end-stage disease and FFI are highly overlapping, which is the difficult point in the differential diagnosis of this case. E200K/M129M/E219E is the most common in gCJD of Chinese population. However, it is rare that sleep disorder is the initial and main manifestations that mimic the clinical phenotype of FFI [Citation25]. Therefore, without PRNP sequencing, it is almost impossible to distinguish E200K gCJD and other types clinically.
5.2. CJD is completely different from WKs in the pathogenesis and pathology. However, rare CJD patients may present simulated clinical manifestations of WKs. The reason for simulating WKs symptoms may be related to the impacted thalamus and papillary body in CJD lesions, which brings difficulties to the diagnosis. We reviewed the CJD mimicking Wernicke encephalopathy cases [Citation16,Citation26–30]. The course from initial symptoms to death ranged from 2 months [Citation16] to 18 months [Citation29]. Two alcoholism patients present typical triad including eye movement disorder, and had cognitive impairment before gait disorder. Among other 6 patients without alcoholism, one patient [Citation30] had mild eye movement disorder, whose dizziness and ataxia occurred before cognitive impairment. One patients [Citation16] had mild recovery in eye movement disorder after thiamine treatment, but the overall condition of all patients continued to deteriorate. Goossens’s case [Citation16] did not present myoclonus, while the rest cases had different degrees of myoclonus at the end of the disease. The imaging findings of the patients may be negative, or show different signal in thalamus [Citation30]. Only one patient [Citation28] has typical periodic triphasic waves at the end of the disease, which is rare in WKs patients. The positive of 14-3-3 protein [Citation16,Citation30] is helpful for the diagnosis. Bertrand et al [Citation31]. large autopsy study found that the incidence of WKs may be higher than reported, 14 of 657 patients diagnosed with CJD were autopsy diagnosed as CJD combined with WKs, which may be due to eating difficulties and malnutrition in CJD patients. CJD combined with WKs make the diagnostic more difficult.
6 Follow-up
The patient died with the company of his family on 10 October 2020. It is a pity that his family did not agree to the autopsy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- He R, Hu Y, Yao L, et al. Clinical features and genetic characteristics of two Chinese pedigrees with fatal family insomnia. Prion. 2019;13(1):116–123. PMID: 31122137.
- Brown K, Mastrianni JA. The prion diseases. J Geriatric Psychiatry Neurol. 2010;23(4):277. PMID: 20938044.
- Capellari S, Strammiello R, Saverioni D, et al. Genetic Creutzfeldt–Jakob disease and fatal familial insomnia insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011;121(1):21–37. PMID: 20978903.
- Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442–455. PMID: 17434099.
- D’Aprile P, Tarantino A, Santoro N, et al. Wernicke’s encephalopathy induced by total parenteral nutrition in patient with acute leukaemia: unusual involvement of caudate nuclei and cerebral cortex on MRI. Neuroradiology. 2000;42(10):781–783. PMID: 11110087.
- Zuccoli G, Siddiqui N, Bailey A, et al. Neuroimaging findings in pediatric Wernicke encephalopathy: a review. Neuroradiology. 2010;52(6):523–529. PMID: 19844698.
- Stone R, Archer JS, Kiernan M. Wernicke’s encephalopathy mimicking variant Creutzfeldt-Jakob disease. J Clin Neurosci. 2008;15(11):1308–1310. PMID: 18829327.
- Day E, Bentham P, Callaghan R, et al. Thiamine for Wernicke-Korsakoff syndrome in people at risk from alcohol abuse. Cochrane Database Syst Rev. 2004;(1):CD004033. PMID: 14974055. DOI:https://doi.org/10.1002/14651858.CD004033.pub2
- Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology. 1998;50(3):652–657. PMID: 9521251.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc. 2010;85(10):881–897. PMID:PMC2947960.
- Mittal MK, Rabinstein AA, Hocker SE, et al. Autoimmune encephalitis in the ICU: analysis of phenotypes, serologic findings, and outcomes. Neurocrit Care. 2016;24(2):240–250. PMID: 26319044.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65(4):424–434. PMID:PMC2677127.
- Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97–108. PMID:PMC2647859.
- Geschwind MD. Rapidly progressive dementia. Continuum (Minneap Minn). 2016;22(2Dementia):510–537. PMID:PMC4879977.
- Krasnianski A, Sanchez Juan P, Ponto C, et al. A proposal of new diagnostic pathway for fatal familial insomnia. J Neurol Neurosurg Psychiatry. 2014;85(6):654–659. PMID:PMC4033028.
- Goossens K, van Bruchem-Visser RL. A patient with a ‘typical presentation’ of Wernicke encephalopathy was found to have sporadic Creutzfeldt-Jakob disease. Neth J Med. 2017;75(5): 211–214. PMID: 28653943.
- Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American academy of neurology. Neurology. 2012;79(14):1499–1506. PMID:PMC3525296.
- Kim MO, Geschwind MD. Clinical update of Jakob-Creutzfeldt disease. Curr Opin Neurol. 2015;28(3):302–310. PMID: 25923128.
- Coulthart MB, Jansen GH, Olsen E, et al. Diagnostic accuracy of cerebrospinal fluid protein markers for sporadic Creutzfeldt-Jakob disease in Canada: a 6-year prospective study. BMC Neurol. 2011;11:133. PMID:PMC3216246.
- Van Harten AC, Kester MI, Visser PJ, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med. 2011;49(3):353–366. PMID: 21342021.
- Forner SA, Takada LT, Bettcher BM, et al. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease. Neurol Clin Pract. 2015;5(2):116–125. PMID:PMC4404282.
- Gambetti P, Kong Q, Zou W, et al. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. PMID: 14522861.
- Kim MO, Cali I, Oehler A, et al. Genetic CJD with a novel E200G mutation in the prion protein gene and comparison with E200K mutation cases. Acta Neuropathol Commun. 2013;1:80. PMID:PMC3880091.
- Sun L, Li X, Lin X, et al. Familial fatal insomnia with atypical clinical features in a patientwith D178N mutation and homozygosity for Met at codon 129 of the prion protein gene. Prion.2015;9(3):228-235. PMID: 26074146.
- Gao L P , Shi Q , Xiao K , et al. The genetic Creutzfeldt-Jakob disease with E200Kmutation: analysis of clinical, genetic and laboratory features of 30 Chinese patients[J]. Scientific Reports, 2019;9(1). PMID: 30755683.
- Gaytan-Garcia S, Gilbert JJ, Deck JH, et al. Jakob-Creutzfeldt disease associated with Wernicke encephalopathy. Can J Neurol Sci. 1988;15(2):156–160. PMID: 3289705.
- Pietrini V. Creutzfeldt-Jakob disease presenting as Wernicke-Korsakoff syndrome. J Neurol Sci. 1992;108(2):149–153. PMID: 1517746.
- Nagashima T, Okawa M, Kitamoto T, et al. Wernicke encephalopathy-like symptoms as an early manifestation of Creutzfeldt-Jakob disease in a chronic alcoholic. J Neurol Sci. 1999;163(2):192–198. PMID: 10371084.
- Bielewicz J, Szczepanska-Szerej A, Ogorek M, et al. Wernicke-Korsakoff syndrome as a rare phenotype of sporadic Creutzfeldt-Jakob disease. Prion. 2018;12(2):143–146. PMID:PMC6016515.
- Iwasaki Y, Hashimoto R, Saito Y, et al. An autopsied case of MM1-type sporadic Creutzfeldt-Jakob disease with pathology of Wernicke encephalopathy. Prion. 2019;13(1):13–20. PMID:PMC6422394.
- Bertrand A, Brandel JP, Grignon Y, et al. Wernicke encephalopathy and Creutzfeldt-Jakob disease. J Neurol. 2009;256(6):904–909. PMID: 19252796.