
ABSTRACT
Prion diseases are a group of incurable zoonotic neurodegenerative diseases (NDDs) in humans and other animals caused by the prion proteins. The abnormal folding and aggregation of the soluble cellular prion proteins (PrPC) into scrapie isoform (PrPSc) in the Central nervous system (CNS) resulted in brain damage and other neurological symptoms. Different therapeutic approaches, including stalling PrPC to PrPSc conversion, increasing PrPSc removal, and PrPC stabilization, for which a spectrum of compounds, ranging from organic compounds to antibodies, have been explored. Additionally, a non-PrP targeted drug strategy using serpin inhibitors has been discussed. Despite numerous scaffolds being screened for anti-prion activity in vitro, only a few were effective in vivo and unfortunately, almost none of them proved effective in the clinical studies, most likely due to toxicity and lack of permeability. Recently, encouraging results from a prion-protein monoclonal antibody, PRN100, were presented in the first human trial on CJD patients, which gives a hope for better future for the discovery of other new molecules to treat prion diseases. In this comprehensive review, we have re-visited the history and discussed various classes of anti-prion agents, their structure, mode of action, and toxicity. Understanding pathogenesis would be vital for developing future treatments for prion diseases. Based on the outcomes of existing therapies, new anti-prion agents could be identified/synthesized/designed with reduced toxicity and increased bioavailability, which could probably be effective in treating prion diseases.
Introduction
Prion diseases (Transmissible spongiform encephalopathies: TSEs) are a set of rare and deadly zoonotic neurodegenerative diseases (NDDs), affecting both humans (Creutzfeldt-Jackob disease: CJD, variant CJD: vCJD, Fatal familial insomnia: FFI, Gerstmann-Straussler-Scheinker syndrome: GSS, Kuru) and animals (Bovine spongiform encephalopathy: BSE, Chronic wasting disease: CWD, Scrapie, Feline spongiform encephalopathy: FSE, Transmissible mink encephalopathy: TME, Ungulate spongiform encephalopathy: USE).
PrPC is a 32 kDa glycophosphatidylinositol (GPI)-anchored glycoprotein expressed in many tissues and abundantly in the nervous tissues [Citation1]. The molecular pathogenesis of prion diseases included the collection of misfolded prion protein (PrPSc) [Citation2]. The normal PrP (PrPC) exists as a monomeric, α-helical, soluble, and protease-sensitive protein [Citation3]. On the contrary, scrapie isoform (PrPSc) is a multimeric, β-sheet rich [Citation4], insoluble, protease-resistant [Citation5,Citation6], and infectious form [Citation7]. The progressive abnormal protein aggregation in the Central nervous system (CNS) affects normal brain structure (formation of spongiform, like holes in the brain tissue) with reduced functions (cognition, memory, movement).
Even though CNS is the key target of prion pathology nevertheless many prion diseases are complemented by prion replication at extra cerebral sites (viz. secondary lymphoid organs, blood, muscle) [Citation8]. PrPC is the responsible molecule for transport via the peripheral nervous system (PNS) into the brain. However, the transport mechanism of neuronal prion from the PNS to the CNS and/or vice versa is poorly understood. Two hypotheses were proposed for prion propagation: the ‘Domino hypothesis’ (prion propagation occurring on the neuronal cell) [Citation9] and the ‘Streetcar hypothesis’ [Citation10] where the nerve endings take up PrPSc and transport them back to the CNS.
Prion diseases can be acquired (from contaminated foods or medical procedures), sporadic (unknown cause), or genetic (from mutations in the PrP gene: PRNP). Truncated mutation of the PrP results in PrP systemic amyloidosis characterized by late-onset sensory and autonomic neuropathy. The symptoms of this disease are comparable to an autonomic dominant NDD, familial amyloid polyneuropathy (FAP), which results from mutations in the transthyretin (TTR) gene [Citation11]. The two diseases can be clinically differentiated by the length of the disease history, FAP being rapidly progressing compared to PrP systemic amyloidosis [Citation12].
Approximately 1–2 people per million are annually affected by prion diseases [Citation13], out of which 85–90% of cases are of sporadic CJD (sCJD) [Citation14]. The major common clinical symptoms of CJD patients include prompt and progressive cognitive debility, difficulty in balance, lack of coordination, visual impairment, and behavioural changes. Some of these symptoms are similar to AD and HD however, CJD patients have a characteristic sponge-like appearance of the brain tissue. The average survival time and survival rate of patients with sCJD in China and other Western countries are 7.1 months with 78.5% mortality within a year of the disease onset [Citation15]. On the other hand, the survival statistics are better in Japan, where the survival period is at the highest of 17.4 months with 46% mortality [Citation16]. The diagnostic tests for CJD are; the detection of periodic synchronous discharge (PSD) or periodic sharp wave complex (PSWC) in electroencephalography (EEG) [Citation17–19], cerebrospinal fluid (CSF) analysis [Citation20], including real-time quaking-induced conversion (RT-QuIC) assays of CSF and nasal-brushing [Citation21,Citation22], cranial magnetic resonance imaging (MRI) [Citation23] and immunohistochemistry of prions in brain tissues from the biopsy.
Numerous reports on prospective targets for treating the prion disease have been investigated. An ample amount of literature is available from various research methodologies, like cell therapy, immunotherapy, pharmacotherapy, and compounds ranging from chemicals to proteins. Compounds destabilizing PrPSc and reducing infection have also been identified. Many of them revealed encouraging results in vitro, while a few in vivo studies were performed without success in the clinical trials. As a result, unfortunately, no conclusive treatment for prion diseases existed, hence, only medicines for easing the disease-associated symptoms are being used. For example, antidepressants and sedatives are prescribed for psychological symptoms; myoclonus can be treated with clonazepam and sodium valproate; donepezil, galantamine, rivastigmine, and memantine are used to treat dementia in CJD patients [Citation24,Citation25]. Recently, a prion protein monoclonal antibody (PRN100) has shown promising results in a clinical trial study on CJD patients [Citation14].
The present review provided an updated and comprehensive outlook on various classes of anti-prion agents, the mode of action, and some other important aspects that would be helpful for further research in this field.
Anti-Prion agents in therapeutics
Nuclear magnetic resonance (NMR) and X-ray crystallography of the PrPC domain revealed a small β-sheet content and α-helical conformation at the C-terminal (amino acid residues 128–231) [Citation26] whereas, the N-terminal region (amino acid residues 23–127) lack proper folding [Citation27] and it is composed octapeptide repeats (OR) (residues 60–91), each repeat with histidine and tryptophan which coordinate up to four copper (Cu II). The fifth copper binding site is located in the non-OR region (His 92 and His111) [Citation28,Citation29]. A palindromic motif (AGAAAAGA) exists between residues 113–120 which is believed to be responsible for prion conversion [Citation30]. The closeness of the palindromic motif to the non-OR region indicates a link between Cu(II) binding and prion conversion [Citation31]. Giachin et al. proposed a model in which PrPC coordinating copper with one His more prone to conversion at acidic conditions suggesting the non-OR region is a key regulator of prion conversion [Citation32]. A recent study used biophysical techniques to demonstrate that the binding of Cu(II) to non-OR region result in compacted conformation and hence affect the structural plasticity of the region. Additionally, Cu coordinate geometries identified as: Type I (closed) and Type 2 (open) for PrPC and PrPSc, respectively [Citation33].
In the pathogenesis of prion diseases, PrPSc proliferates by seeding its altered conformation in the presence of PrPC as a substrate [Citation34]. Thus, besides acting as a substrate, PrPC oligomers are transducers of neurotoxicity [Citation35]. Hence, the anti-prion agents that can restrict the conversion from PrPC to PrPC oligomers or PrPSc will be of therapeutic importance. These interactions or polymerization inhibitions can be achieved by binding to PrPC and/or PrPSc. As the plasma membrane is the primary site for the conversion of PrPC to PrPSc [Citation36, Citation37
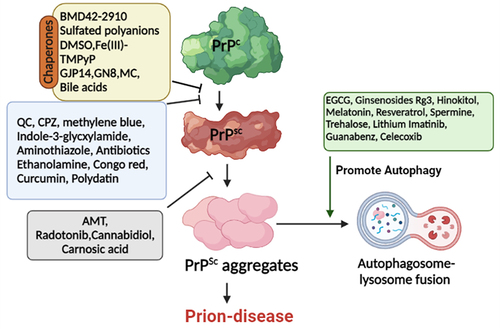
], PrPC redistribution is another key target of anti-prion agents. Additionally, subduing PrPC expressions and targeting other pathways to reduce disease-associated neurotoxicity have also been explored. Henceforward, the anti-prion agents work by either of these mechanisms i.e. specific conformational stabilization of PrPC; non-specific stabilization of PrPC; prevention of PrPSc accumulation, and preventing proliferation by the interaction of PrPSc with molecules other than PrPC [Citation38]. Other approaches include the autophagy regulations and the use of chaperones to assist cellular trafficking of PrPSc and stabilization of PrPC, respectively. The mode of action of several anti-prion agents is illustrated in .
Figure 1. Schematic representation of some anti-prion agents targeting prion proteins. PrPC, PrPSc, and the conversion of PrPC to PrPSc are the main targets of these anti-prion agents. Chaperones directly interact with PrPC thereby stabilizing it and preventing/reducing its conversion to PrPSc. Another set of compounds clears the toxic aggregates by promoting autophagy. Abbreviations: PrPC: normal prion protein; PrPSc: scrapie prion protein; EGCG: Epigallocatechin-3-gallate; AMT: Aminothiazoles; CPZ: Chlorpromazine; DMSO: Dimethylsulfoxide; QC: Quinacrine.(Prepared by Biorender.com).
1. Chemical compounds
In this section, various important scaffolds used for anti-prion studies have been discussed. Information on the binding of these compounds to prion protein and the structure-activity relation (SAR) of synthetic derivatives has also been mentioned. summarizes various aspects covered in this section.
Table 1. Chemical-based anti-prion compounds.
2-Aminothiazoles (2-AMT)
2-AMTs are a class of small molecules with anti-prion activity. Initially, it was proposed that 2-AMTs do not affect PrPC expression, but they can influence PrPSc formation/removal [Citation39,Citation40]. The improved 2-AMT analogues displayed an effective anti-prion activity (EC50 81 nM) in scrapie’s infected neuroblastoma cells (ScN2a-cl3) through oral administration in the animal model. It readily attained therapeutic steady-state concentration in the brain [Citation41] indicating its ability to cross the blood-brain-barrier (BBB) easily. 2-AMT derivatives, IND24 and IND81, considerably extended the life span of scrapie’s infected mouse or with CWD prion models. However, the mice exhibited advanced neurologic dysfunction in due course, showing the limited efficacy of these 2-AMTs [Citation42]. Another 2-AMT derivative, IND125, can prevent both PrPSc accumulation and astrocytic gliosis in mice cerebrum, but the progressive CNS dysfunction could not be prevented, probably due to the build-up of PrPSc in the brain stem [Citation43]. Despite good in vitro profile, bioavailability, and extension of survival time in mice, 2-AMTs failed in other animal experiments. Furthermore, combination therapy with IND24 and Anle138b had no significant effect in extending the infection time than monotherapy, proposing the inability of a combination therapeutic approach [Citation44] in the treatment of prion diseases.
Benzoxazole derivative
BMD42-2910 is a recently discovered anti-prion agent, which exhibited low EC50 in vitro without toxic effects in the mouse model [Citation45,Citation46]. The compound significantly reduced PrPSc in prion-infected mice brains and extended the survival time. Molecular docking experiments predicted the binding interactions between PrPC and BMD42-2910 at 4 amino acid residues, namely Asn159, Gln160, Lys194, and Glu196 [Citation46]. These results indicated that BMD42-2910 is a promising novel anti-prion compound, which should be investigated further for keeping clinical trials in mind.
Carbazole derivatives
GJP14 or [2,3,4,9-Tetrahydro-9-[2-hydroxy-3-(1-piperidinyl) propyl]-6-methyl-1 H-carbazol-1-one] has inhibited PrPSc accumulation (IC50 8.54 µM) in cellular assay by GT+FK cells [mouse neuronal cells (GT1-7) persistently infected with mouse-adapted GSS agent (Fukuoka-1 strain)] [Citation47]. Taking it as a lead compound, various derivatives have been synthesized and screened for anti-prion activity. The SAR revealed the importance of the tricyclic aromatic ring, an amino group at the 3-position, and a hydroxy group at the 2-position of the N-propyl group for displaying anti-prion activity. As a result, the N-ortho-fluorobenzyl analogues (IC50 1.11 µM) was found to be the most effective approximately 8 times stronger than the parent compound [Citation47]. The binding properties of GJP14 were elucidated by surface plasmon resonance (SPR) and NMR spectroscopy, which displayed a ligand-binding pocket at the C-terminal domain of PrPC [Citation48]. Nevertheless, reports on in vivo studies of GJP14 on prion-infected models are pending.
Cyclic tetrapyrroles
Cyclic tetrapyrroles are planar aromatic ring structures with metal ions, defined by peripheral substituent groups of different chemical nature. Porphyrins (deuteroporphyrin IX 2,4-bis-(ethylene glycol) iron(III) (DPG2-Fe3+), meso-tetra (2- N-methylpyridyl)porphine iron(III) (TMPP-Fe3+), tetra (4- N,N,N-trimethylanilinium)porphine (Fe-TAP), tetra (4- sulphonatophenyl)porphine (Fe-TSP), haemin) and phthalocyanines belong to this group. Tetrapyrroles have been reported to inhibit PrPSc oligomerization in vitro [Citation49] and increase the survival time in vivo [Citation50–52]. SPR revealed the direct interactions of cyclic tetrapyrroles with PrPC or PrPSc [Citation53], which bind to PrPC by stacking and interrelating through the N-terminal of PrP [Citation54,Citation55]. NMR studies identified residues at the C terminal of helix 3 and in the loop (between residues 160–180) on human PrP as the binding site of Fe(III)-TMPyP (KD 4.52 µM) responsible for inhibition of prion replication (EC50 1.6 µM) [Citation56] and PrPC-mediated toxicity in the cell-based assay [Citation57]. Binding of Fe(III)-TMPyP seemed to stabilize the folded PrP conformation both thermodynamically and kinetically [Citation58]. Tetrapyrroles proved as the most potent pharmacological chaperones of PrPC in vitro and cell-based assays, but disappointingly, the therapeutic capabilities of porphyrins could not be approved in clinical trials, owing to poor bioavailability and non-specific interactions with the plasma proteins [Citation58].
Diazo dyes
Congo red (CR) is a conventional diazo dye used for histological studies with binding affinity to fibril proteins rich in β-sheet conformation [Citation59,Citation60]. CR has anti-prion activity and inhibits PrPSc formations [Citation61] by altering the cellular levels of PrPC, stabilizing PrPSc, and preventing the further conversion of PrPC [Citation62–65]. SPR displayed CR binding to human recombinant PrP with KD value of 1.6 µM [Citation66]. A few other related dyes (Sirius Red and other sulphonated dyes) were also reported to block PrPSc generation in vitro [Citation67]. However, despite its strong in vitro anti-prion activity, CR is considered potentially toxic (as benzidine derivative is released during catabolism of CR) and displays poor bioavailability in vivo [Citation68,Citation69] with high non-specific binding [Citation38]. To increase the bioavailability (by replacing sulphonate of core moiety with carboxylic acid) and decrease toxicity (replacing benzidine), a series of CR analogues were synthesized and screened for anti-prion activity [Citation70]. Different analogues of CR inhibited PrPSc aggregation in the nano-molar range. The most active compound, 2a, potentially inhibited (EC50 25–50 nM) PrPSc formation in scrapie mouse brain cells (SMB) [Citation71] and scrapie-infected hamsters [Citation72]. However, no conclusive report is available on its interactions with PrPC.
Diphenylmethane derivatives
Evidence from the in silico-based drug screening revealed that GN8, a diphenylmethane derivative, stabilizes PrPC conformation by acting as a chaperone and decreasing PrPSc levels (IC50 ~ 1.35 µM) considerably in a mouse neuronal cell line, prolonging the survival of TSE-infected mice (subcutaneous injection) [Citation73]. SPR confirmed the explicit binding of GN8 to PrPC. Most of the residue involved in the binding is located at the C-terminal domain i.e. placed in the S2-A loop, the B-helix, or the B-C loop. The intercalation of the A-S2 loop and B-C loop seems to be crucial for stabilizing PrPC conformation. GN8 connected distant Asp159 and Glu196 (36 amino acids apart in PrPC) by hydrogen bonds, due to which further conformational changes are blocked. The free energy of the PrPC–GN8 complex (∆∆H = 6.7 kcal/mol) is considerably lower than that of PrPC alone, which ultimately reduced the chances of the formation of a transition state, stabilizing PrPC conformation [Citation73]. Later, additional GN8 derivatives were synthesized and analysed for anti-prion activity in TSE-infected mouse neuronal cells. SAR indicated better anti-prion activity (IC50 0.51–0.83 μM) of derivatives bearing substituents at the benzylic position [Citation47,Citation74]. Positron emission tomography (PET) images from the 11C-labelled compound established that the GN8 derivative can cross BBB and enter the brain [Citation48]. Additionally, the nonclinical safety assessment of GN8 did not show any severe adverse effects at doses sufficient for anti-prion activity [Citation75]. In further studies for better active chaperone, researchers modified GN8 to a medical chaperon (MC: N,N'-([cyclohexylmethylene]di-4,1-phenylene)bis(2-[1-pyrrolidinyl]acetamide). MC binding to PrPC reduced the fluctuations in the binding site (amino acid residues 186–196) and increased the instability in other regions (amino acid residues 144–154) of helix A. Moreover, it inhibited PrPSc oligomerization and favoured its removal in the infected cells. Interestingly, MC exhibited effectiveness in a variety of strains (Fukuoka-1, 22 L, RML) and hosts (mice, macaque), preventing the generation of drug resistance [Citation76]. Due to the encouraging results shown by MC in prion disease, scientists are working towards human clinical trials for prion diseases.
Diphenylpyrazole derivatives (DPP)
Diphenylpyrazole analogues are potent inhibitors of PrPSc oligomerization in vitro. The best efficient inhibitor, named DPP-1, displayed an IC50 value of 0.6 μM and 1.2 μM in SMB and ScN2a cells, respectively [Citation77]. In a SAR study, the methyl group was documented as a significant motif for the improved activity in SMB cells, and not in ScN2a cells; on contrary, the activity dropped in both cell lines when fluorine was replaced by bromine [Citation78]. In ScN2a cells, DPP-1 neither affected PrPC expression nor proteasome activity and also failed to show any effect in a cell-free assay. Nevertheless, it delayed the clinical onset of the disease in mice with no reported toxicity in vitro or in vivo [Citation77].
Anle138b, 3-(1,3-Benzodioxol-5-yl)-5-(3-bromophenyl)-1 H-pyrazole, inhibited PrPSc formation in a cell-free protein misfolding cyclic amplification (PMCA) and mice infected with different PrPSc strains (EC50 7.1 μM for the vCJD; 7.3 μM for the murine RML). Impressively, it also prolonged the survival in late prion infection [Citation79]. Excitingly, Anle138b also inhibited α-synuclein aggregation [Citation80] for its potential therapeutic role in the pathophysiology of Parkinson’s disease (PD). Therefore, Anle138b can be a promising new lead against other protein misfolding diseases. When the mode of action of Anle138b was elucidated using all-atom molecular dynamic stimulations, it interacted with the unorganized protein through its pyrazole moiety, blocking inter-peptide interactions and spontaneous formation of β sheets. These interactions helped in reducing intramolecular hydrogen bonding and remodelling from the allosteric induction. However, another analog, Anle234b, did not affect aggregation in this study [Citation81]. No doubt Anle138b is a non-toxic promising lead compound with the potential for oral administration. Still, it is not a cure as it only delays the disease progression and is not a disease-modifying candidate. A ‘First-in-human study to assess the safety, tolerability, and pharmacokinetics of Anle138b in healthy male and female subjects’ cleared Phase 1 [Citation82]. The outcome of the study indicated that the drug’s half-life is 12 h, and plasma levels significantly exceeded those required for the efficacy. The doses up to 300 mg/day have been considered safe. The company (MODAG GmbH) started a Phase 1b study for PD with Anle138b [Citation81]. The trial will evaluate the well-being, acceptability, and pharmacokinetics of 150 mg and higher daily doses. The study will be completed this year [Citation83].
dimethyl sulphoxide (DMSO)
DMSO is an aprotic solvent frequently used in laboratories. It has been used in the management of many peripheral amyloidotic diseases since it could block intermediate β-sheet formation [Citation84,Citation85]. DMSO administration has significantly increased the survival time in prion-infected hamsters and decreased PrPSc accumulation in the brains of infected animals which was later detected in the urine [Citation86]. Although DMSO is unable to solubilize preformed PrPSc aggregates, it can successfully subdue the PrPSc accumulation arising during the extraction of prion-infected membranes by detergents [Citation87], suggesting its role as a chemical chaperone [Citation88].
Ethanolamine
Ethanolamine is a newly discovered anti-prion compound, which effectively reduced PrPSc levels in prion-infected N2aC24L1-3 cells in a dose-dependent manner. In the same study, oral administration of ethanolamine proved effective against RML prion-infected mice [Citation89]. Ethanolamine neither affected the subcellular localization of PrPC nor disturbed the in vitro PrPC to PrPSc conversion. Ethanolamine is required for phosphatidylethanolamine biosynthesis, a key lipid component of the cellular membranes [Citation90], which in turn facilitated the conversion of PrPC into PrPSc [Citation91]. Contrarily, in yet another study phosphatidylethanolamine blocked the conversion of PrPC into PrPSc [Citation92]. Therefore, more investigations should be performed to confirm the role of ethanolamine in prion diseases [Citation89].
Indole-3-Glyoxylamides
Indole-3-glyoxylamide scaffold is of medicinal significance and is being used in the treatment of atopic dermatitis [Citation93], HIV [Citation94], some cancers [Citation95], and acute coronary syndrome [Citation96]. A series of indole-3-glyoxylamides have been synthesized and assessed for anti-prion activity in SMB cells, where the two most potent compounds, 1 (EC50 1.5 μM) and 2 (EC50 6.4 μM) were identified as anti-prion agents. However, it is interesting to note that indole-3-glyoxylamides did not interact directly with PrPC up to 40 μM [Citation97].
The SAR studies revealed that glyoxylamides derived from primary aromatic amines and bearing a para-substituent are the key requirements for optimum potency in the SMB cell line assay. Based on these results further modifications have been carried out, where indole-3-glyoxylamides unsubstituted at N-1 exhibited higher activity, and there must be no substituent at C-2 for anti-prion activity. The best effective compounds possessed a p-amino substituent: p-piperidino > p-NMe2 > p-morpholino > p-pyrrolyl > p-1 H-pyrazolyl with IC50 in nanomolar range (72 nM >26 nM >9 nM >6 > 1 nM, respectively) [Citation97,Citation98]. Additionally, substitution at C-6 (R3 = 6-Me, 6-CN or 6-NO2) resulted in improved stability, low toxicity, and better EC50 values in comparison to the unsubstituted parents [Citation99]. However, no report of in vivo studies on prion-infected models is available.
Lithium (Li)
Lithium is a silvery-white alkali metal with potential neuroprotective activity in the NDDs [Citation100,Citation101]. Furthermore, the role of Li as an anti-prion agent has been assessed in prion-infected cell cultures, where Li reduced PrPSc aggregation through the autophagy mechanism [Citation102]. Li is reported to block the activation of glycogen synthase kinase 3 (GSK-3), a critical enzyme involved in prion-induced neurodegeneration [Citation103]. Song et al. [Citation104,Citation105] suggested that Li exhibited the neuroprotective effect by restoring survival-associated proteins (RE1 Silencing Transcription Factor: REST & Wingless-related integration site: Wnt), reducing reactive oxygen species (ROS), and repairing synapses. However, it had a narrow therapeutic window (0.5–1.2 mEq/L plasma concentration) and is toxic beyond the above concentration [Citation105]. Administration of Li microemulsion (NP03) in prion-infected mice at a wide range of doses (40 µg/kg/day-16 mg/kg/day) extended the survival of infected animals, even at the lowest dose with a slight reduction in PrPSc level [Citation106]. The encouraging results of NP03-Li micro emulsion in improving the neuropathology are prominent in prion mouse models, even at late stages of the disease in comparison with Li alone, suggesting its possible use in the chronic treatment of prion disease.
Phenothiazine derivative
Methylene Blue (MB), a phenothiazine derivative, can bind to PrPC and affect oligomerization kinetics [Citation107] in a study with human, ovine, and murine recombinant PrP. NMR results indicated that MB can bind to Asn146, 156; Tyr160; Lys188; Thr191,194, 195; Val192, and Gln215 residues in helix 1–3 [Citation107]. However, the non-specific binding of MB to other proteins could limit its use [Citation108].
Sulphated polyanions
Pentosan polysulfate (PPS) is one of the important drugs in prion disease after quinacrine. The polyanionic compounds interact with N-terminal His, while the planar aromatic compounds form π-stacks and intercalate within Trp side chains [Citation55,Citation109]. The binding is responsible for down-regulating PrPC expression [Citation110] or PrPC internalization [Citation111]. Isothermal titration calorimetry (ITC) showed that PPS has two binding sites on PrP, which can bind to multiple PrP molecules, delaying refolding. Force spectroscopy measurements revealed that PPS stabilizes both native PrP and other incompletely folded intermediates of PrP, as a chaperone [Citation112]. PPS has also displayed its anti-prion potential in cell cultures by inhibiting prion proliferation [Citation113] and prolonging the survival times prophylactically and therapeutically in prion-infected mice [Citation114,Citation115]. It was speculated that PPS may competitively affect the binding of PrPC and PrPSc by endogenous glycosaminoglycans (GAG) [Citation113] or fragment PrPSc at the cell surface, like heparin-mimetic compounds [Citation116]. In mouse neuroblast cells overexpressing chicken PrPC, PPS can enhance the PrPC internalization and redistribution into late endosomes and/or lysosomes [Citation111], in addition to reducing PrPC levels at 100 µg/ml [Citation117]. On the contrary, PPS decreased the PrPSc levels promptly in N2a-3 cells (a sub clone of mouse neuroblastoma cell line Neuro 2a), without affecting the intracellular distribution of PrPC up to 10 µg/ml [Citation118]. PPS treatment also inhibited PrPSc aggregation in a CWD-infected deer-cell model with similar efficacy as reported in RML-infected N2a cells [Citation119]. PPS extended the survival in vCJD subjects although no clinical benefit is reported [Citation120]. The poor BBB permeability of PPS can be improved by intraventricular delivery [Citation115,Citation121], but there was no considerable change in the pathology of treated and untreated groups, unfortunately [Citation122].
Dextran sulphate (DS) displayed minimal cytotoxicity with powerful anti-prion activity by blocking the conversion of PrPC [Citation123]. In N2a-58 cells, DS specifically inhibited PrPSc attachment to the membrane and its propagation [Citation124]. Its administration also delayed the onset of the disease in mice [Citation125,Citation126] and hamsters [Citation127] within 2 h of infection, afterwards, it became ineffective [Citation125].
Cyclodextrans are used as additives for various purposes in the food industry [Citation128]. The β-form of cyclodextrin was found to be more efficient (IC50 75 µM) in removing PrPSc from scrapie-infected neuroblastoma cells than the α-form (IC50 750 µM). The SAR discovered that anti-prion activity is reliant on the size of the cyclodextrin. The structural orientation of glucopyranose units in cyclodextrin creates a hydrophobic cavity, which can encapsulate the hydrophobic moieties [Citation129]. The anti-prion activity of cyclodextrin can be enhanced on sulphation due to its chaperoning function for assisting the proper folding of PrPC, and not due to its anionic character [Citation130].
2. Repurposed drugs
Even though the drugs are also chemical compounds, they will be discussed under a separate heading. In this section, drugs with United States Food and Drug Administration (US-FDA) approval will be discussed, which are already in the market for the treatment of a range of other diseases with acceptable pharmacokinetics and safety profiles. These drugs have been repurposed to study their efficacy in treating prion diseases ().
Table 2. Repurposed drugs as anti-prion compounds.
Celecoxib
Celecoxib is a FDA-approved non-steroidal anti-inflammatory drug (NSAID) [Citation131] for inhibiting cycloxygenase (COX-2). It can also block PrPSc-induced microglia activation and releases prostaglandin (PGE2) and nitric oxide (NO) [Citation132]. On the other hand, ketoprofen (COX-1 inhibitor) was not able to inhibit microglial activation and the associated reactions caused by PrPSc, suggesting the role of PrPSc-induced inflammation in prion progression [Citation132]. An anticancer agent, celecoxib derivative: AR-12 (OSU-03012), was assessed by FDA as an approved investigational new drug (IND) in phase I of clinical trials for advanced or recurrent solid tumours or lymphoma patients [Citation133]. Abdurahman et al. evaluated the effect of AR-12 and its derivative, and AR-14 revealed anti-prion properties in neuronal and non-neuronal cell culture models [Citation134]. AR-12 showed a significant reduction of PrPSc in prion-infected ScN2a and ScMEF cells. Prolonged treatment eliminated PrPSc in a sensitive RT-QuIC analysis, even when the treatment was discontinuation. It has been suggested that both AR-12 and AR-14 can induce autophagy in cells and eradicate infection [Citation134]. Notably, AR-12 can cross BBB successfully [Citation135], which is an essential requirement for an anti-prion drug. Autophagy is the natural cellular degradation and reprocessing pathway, in which the dysfunctional components from the cytoplasm are packed in vesicles and delivered to the lysosomes for degradation. Thus, by enhancing the autophagic flux, PrPSc lysosomal degradation can also be enhanced, providing a therapeutic approach for prion diseases [Citation136].
Chlorpromazine (CPZ)
CPZ, a phenothiazine derivative, is a US-FDA-approved antipsychotic drug [Citation137], which can cross BBB. CPZ inhibited PrPSc formation (EC50 3 μM) [Citation138] by binding to human recombinant PrP by acting as a pharmacological chaperone. However, its interactions are weaker than quinacrine (antimalarial drug) [Citation53,Citation139,Citation140]. Unfortunately, CPZ failed to show results in protein misfolding cyclic amplification (PMCA) assay [Citation141] and was unable to inhibit PrPSc build-up in the cellular assay [Citation142]. It was suggested that CPZ may induce PrPSc reallocation in late endosomes/lysosomes [Citation143]. CPZ was found to be 10 times less active than quinacrine, which completely inhibited PrPSc formation at 400 nM [Citation45]. Administration of CPZ prolonged the incubation duration and somewhat reduced the morbidity in mice infected intracerebrally with prions [Citation144].
Efavirenz (EFV)
EFV is a US-FD- approved oral non-nucleoside reverse transcriptase inhibitor (NNRTI) medication to treat the human immunodeficiency virus (HIV). Importantly, EFV could effectively cross BBB with a significant reduction in Cytochrome P450 Family 46 Subfamily A Member 1 (CYP46A1) levels in prion disease [Citation145]. Therefore, being a CYP46A1 activator, EFV can be used on prion-infected neuronal cells and mice with considerably alleviated PrPSc proliferation, without affecting PrPC [Citation145]. Hence, EFV might prove to be an effective drug in prion diseases.
Flupirtine (FLU)
FLU, a triaminopyridines, is a famous, well-tolerated, non-opiate analgesic drug with additional myorelaxant properties [Citation146]. It does not have US-FDA approval but is permitted in Europe [Citation147]. Earlier studies demonstrated that FLU might act as an antagonist of N-methyl-D-aspartate receptor (NMDAR) without binding to PrPC [Citation148], but can interact with NMDARs on neurons and modulate NMDAR-dependent neuronal excitability and excitotoxicity [Citation149]. The binding of PrPSc to PrPC on the cell surface could initiate a signalling cascade, which leads to the activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and NMDAR, the inflow of calcium ions, and stimulation of p38 mitogen-activated protein kinase (MAPK): a significant modulator of microglial neuroinflammation. Eventually, the actin cytoskeleton breaks down leading to the degradation of dendritic spines, thus the excitatory neurotransmission decreases [Citation150].
FLU protected the neuronal cells from neurotoxicity, which could be induced by PrP fragmentations and amyloid beta induction in vitro [Citation151]. A double-blind study on CJD patients [Citation152] with FLU treatment revealed improved cognition, supporting its ability to cross the BBB [Citation153]. However, FLU failed to show any impact on disease progression or survival period in the same study. The neuroprotective potential of FLU is possibly by its anti-oxidant nature (maintain glutathione levels); defence against glutamate-mediated excitotoxicity and role in apoptotic pathways (increasing anti-apoptotic B-cell lymphoma 2 protein: Bcl-2) [Citation154].
Guanabenz (GA)
GA: α2-adrenergic receptor (α2-AR) agonist [Citation155] is FDA approved antihypertensive drug in use for a long. In a screening study with anti-prion properties, GA stimulated the clearance of PrPSc in vivo against both yeast and ovine prions. GA is not directly involved in the PrP conversion, instead, it alters the protein folding activity of the ribosome (PFAR) [Citation156]. Moreover, GA is non-toxic and can easily cross BBB [Citation157,Citation158]. To overcome any side effects (anti-hypertensive) of GA in treating prion diseases, removing α2-AR agonist activity would be essential. Hence, based on SAR, modification of the positions of chlorine on benzyl moiety and guanidine was performed. These changes led to derivatives 6 and 7, which possessed a strong anti-prion activity in comparison to GA (IC50 2.8 ± 1.3 μM;1.1 ± 0.5 μM and 12.5 ± 2.7 μM for 6, 7, and GA, respectively), independent of α2-AR agonist activity. Like GA, these derivatives exhibited an anti-prion effect by inhibiting PFAR [Citation159]. Therefore, these two GA derivatives would be worth it for the next in vivo and clinical studies.
Glimepiride
Glimepiride is a sulphonylurea derivative, approved by the US FDA for the treatment of type 2 diabetes [Citation160]. The effect of glimepiride was investigated on prion-infected neuronal cells (ScN2a, SMB, and ScGT1 cells). The results indicated that the drug decreased PrPC expression and formation of PrPSc by activating endogenous glycosylphosphatidylinositol (GPI)-phospholipase C, which reduced the PrPC expressions on the surface of neurons. Additionally, glimepiride also displayed a neuroprotective effect against prion-derived peptide PrP82-146 [Citation161]. This peptide is known to induce the activation of cytoplasmic phospholipase A2 (cPLA2) and the production of prostaglandin E2 (PGE2), linked with neuronal injury in prion diseases [Citation162,Citation163]. In addition, glimepiride exerted anti-inflammatory activity by reducing the expression of CD14 (cluster of differentiation 14) and other inflammatory cytokines in the macrophages [Citation164]. From the animal data, glimepiride has been shown to pass through the placenta, but its BBB permeability is low [Citation165].
Imatinib
Imatinib is a tyrosine kinase inhibitor approved by US FDA as oral chemotherapy medication. Imatinib can induce autophagy [Citation166,Citation167] and accelerate the lysosomal clearance of PrPSc in prion-infected ScN2a cells and scrapie-infected mouse spleens [Citation166,Citation168]. However, no such effect of clearance was observed in CNS by either intraperitoneal (i.p.) or intracerebroventricular (i.c.) delivery due to its poor BBB penetration [Citation169,Citation170].
Quinacrine (QC)
QC is an acridine derivative that inhibited the PrPSc formation (EC50 0.3 µM) effectively in the cell-based assays [Citation171]. US FDA has approved the repurposing QC [Citation172], which is an antimalarial drug with the ability to cross the BBB [Citation173]. As observed by NMR spectroscopy, QC was able to bind to the Tyr226, and Gln227 of helix- 3 of human recombinant PrP globular domain (amino acid residues 121–230), Tyr225 [Citation174] and act as a pharmacological chaperone [Citation140]. QC is also involved in PrPSc clearance in vitro [Citation175]. However, it was unsuccessful with ScGT1 cells (chronically scrapie-infected murine neuronal cells), suggesting its therapeutic potential only after long-term treatments [Citation172]. The cytotoxicity of QC was above 2 μM [Citation45,Citation171] in the cellular assay, and the optimal concentration to prevent PrPSc formation was ~ 4 μM [Citation172]. Combination therapy using QC with simvastatin or desipramine was found to be more effective than QC alone [Citation176].
The SAR-based studies indicated that (S)- QC exhibits a superior anti-prion activity in comparison to (R)-enantiomer [Citation171]. Another study has shown the importance of nitrogen at position 7 of the tricyclic scaffold and aliphatic side chains for increasing anti-prion activity [Citation138]. Compounds with dimethylaminopropyl side chain (promazine, chlorpromazine, acepromazine) from the ring nitrogen at the 9 position of the tricyclic skeleton exhibited better binding activity (EC50 5 μM). Whereas, promethazine with a dimethylamino-2-methylpropyl side chain was relatively less active (EC50 ≈ 8 μM) [Citation138]. Despite the encouraging in vitro anti-prion activity of QC, no beneficial effect was observed in vivo [Citation172,Citation177]. Moreover, no positive outcome was observed, when QC was assessed in phase 2 clinical trials in CJD patients, rather it led to liver dysfunction [Citation177–180]. Another result from the continuous QC treatment seemed to create drug resistance prions. The transitory build-up of these drug-resistant prions might be responsible for the ineffectiveness of QC in vivo [Citation39].
Radotinib
Radotinib is an oral tyrosine kinase inhibitor that is approved by the Korean FDA for treating Chronic myeloid leukaemia–chronic phase (CML-CP) [Citation181]. It is also considered to treat PD patients [Citation182]. It is structurally similar to Imatinib and demonstrated superior anti-cancer activity in comparison to Imatinib in the pre-clinical studies [Citation183], without toxicity up to 1 g/day in phase I [Citation184]. Radotinib (100 mg/Kg p.o.) has shown promising results in the prion-infected hamster model by decreasing PrPSc deposition in the brain and extending the survival time in the treatment group [Citation185]. Other details related to the mechanism of action are not available yet.
Simvastatin
As membrane cholesterol has an important role in the conversion of PrPC to PrPSc [Citation186,Citation187], cholesterol inhibitors have been explored for their anti-prion activity. Simvastatin is approved by US FDA as a cholesterol-lowering medication. Simvastatin treatment (1 mg/Kg) significantly delayed the progression of prion disease and lengthened the survival times of prion-infected mice [Citation188] without any antagonistic effect of statin in the treatment group [Citation189]. It was proposed that simvastatin could exert its beneficial effects in prion infection through protein prenylation, as statins are already known to inhibit both protein farnesylation and geranylation by inhibiting upstream mevalonate/isoprenoid pathway [Citation190]. Convincing data suggested that the inhibition of isoprenoid synthesis and protein prenylation can reduce Aβ accumulations in vivo [Citation191], which will be a better strategic mechanism for facilitating statin-induced neuroprotection [Citation192,Citation193]. Additionally, anti-inflammatory and antioxidant effects will have positive synergies in treating prion diseases [Citation186,Citation194].
Antibiotics
The resistance to proteinase K digestion is an important characteristic of PrPSc [Citation27]. Studies revealed that when prions are exposed to antibiotics, they become more prone to enzymatic digestion, accompanied by a reduction in prion infection [Citation195]. As a result, antibiotics are also being reconsidered for brain-wasting diseases, apart from antibiotic activity.
Tetracycline is a well-characterized, safe, and US FDA-approved antibiotic. It protected the cells from prion infection in vitro [Citation196,Citation197] and ex vivo conditions. Preincubation of prion-infected brain homogenates with tetracycline delayed the onset of clinical symptoms and extended survival in animals. Forloni et al. demonstrated that tetracycline is a potent therapeutic drug for prion diseases, as it targeted fibril formation and stopped neuronal loss and astrocyte proliferation [Citation195]. NMR spectroscopy revealed that tetracycline interacted with amino acid (residues 100–126), a key region in the conformational changes [Citation196]. Docking and molecular simulations suggested a strong interaction of the tetracycline with functional groups of Thr (190–193) in the helix-2 of PrP [Citation198]. Tetracycline might also display indirect neuroprotective effects through antioxidant, anti-inflammatory, and anti-apoptotic mechanisms [Citation199]. A similar molecule, the anthracycline idodoxorubicin, was reported to have an affinity for PrP for reduced prion infection in a rodent model [Citation200,Citation201]. Doxycycline is a second-generation tetracycline antibiotic with better BBB permeability and safety profile [Citation202] and has been reported to reduce PrPSc levels in vitro and extended the survival of prion-infected animals [Citation203]. However, in a randomized, double-blind, placebo-controlled phase 2 clinical trial on CJD, Doxycycline (100 mg/day) was well tolerated without any significant effect in the late stages of CJD [Citation199,Citation204].
Amphotericin B (AmB) is US-FDA approved polyene antibiotic, which also exhibited anti-prion activity [Citation205] but its use is limited because of its toxicity. As a result, derivatives (carboxylic groups replaced by methyl) of AmB were tested for anti-prion potentials and found to be less toxic but equally potent in comparison to AmB [Citation206,Citation207]. While, another derivative of AmB, MS-8209, failed to show a direct effect on PrPSc [Citation208]. Recently, aminoglycoside G418 (Geneticin) has been reported to interfere with the de novo processes involved in earlier stages of prion infection, without affecting the level or localization of PrP [Citation209].
3. Natural compounds
Several natural polyphenols have already been reported for their anti-inflammatory, antioxidative, neuroprotective, and pro-autophagic functions [Citation210]. Polyphenols inhibited aggregation of various pathological proteins, like β-amyloid and α-synuclein [Citation211,Citation212] through direct interactions with β-sheets by inhibiting abnormal conformational changes [Citation213,Citation214] or by tilting the metal ion interactions of protein aggregation [Citation215,Citation216]. Apart from natural compounds from plants, other natural compounds with anti-prion potentials, like bile acids, hormones and disaccharides have also been discussed ().
Table 3. Natural compounds as anti-prion agents.
Baicalein
Baicalein, a flavonoid obtained from the mint family herb Scutellaria lateriflora (Blue skullcap), is known to be effective in neuroprotection [Citation217]. It has been reported to exert a neuroprotective mechanism and reduce PrPSc accumulation in SH-SY5Y and SK-N-SH cells in vitro [Citation218] by modulating ROS-induced cell death through inhibiting the phosphorylation in the c-Jun N-terminal kinase (JNK) pathway [Citation219]. Moreover, oral administration of S. lateriflora (water extract) increased the incubation times in prion-infected mice. It was observed that both baicalein-hydrate and baicalein have almost the same inhibitory effects on prion proliferation and removal of remaining fibrils [Citation220].
Bile acids
The bile acids, tauroursodeoxycholic acid (TUDCA) and ursodeoxycholic acid (UDCA) are neuroprotective in vitro, ex vivo, and in vivo by preventing the conversion of PrPC to PrPSc in ScN2a cells from the reduced neural loss, and increased survival of male mice in prion animal model [Citation221], referring as chemical chaperones [Citation222]. In addition to the promising results, these bile acids are relatively non-toxic, can cross the BBB, and are orally bio-available. Moreover, TUDCA has already been approved by FDA for the treatment of primary biliary cirrhosis [Citation223].
Cannabidiol (CBD)
CBD, a non-psychoactive compound from Cannabis plant, has been reported to inhibit the cerebral accumulation of PrPSc in prion-infected sheep and mice [Citation224]. Unfortunately, two structurally related cannabinoid analogues, Δ9-tetrahydrocannabinol (THC) and endocannabinoid arachidonoyl glycine (AG) proved to be ineffective. The mechanism of action is not conclusive, since CBD did not affect the cellular trafficking of PrPSc nor it disrupted the pre-existing PrPSc aggregates. It was suggested that CBD may exhibit neuroprotection by some other mechanism, which reduces PrPSc levels. Since CBD can cross BBB without drug tolerance from prolonged CBD therapy, it may be a promising anti-prion candidate [Citation224].
Carnosic acid (CA)
CA, a catechol isolated from Rosmarinus officinalis (Rosemary), has been reported for its potential antioxidant, anti-inflammatory, and neuroprotective activities [Citation225]. CA and its metabolite, carnosol (CS), have been shown to exert its potent antioxidant and anti-prion activity in N2a22L and N2a58 cells by reducing de novo generation of PrPSc aggregates, as well as favouring removal of already formed PrPSc clusters [Citation226]. In addition, CA can cross BBB after oral administration [Citation227], which was also confirmed by Light-BBB computational prediction [Citation228]. Therefore, the direct inhibition of PrPSc and antioxidant effects could be of specific therapeutic significance, not only in prion diseases but also in others with protein misfolding. Several in vivo studies are in progress.
Curcumin
Curcumin, 1,7-bis (4- hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione, is the yellow coloured pigment from turmeric (Curcuma longa) rhizome. Curcumin has been suggested as an anti-prion compound from partial inhibition of the cell-free conversion of PrPC to PrPSc and prevention of PrPSc accumulation in scrapie-infected neuroblastoma cells (IC50 ~ 10 nM) [Citation229]. Yet, the in vivo studies revealed that dietary administration of curcumin has no substantial effect on scrapie onset in hamsters [Citation229]. In a mouse model, curcumin was able to cross BBB and bind β-amyloid aggregates in the brain [Citation230]. Plasma proteins have been suggested as a carrier of curcumin transport to the brain [Citation231]. Researchers used the circular dichroism (CD) to identify the curcumin binding site on PrP and observed that the binding to the α-helix intermediate blocked the conformational conversion to β-sheet [Citation232]. In a cell-free system, curcumin significantly decreased the fibril formation of the mouse prion protein. In addition, co-incubation with prion amyloid fibrils and curcumin protected the cells against autophagy dysfunction and also decreased the level of ROS in mouse neuroblastoma cells [Citation233]. Since the structure of curcumin has similarities to Congo red, both compounds seemed to compete for binding to the α-helix intermediate. Interestingly, unlike Congo red, curcumin is non-toxic. However, water insolubility (< 0.6 μg/mL) [Citation234] and poor bio-availability are its limitations, which became the restrictions in the clinical trials. Therefore, curcumin in combination with other drugs, the effective drug delivery could be the key in its therapeutic applications.
Epigallocatechin gallate (EGCG)
EGCG from the green tea extract may be used as a potential therapeutic compound for treating NDDs. In primary neuronal cells, EGCG displayed neuroprotection against prion infection by Bax inhibition, translocation of cytochrome C, activating class III histone deacetylase, and activating autophagy by Sirtulin-1 (sirt-1) induction [Citation235]. The involvement of sirt-1 in prion diseases suggested the maintenance of mitochondrial homoeostasis as a major neuroprotective mechanism [Citation236]. The beneficial effects of sirt-1 overexpression have already been observed in various NDDS [Citation237–239]. Moreover, EGCG promotes PrPC internalization and degradation in lysosomes.
Ginsenoside-RG3
Ginsenosides Rg3, a saponin from steamed ginseng, is known to have neurogenic potentials [Citation240] by promoting autophagy functions [Citation241]. In a study with ginsenosides Rg3, it protected primary neurons and SK-KSH cells from the induced neurotoxicity of prion peptide (106–126) by promoting autophagy activities [Citation242]. The mechanism of enhanced autophagy functions by ginsenoside-RG3 from prion-induced toxicity is yet to be determined.
Hinokitiol
Hinokitiol (β-thujaplicin) chemically known as 2-hydroxy-4-isopropylcyclohepta-2,4,6- trien-1-one, is a tropolone derivative from the Cypressus trees with neuroprotective pharmacological properties [Citation243,Citation244]. Moon et al. [Citation245] observed that hinokitiol can protect the neuronal cells against PrP106−126-induced neurotoxicity through enhanced autophagy functions. Electron microscopy disclosed that hinokitiol boosted the formation of autophagic vacuoles in neurons, where the results advocated the induced autophagy through the hypoxia-inducible factor-1α (HIF-1α) pathway, the key neuroprotective mechanism against PrP-induced toxicity [Citation246].
Melatonin
Melatonin, a neuroendocrine hormone produced by the pineal gland, is a potent antioxidant molecule [Citation247] linked with various neurodegenerative disorders [Citation248]. Melatonin-induced autophagy exhibited neuroprotective activity in prion-infected cells by modulating mitochondrial functions [Citation249,Citation250]. The pre-treatment with melatonin inhibited the mitochondrial-mediated apoptosis in vitro by protecting/modulating the mitochondrial morphology from PrP106−126 toxicity by interacting with dynamin-related protein 1 (DRP1) in the mitochondrial fission [Citation251] and optic atrophy 1 (OPA1) in the fusion pathways [Citation252,Citation253]. However, the study of administrating low and high doses of melatonin in animal studies presented the controversial results of stimulating or inhibiting prion activities [Citation254].
Polydatin
Polydatin, 3,4',5-trihydroxystilbene-3-β-D-glucoside, from roots of Japanese knotweed (Polygonum cuspidatum) is a natural precursor of resveratrol [Citation255]. Apart from various pharmaceutical properties [Citation256], polydatin also inhibited amyloid formation [Citation257,Citation258]. In a recent biophysical study, polydatin revealed its anti-prion activity in vitro and in silico [Citation259] by binding to PrPC and inhibiting the conversion to PrPSc. The detailed structural analysis using molecular dynamics simulations explained that polydatin could suppress the oligomerization by abolishing the unfavourable structural transitions in the β2–α2 loop, α3-helix, and N-terminal amyloidogenic region, favouring α-helical and random coils formation, thereby disabling the prion aggregations and pathogenesis [Citation259].
Resveratrol
Resveratrol, a polyphenol with antioxidant, anticancer, and neuroprotective properties, is present in a variety of fruits, like grapes and berries [Citation260]. Resveratrol has been reported to decrease the accumulations of PrPSc (EC50 0.61 μM) in prion-infected SMB-S15 cells in a dose-dependent manner. In addition, one-week treatment with resveratrol significantly reduced the prion infectivity in mice [Citation261], indicating its potential as an anti-prion agent. It is also reported that resveratrol can remove PrPSc by decreasing sirt-1 (regulates prion protein-induced neuronal cell death) levels in the brain [Citation262]. Structurally, resveratrol interacted with Tyr128 by π-π stacking and stabilized PrPC [Citation263] and thus prevented the aggregation. Another neuroprotective potential of resveratrol against prion neurotoxicity is through resveratrol-mediated and enhanced autophagy signals [Citation264].
Spermine
Spermine. a biologically active polyamine, is essential for cell growth and differentiation. Since the unstable microtubules and influences of atypical protein folding could influence the neurodegenerative processes, the spermine seemed to increase the expressions of proteins of microtubule acetylation (Tubulin alpha1B/1C, Tubulin beta-4B, Tubulin beta-5) for the stabilization of microtubules. Acetylated microtubules play an essential role in trafficking autophagosomes to the lysosomes [Citation265]. Using affinity experiment microtubule, Tubb6 has been identified as a binding partner to spermine, which facilitated the autophagic degradation of PrPSc in cells [Citation266].
Trehalose
Trehalose, a non-reducing disaccharide with α, α-1,1-glucosidic bond between two α- glucose units, is present in almost all organisms, except vertebrates, with cellular defence functions against different types of stress conditions [Citation267]. Trehalose can remove protein aggregates, like α-synuclein by activating autophagy in the mammalian target of rapamycin (mTOR) independent pathway [Citation268]. Aguib et al. [Citation269] reported the autophagy induction and simultaneous reduction of PrPSc in prion-infected cultured cells from the trehalose treatment. In the mice model (intraperitoneally infected with prion), oral administration of trehalose reduced PrPSc accumulation slightly without affecting the survival of the animals. It is suggested that the trehalose levels in CNS are not high enough to show the desired effects from limited BBB crossing [Citation269]. Trehalose can also protect the prion-infected cells against induced oxidative stress by modifying the subcellular localization of PrPSc [Citation270]. It is speculated that the neuroprotective effects of trehalose are exhibited through direct or indirect mechanisms, or with involvements of gut microbiota [Citation271].
4. Immunotherapy
Immunotherapy comprises both active (vaccine-mediated) and passive (antibody-mediated) immunizations. The proper identification of prion epitope is essential to develop strong specificity and fewer side effects of the therapy [Citation272]. Various encouraging results have been reported from immunotherapy studies with prion-infected animal models.
Immunization with small prion fragments is capable to fit in the furrows of major histocompatibility complex class II (MHC II) by stimulating anti-PrPC immunity for the PrPSc reduction [Citation273]. However, as the immune system is tolerant to self-antigens, the antibodies gained by immunizations were not frequently effective [Citation274].
As discussed above, PrPC stabilization is quite important to block the conversion of PrPC to PrPSc hence, a therapy targeting PrPC will have limited neuroprotective effects in prion diseases. However, due to structural differences between PrPSc and PrPC, PrPSc can also be recognized as an antigen by the immune system and provoke the immune response. This type of immune response has been reported in the terminal stage of prion-infected mice [Citation275]. To develop PrPSc-specific antibody response, the exclusive epitopes on PrPSc were sought, for which tyrosine-tyrosine-arginine (YYR) repeat motif, a tyrosine-methionine-leucine (YML) motif in β-sheet 1, and the rigid loop (RL) connecting β-sheet 2 to α-helix 2 were identified [Citation276]. The multivalent PrPSc-specific vaccines were prepared using these epitopes and analysed for immunogenicity and safety [Citation277]. The encouraging results of the YYR-based vaccine have been reported in a study on prion-infected deer [Citation278]. Surprisingly, the same vaccine increased the disease onset in elk with the 132 MM genotype and had no effect in elk (132ML genotype), however, the reason was not clear [Citation279].
The binding of PrPC-antibodies can block the conversion of PrPC to PrPSc, hence several studies with various antibodies have been carried out extensively.
POM1 and POM6 are globular domain (GD) binding monoclonal antibodies (mAbs) with a distinct biological response as POM1 triggers neurotoxicity while POM6 is neuroprotective. However, it was observed that POM1 toxicity is independent of prion replication [Citation280]. Investigating the link between the PrPC binding of POM1 and the resulting toxicity, ‘H-latch’ (intramolecular hydrogen bond between His140 & Arg208 of GD) formation was identified as an early reporter of toxicity. Hence designing antibodies that prevent H-latch formation would protect against the damaging effects of prion infection [Citation281].
The simulations studies disclosed that the POM1/PrP interface is more stable compared to POM6/PrP and the binding of antibodies enhances GD flexibility largely (except in β1-α1 loop), signifying the contribution of this loop in the pathological alteration [Citation282]. To address the toxicity concern raised by some anti-PrP antibodies, a bi-specific antibody, scPOM-bi, has been designed as a molecular prion tweezer. It is comprised of POM1 and POM2 complementarity determining regions that simultaneously target GD and flexible tail (FT) resulting in strong neuroprotection and might be used as a promising strategy to develop anti-prion immunotherapy [Citation283].
A potent antibody recognizing multiple epitopes in a different region of PrP revealed its inhibitory mechanisms with different effectivity [Citation284]. Monoclonal antibodies (mAbs) significantly restricted the conversion of PrPC in prion-infected cells [Citation285–288] through four epitopes (octarepeat region, residues 90–110, residues 145–160 in helix-1, and residues 210–220 in the C terminal) [Citation289]. Additionally, mAbs directed against regions 90–110 or 145–160 displayed in vivo anti-prion activity after intraperitoneal prion infection but not in case of intracerebral infection as these mAbs are inaccessible to BBB.
The anti-PrP antibodies have also been assessed in prion-infected mice models. The 8B4 (recognizing residues 34–52) and 8H4 (recognizing residues 175–185) antibodies were more effective in reducing the prion infection in comparison to 8F9 (recognizing residues 205–233), due to the better affinity for PrPC and PrPSc [Citation290]. Short-term intraperitoneal treatment with 6D11 also extended the survival in prion-infected mice [Citation291]. It was determined that PrPC and PrPSc were sequestered by 6D11 from the cell surface and formed complexes (PrPC/6D11 and PrPSc/6D11), which would be then exported to lysosomes for degradation [Citation292]. The most efficient antibodies were identified as ICSM 18 (recognizing residues 146–59) and ICSM 35 (recognizing residues 91–110), which increased the survival time considerably in mice, but unfortunately proved ineffective at later stages of the infection or when the mice were infected through the intracerebral route [Citation293], suggesting that these antibodies could not stop the disease progression in CNS, owing to BBB impermeability.
Recent important progress in antibody treatment is the development and clinical efficacy of a humanized anti-PrPC monoclonal antibody (an IgG4κ isotype; PRN100), which increased survival in prion-infected mice [Citation293]. PRN100 has been developed by researchers at the Medical Research Council (MRC) Prion Unit, University College London (UCL), after a battery of mouse monoclonal antibodies developed against human PrP [Citation294] for their binding and stabilizing PrPC properties. In a recent clinical study, six CJD patients received PRN100 intravenously every 2 weeks till death or withdrawal from the programme. Through i.v. administration, PRN100 was able to reach the target CSF drug concentration (50 nM), and additionally, it was well tolerated without any adverse effects for up to 8 months [Citation14]. After such promising results, PRN100 will be evaluated for Phase-II trials.
5. Clinical trials
Anle138b clinical study was conducted in 2020 with 68 healthy male and female subjects and passed Phase 1 evaluation of safety, tolerability, and pharmacokinetics [Citation82]. Adverse events were considered to be mild and patients fully recovered during this study. In the future, longer and more large-scale evaluations could be conducted to access the full safety profile of Anle138b. Since Anle138b was initially started for the multiple system atrophy treatment and previously hampered α-synuclein aggregation, a Phase 1b trial for 48 patients with PD was initiated in 2020 [Citation81,Citation83]. The trial will assess Anle138b administrated orally at a daily dose of 150 mg and higher for 7 days in PD patients including CSF tests and motor function improvement and is expected to conclude in 2022.
QC has been utilized as an antimalarial drug [Citation173] and also approved as a repurposing anticancer agent [Citation172]. Anaplastic anaemia is the most harmful side effect of QC, which occurs in a lower percentage (0.003%) of patients at high dosages than indicated [Citation295]. A dosage of 300 mg has been reported to be well tolerated in recent clinical trials on QC, but no improvement in survival in CJD patients was observed [Citation177–180]. QC was tolerated at a daily dose of 300 mg but did not affect prion disease in this preference clinical trial [Citation177]. Oral administration of QC for 2 months did not prolong the survival of sCJD patients [Citation178]. Despite the negative outcome, previous clinical trials for CJD showed that proper randomized controlled trials could be conducted in rare, fast progressive, and fatal neuronal disorders.
PPS in humans does not have a proven safe or effective dose regimen. Bone et al. showed that PPS was tolerated up to 110 μg/kg/day doses for 6 months [Citation120]. From two studies, PPS was treated in sporadic, iatrogenic, inherited, and vCJD patients and prolonged mean survival of patients than natural history prion patients with the reduction of abnormal prion protein in the brain [Citation296]. However, treatment with PPS in other studies did not achieve clinical improvement [Citation121], so further studies are needed to clarify the effectiveness of PPS against prion diseases.
FLU was permitted in Europe and Asian countries [Citation147], but later in March 2018, it was withdrawn from the European market. FLU, the first randomized research of a CJD treatment, revealed no effectiveness in survival expansion but did potentially delay cognitive deterioration [Citation152]. Positive results of FLU on cognitive improvement may imply therapeutic benefits in other neurodegenerative disorders.
In a study with a small number of CJD patients, the treatment of doxycycline in the early stage of the disease revealed slightly positive effects [Citation204], but no significant effects were observed in a randomized trial [Citation199]. A doxycycline trial for FFI patients with Asp178Asn mutation in the PRNP gene is under progress [Citation297].
Conclusion and future perspectives
Various classes of compounds have been assessed for anti-prion activity in vitro and in vivo. Different scaffolds displayed anti-prion properties by either direct or indirect interactions with PrPC, stabilizing PrPSc, reducing PrPSc accumulation, or by other neuroprotective mechanisms. Membrane-bound PrPC are largely present in the lipid rafts, consequently, the drugs for inhibiting cholesterol biosynthesis or disturbing raft membranes can redistribute surface PrPC, and in that way can reduce accumulation or increase the clearance of PrPSc.
Apart from various strategies that target PrpC or PrPSc, a novel non-PrP targeted drug strategy emerged as an effective approach to treat prion disease [Citation298]. Upregulation of SerpinA3/SerpinA3n has been associated with NDDs, including prion disease [Citation299] as a serine protease inhibitor, it retards the protease activity responsible for clearing prion protein aggregates. With this idea, a small molecule serpin inhibitor (ARN1468) has been designed which effectively reduced prion load in scrapie’s infected ScN2a RML and ScN2a 22 L cell lines. Unfortunately, the low bioavailability of the compound limited the in vivo studies [Citation298]. Analogues of ARN1468 with enhanced bioavailability could be promising strategies in treating prion disease.
The ubiquitin-proteasome system (UPS) with an important function in degrading intracellular proteins in eukaryotes seemed to be closely involved in the clearance of protein aggregates. In certain neurodegenerative disorders, this activity has been compromised and led to the accumulation of toxic aggregates of misfolded proteins [Citation300]. Thus, augmenting UPS activity by increasing ubiquitin [Citation301], overexpressing specifically related genes [Citation302], or by inhibiting p38 mitogen-activated protein kinase (MAPK) pathways could also be good candidates for therapies through UPS activations [Citation303].
Chaperones have a key role in the re-folding of cellular protein thereby maintaining proteostasis. However, the proteostasis capacity decreases with age and hence enhances the manifestation of NDDs. Additionally, since the pharmaceutical chaperones usually have a short-half life, maintaining their high concentrations may prevent the protein misfolding as well as reduce the toxicity of protein aggregates. On the other hand, hydrophobic chaperones are less toxic and more suitable for the therapeutic purpose [Citation222]. Literature also suggested the important functions of autophagy in prion disease, where much evidence of autophagosomes in the infected brains would degrade protein aggregates [Citation136]. Yet, the enhanced activity of autophagosomes by potential compounds may not be adequate to eradicate prion infection through induced/enhanced autophagy-flux. Many natural compounds and tyrosine kinase inhibitors could work through this mechanism in ameliorating prion pathology. Due to abnormal protein aggregation, a dramatic increase in oxidative stress and loss of antioxidant defence has been observed in prion diseases. Therefore, compounds reducing oxidative stress could be also beneficial in reducing neurotoxicity and neuroinflammation associated with the disease.
Lately, the encouraging results of a monoclonal antibody (PRN100) in CJD patients could pave the way for new anti-prion agents and will be helpful in the treatments of other NDDs. The success story did not end here, as another promising molecule MC and GN8 would be ready for human clinical trials on prion diseases, while Anle138b was in Phase 1b for PD patients.
As mentioned earlier, quinacrine, pentosan polysulfate, flupirtine, and doxycycline failed in clinical trials on CJD patients. The ineffectiveness of most of the molecules tested was either due to the inability to cross BBB, toxicity, or transitory accumulations of drug-resistant prions. Hence, it would be incredibly important to learn from the backstory. By understanding what was already investigated, what would be the mechanism of drug action, and why the clinical trials failed, we could comprehend prion diseases better and chalk out the right direction for more effective treatments. Lastly, targeting more than one pathway involved in prion diseases may provide synergistic benefits.
Author Contributions
Conceptualization, K.H.S, N.S., and S.S.A.A.; writing—original draft preparation, N.S.; writing—review and editing, K.H.S, N.S. and S.S.A.A.; funding acquisition, S.S.A.A. All authors read and agreed to the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bendheim P, Brown HR, Rudelli RD, et al. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology. 1992;42(1): 149-149. DOI:10.1212/WNL.42.1.149.
- Prusiner SB, McKinley MP, Bowman KA, et al. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35(2):349–358.
- Riek R, Hornemann, S, Wider G, Glockshuber R, Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP (23–231). FEBS letters 08 181997. doi:10.1016/S0014-5793(97)00920-4.
- Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences (PNAS), USA, 1993. Vols. 90(23): p. 10962–10966.
- Caughey B, Kocisko DA, Raymond GJ, et al. Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem Biol. 1995;2(12):807–817.
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3(1):a006833.
- Silveira JR, Raymond GJ, Hughson AG, et al. The most infectious prion protein particles. Nature. 2005;437(7056):257–261.
- Heikenwalder M, Julius C, Aguzzi A. Prions and peripheral nerves: a deadly rendezvous. J Neurosci Res. 2007;85(12):2714–2725.
- Glatzel M. Sympathetic prions. The Scientific World JOURNAL. 2001;1:555–556.
- Aguzzi A. Prions and the immune system: a journey through gut, spleen, and nerves. Adv Immunol. 2003;81:123–172.
- Costa PP, Figueira AS, Bravo FR, Amyloid fibril protein related to prealbumin in familial amyloidotic polyneuropathy. Proceedings of the National Academy of Sciences (PNAS), USA, 1978. Vols. 75(9): p. 4499–4503.
- Mead S, Reilly MM. A new prion disease: relationship with central and peripheral amyloidoses. Nat Rev Neurol. 2015;11(2):90–97.
- Chen C, Dong X-P. Epidemiological characteristics of human prion diseases. Infect Dis Poverty. 2016;5(1):1–10.
- Mead S, Khalili-Shirazi A, Potter C, et al. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt–Jakob disease: evaluation of a first-in-human treatment programme. Lancet Neurol. 2022;21(4):342–354.
- Chen C, Wang J-C, Shi Q, et al. Analyses of the survival time and the influencing factors of Chinese patients with prion diseases based on the surveillance data from 2008–2011. PLoS One. 2013;8(5):e62553.
- Nagoshi K, Sadakane A, Nakamura Y, et al. Duration of prion disease is longer in Japan than in other countries. J Epidemiol. 2011; 1105250231-1105250231.
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(10):2659–2668.
- Kulanthaivelu K, Sinha S. EEG observations in probable sporadic CJD. Ann Indian Acad Neurol. 2020;23(6):761.
- Matsubayashi T, Akaza M, Hayashi Y, et al. Focal sharp waves are a specific early-stage marker of the MM2-cortical form of sporadic Creutzfeldt-Jakob disease. Prion. 2020;14(1):207–213.
- Zerr I, Bodemer M, Gefeller O, et al. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol. 1998;43(1):32–40.
- Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17(2):175–178.
- Orrú CD, Groveman BR, Hughson AG, et al. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6(1)
- Meer B, Kallenberg K, Sanchez-Juan P, et al. MRI and clinical syndrome in dura materrelated Creutzfeldt-Jakob disease. J Neurol. 2009;256(3):355–363.
- Zafar S, Noor A, Zerr I. Therapies for prion diseases. Handb Clin Neurol. 2019;165:47–58.
- De Vries K, Cousins E, Dening KH. Palliative care in Creutzfeldt-Jakob disease: looking back, thinking ahead. BMJ Support Palliat Care. 2021; DOI:10.1136/bmjspcare-2020-002799
- Legname G, Giachin G, Benetti F. Structural studies of prion proteins and prions, in non-fibrillar amyloidogenic protein assemblies-common cytotoxins underlying degenerative diseases. Dordrecht: Springer; 2012. p. 289–317.
- Zhang Y, Ge B, Duncan I. Restriction fragment length polymorphism linkage map for Arabidopsis thaliana. Proc Natl Acad Sci USA. 1988;85(18):6856. 2000
- Walter ED, Stevens DJ, Spevacek AR, et al. Copper binding extrinsic to the octarepeat region in the prion protein. Current Protein Pept Sci. 2009;10(5):529–535.
- Walter ED, Chattopadhyay M, Millhauser GL. The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity. Biochemistry. 2006;45(43):13083–13092.
- Kuwata K, Matumoto T, Cheng H, et al. NMR-detected hydrogen exchange and molecular dynamics simulations provide structural insight into fibril formation of prion protein fragment 106–126. Proceedings of the National Academy of Sciences (PNAS), USA, 2003. Vols.100(25): p. 14790–14795.
- Migliorini C, Sinicropi A, Kozlowski H, et al. Copper-induced structural propensities of the amyloidogenic region of human prion protein. J Biol Inorg Chem. 2014;19(4–5):635–645.
- Giachin G, Mai PT, Tran TH, et al. The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci Rep. 2015;5(1):1–14.
- Salzano G, Brennich M, Mancini G, et al. Deciphering copper coordination in the mammalian prion protein amyloidogenic domain. Biophys J. 2020;118(3):676–687.
- Lansbury PT. Mechanism of scrapie replication. Science. 1994;265(5178): 1510-1510.
- Sigurdson CJ, Bartz JC, Glatzel M. Cellular and molecular mechanisms of prion disease. Annu Rev Pathol. 2019;14(1):497–516.
- Harris DA. Cell biological studies of the prion protein. Curr Issues Mol Biol. 1999;1(1–2):65–76.
- Goold R, Rabbanian S, Sutton L, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun. 2011;2(1):1–11.
- Kamatari YO, Hayano Y, Yamaguchi K-I, et al. Characterizing antiprion compounds based on their binding properties to prion proteins: implications as medical chaperones. Protein Sci. 2013;22(1):22–34.
- Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5(11):e1000673.
- Ghaemmaghami S, May BCH, Renslo AR, et al. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol. 2010;84(7):3408–3412.
- Gallardo-Godoy A, Gever J, Fife KL, et al. 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem. 2011;54(4):1010–1021.
- Berry D, Giles K, Oehler A, et al. Use of a 2-aminothiazole to treat chronic wasting disease in transgenic mice. J Infect Dis. 2015;212(suppl 1):S17–S25.
- Giles K, Berry DB, Condello C, et al. Different 2-aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. J Pharmacol Exp Ther. 2015;355(1):2–12.
- Burke CM, Mark KMK, Kun J, et al. Emergence of prions selectively resistant to combination drug therapy. PLoS Pathog. 2020;16(5):e1008581.
- Hyeon JW, Choi J, Kim S, et al. Discovery of novel anti-prion compounds using in silico and in vitro approaches. Sci Rep. 2015;5(1):1–11.
- Hyeon JW, Noh R, Choi J, et al. BMD42-2910, a novel benzoxazole derivative, shows a potent anti-prion activity and prolongs the mean survival in an animal model of prion disease. Exp Neurobiol. 2020;29(1):93.
- Kimura T, Hosokawa-Muto J, Asami K, et al. Synthesis of 9-substituted 2,3,4,9-tetrahydro-1H-carbazole derivatives and evaluation of their anti-prion activity in TSE-infected cells. Eur J Med Chem. 2011;46(11):5675–5679.
- Kimura T, Sako T, Sakaguchi S, et al. Synthesis of an 11 C-labeled antiprion gn8 derivative and evaluation of its brain uptake by positron emission tomography. ChemMedChem. 2013;8(7):1035–1039.
- Caughey WS, Raymond LD, Horiuchi M, et al., Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proceedings of the National Academy of Sciences (PNAS), USA., 1998. Vols.95(21): p. 12117–12122.
- Priola SA, Raines A, Caughey WS. Porphyrin and phthalocyanine antiscrapie compounds. Science. 2000;287(5457):1503–1506.
- Priola SA, Raines A, Caughey W. Prophylactic and therapeutic effects of phthalocyanine tetrasulfonate in scrapie-infected mice. J Infect Dis. 2003;188(5):699–705.
- Kocisko DA, Caughey WS, Race RE, et al. A porphyrin increases survival time of mice after intracerebral prion infection. Antimicrob Agents Chemother. 2006;50(2):759–761.
- Touil F, Pratt S, Mutter R, et al. Screening a library of potential prion therapeutics against cellular prion proteins and insights into their mode of biological activities by surface plasmon resonance. J Pharm Biomed Anal. 2006;40(4):822–832.
- Wong CN, Xiong LW, Horiuchi M, et al. Sulfated glycans and elevated temperature stimulate PrPSc-dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001;20(3):377–386.
- Caughey WS, Priola SA, Kocisko DA, et al. Cyclic tetrapyrrole sulfonation, metals, and oligomerization in antiprion activity. Antimicrob Agents Chemother. 2007;51(11):3887–3894.
- Nicoll AJ, Trevitt CR, Tattum MH, et al. Pharmacological chaperone for the structured domain of human prion protein. Proceedings of the National Academy of Sciences (PNAS), USA, 2010. Vols.107(41): p. 17610–17615.
- Massignan T, Cimini S, Stincardini C, et al. A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein. Sci Rep. 2016;6(1):1–14.
- Rezajooei N, Cappellano TR, Tittle C. Studying the effects of anti-prion compounds on prp folding at the single-molecule level. Masters thesis, Spring 2017, University of Alberta, Canada, 2017;23(9):1376–1384. https://doi.org/10.7939/R3GF0N804
- Frid P, Anisimov SV, Popovic N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res Rev. 2007;53(1):135–160.
- Espargaró Colomé A, Llabrés S, Saupe SJ, et al. On the binding of congo red to amyloid fibrils. Angewandte Chemie International Edition. 2020;59(21):8104–8107.
- Supattapone S, Nguyen H. O. B, Cohen F. E. Elimination of prions by branched polyamines and implications for therapeutics. Proceedings of the National Academy of Sciences (PNAS), USA, 1999. Vols.96(25): p. 14529–14534.
- Caspi S, Halimi M, Yanai A, et al. The anti-prion activity of Congo red: putative mechanism. J Biol Chem. 1998;273(6):3484–3489.
- Caughey B, Brown K, Raymond GJ, et al. Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and Congo red [corrected]. J Virol. 1994;68(4):2135–2141.
- Caughey B, Race RE. Potent inhibition of Scrapie-associated prp accumulation by congo red. J Neurochem. 1992;59(2):768–771.
- Corato M, Ogliari P, Ceciliani F, et al. Doxorubicin and Congo red effectiveness on prion infectivity in golden Syrian hamster. Anticancer Res. 2009;29(7):2507–2512.
- Kawatake S, Nishimura Y, Sakaguchi S, et al. Surface plasmon resonance analysis for the screening of anti-prion compounds. Biol Pharm Bull. 2006;29(5):927–932.
- Demaimay R, Chesebro B, Caughey B. Prion Diseases. Vienna: Springer; 2000. Inhibition of formation of protease-resistant prion protein by trypan blue, sirius red and other congo red analogs; p. 277–283.
- Bos R, Koopman JP, Theuws JLG, et al. The essential role of the intestinal flora in the toxification of orally administered benzidine-based dyes. Internal exposure of rats to benzidine after intestinal azo reduction. Mutat Res. 1987;181(2):327.
- Ingrosso L, Ladogana A, Pocchiari M. Congo red prolongs the incubation period in scrapie-infected hamsters. J Virol. 1995;69(1):506–508.
- Rudyk H, Birkett CR, Hennion RM, et al. Screening Congo Red and its analogues for their ability to prevent the formation of PrP-res in scrapie-infected cells. J Gen Virol. 2000;81(4):1155–1164.
- Sellarajah S, Lekishvili T, Bowring C, et al. Synthesis of analogues of Congo red and evaluation of their anti-prion activity. J Med Chem. 2004;47(22):5515–5534.
- Poli G, Martino PA, Villa S, et al. Evaluation of anti-prion activity of Congo red and its derivatives in experimentally infected hamsters. Arzneimittelforschung. 2004;54(7):406–415.
- Kuwata K, Nishida N, Matsumoto T, et al. Hot spots in prion protein for pathogenic conversion. Proceedings of the National Academy of Sciences (PNAS), USA, 2007. Vols.104(29): p. 11921–11926.
- Kimura T, Hosokawa-Muto J, Kamatari YO, et al. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg Med Chem Lett. 2011;21(5):1502–1507.
- Hosokawa-Muto J, Kimura T, Kuwata K. Respiratory and cardiovascular toxicity studies of a novel antiprion compound, GN8, in rats and dogs. Drug Chem Toxicol. 2012;35(3):264–271.
- Yamaguchi K, Kamatari YO, Ono F, et al. A designer molecular chaperone against transmissible spongiform encephalopathy slows disease progression in mice and macaques. Nat Biomed Eng. 2019;3(3):206–219.
- Leidel F, Eiden M, Geissen M, et al. Diphenylpyrazole-derived compounds increase survival time of mice after prion infection. Antimicrob Agents Chemother. 2011;55(10):4774–4781.
- Leidel F, Eiden M, Geissen M, et al. Piperazine derivatives inhibit PrP/PrPres propagation in vitro and in vivo. Biochem Biophys Res Commun. 2014;445(1):23–29.
- Wagner J, Ryazanov S, Leonov A, et al. Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013;125(6):795–813.
- Guerrero-Muñoz MJ, Castillo-Carranza DL, Kayed R. Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochem Pharmacol. 2014;88(4):468–478.
- Matthes D, Gapsys V, Griesinger C, et al. Resolving the atomistic modes of Anle138b inhibitory action on peptide oligomer formation. ACS Chem Neurosci. 2017;8(12):2791–2808.
- GmbH M A first-in-human study of single and multiple doses of anle138b in healthy subjects. NCT04208152 2020 2022 April 28]; Available from: https://clinicaltrials.gov/ct2/show/NCT04208152.
- GmbH M A study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of anle138b in parkinson’s disease. 2021 2022 April 28]; Available from: https://clinicaltrials.gov/ct2/show/results/NCT04685265?term=Anle138b&draw=2&rank=1&view=results.
- Regelson W, Harkins S. “amyloid is not a tombstone”—A summation: the primary role for cerebrovascular and csf dynamics as factors in alzheimer’s disease (AD): DMSO, Fluorocarbon oxygen carriers, thyroid hormonal, and other suggested therapeutic measures. Ann N Y Acad Sci. 1997;826(1):348–374.
- Shen CL, Murphy RM. Solvent effects on self-assembly of beta-amyloid peptide. Biophys J. 1995;69(2):640–651.
- Shaked GM, Engelstein R, Avraham I, et al. Dimethyl sulfoxide delays PrPsc accumulation and disease symptoms in prion-infected hamsters. Brain Res. 2003;983(1–2):137–143.
- Shaked GM, Fridlander G, Meiner Z, et al. Protease-resistant and detergent-insoluble prion protein is not necessarily associated with prion infectivity. J Biol Chem. 1999;274(25):17981–17986.
- Tatzelt J, Prusiner SB, Welch WJ. Chemical chaperones interfere with the formation of scrapie prion protein. EMBO J. 1996;15(23):6363–6373.
- Uchiyama K, Hara H, Chida J, et al. Ethanolamine Is a New Anti-Prion Compound. Int J Mol Sci. 2021;22(21):11742.
- Calzada E, Onguka O, Claypool SM. Phosphatidylethanolamine Metabolism in Health and Disease. Int Rev Cell Mol Biol. 2016;321:29–88.
- Deleault NR, Piro JR, Walsh DJ, et al. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proceedings of the National Academy of Sciences (PNAS), USA, 2012. Vols.109(22): p. 8546–8551.
- Hoover CE, Davenport KA, Henderson DM, et al. Endogenous brain lipids inhibit prion amyloid formation in vitro. J Virol. 2017;91(9)
- Baumer W, Hoppmann J, Rundfeldt C, et al. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflammation & Allergy-Drug Targets (Formerly Current Drug Targets-Inflammation & Allergy)(Discontinued). 2007;6(1):17–26.
- Wang T, Yin Z, Zhang Z, et al. Inhibitors of human immunodeficiency virus type 1 (HIV-1) attachment. 5. An evolution from indole to azaindoles leading to the discovery of 1-(4-benzoylpiperazin-1-yl)-2-(4, 7-dimethoxy-1 H-pyrrolo [2, 3-c] pyridin-3-yl) ethane-1, 2-dione (BMS-488043), a drug candidate that demonstrates antiviral activity in HIV-1-infected subjects. J Med Chem. 2009;52(23):7778–7787.
- Oostendorp RL, Witteveen PO, Schwartz B, et al. Dose-finding and pharmacokinetic study of orally administered indibulin (D-24851) to patients with advanced solid tumors. Invest New Drugs. 2010;28(2):163–170.
- Nicholls SJ, Cavender MA, Kastelein JJP, et al. Inhibition of secretory phospholipase A2 in patients with acute coronary syndromes: rationale and design of the vascular inflammation suppression to treat acute coronary syndrome for 16 weeks (Vista-16) trial. Cardiovasc Drugs Ther. 2012;26(1):71–75.
- Thompson MJ, Borsenberger V, Louth JC, et al. Design, Synthesis, and Structure−Activity Relationship of Indole-3-glyoxylamide Libraries Possessing Highly Potent Activity in a Cell Line Model of Prion Disease. J Med Chem. 2009;52(23):7503–7511.
- Thompson MJ, Louth JC, Ferrara S, et al. Structure-Activity Relationship Refinement and Further Assessment of Indole-3-glyoxylamides as a Lead Series against Prion Disease. ChemMedChem. 2011;6(1):115–130.
- Thompson MJ, Louth JC, Ferrara S, et al. Discovery of 6-substituted indole-3-glyoxylamides as lead antiprion agents with enhanced cell line activity, improved microsomal stability and low toxicity. Eur J Med Chem. 2011;46(9):4125–4132.
- Hong N, Choi Y-S, Kim SY, et al. Neuroprotective effect of lithium after pilocarpine-induced status epilepticus in mice. Korean J Physiol Pharmacol. 2017;21(1):125–131.
- Pouladi MA, Brillaud E, Xie Y, et al. NP03, a novel low-dose lithium formulation, is neuroprotective in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2012;48(3):282–289.
- Heiseke A, Aguib Y, Riemer C, et al. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109(1):25–34.
- Pérez M, Rojo AI, Wandosell F, et al. Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem J. 2003;372(1):129–136.
- Song Z, Yang W, Zhou X, et al. Lithium alleviates neurotoxic prion peptide-induced synaptic damage and neuronal death partially by the upregulation of nuclear target REST and the restoration of Wnt signaling. Neuropharmacology. 2017;123:332–348.
- Gitlin M. Lithium side effects and toxicity: prevalence and management strategies. Int J Bipolar Disord. 2016;4(1):1–10.
- Relaño-Ginés A, Lehmann S, Brillaud E, et al. Lithium as a disease-modifying agent for prion diseases. Transl Psychiatry. 2018;8(1):1–11.
- Cavaliere P, Torrent J, Prigent S, et al. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim Biophys Acta Mol Basis Dis. 2013;1832(1):20–28.
- Mori T, Koyama N, Segawa T, et al. Methylene blue modulates β-secretase, reverses cerebral amyloidosis, and improves cognition in transgenic mice. J Biol Chem. 2014;289(44):30303–30317.
- Caughey B, Caughey WS, Kocisko DA, et al. Prions and Transmissible Spongiform Encephalopathy (TSE) Chemotherapeutics: a Common Mechanism for Anti-TSE Compounds? Acc Chem Res. 2006;39(9):646–653.
- Karpuj MV, Gelibter-Niv S, Tiran A, et al. Conditional modulation of membrane protein expression in cultured cells mediated by prion protein recognition of short phosphorothioate oligodeoxynucleotides. J Biol Chem. 2011;286(9):6911–6917.
- Shyng S-L, Lehmann S, Moulder KL, et al. Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J Biol Chem. 1995;270(50):30221–30229.
- Petrosyan R, Patra S, Rezajooei N, et al. Unfolded and intermediate states of PrP play a key role in the mechanism of action of an antiprion chaperone. Proceedings of the National Academy of Sciences (PNAS), USA, 2021. Vols.118(9).
- Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol. 1993;67(2):643–650.
- Diringer H, Ehlers B. Chemoprophylaxis of scrapie in mice. J Gen Virol. 1991;72(2):457–460.
- Doh-Ura K, Ishikawa K, Murakami-Kubo I, et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78(10):4999–5006.
- Rouvinski A, Karniely S, Kounin M, et al. Live imaging of prions reveals nascent PrPSc in cell-surface, raft-associated amyloid strings and webs. J Cell Biol. 2014;204(3):423–441.
- Yamaguchi S, Nishida Y, Sasaki K, et al. Inhibition of PrPSc formation by synthetic O-sulfated glycopyranosides and their polymers. Biochem Biophys Res Commun. 2006;349(2):485–491.
- Yamasaki T, Suzuki A, Hasebe R, et al. Comparison of the anti-prion mechanism of four different anti-prion compounds, anti-PrP monoclonal antibody 44B1, pentosan polysulfate, chlorpromazine, and U18666A, in prion-infected mouse neuroblastoma cells. PLoS One. 2014;9(9):e106516.
- Raymond GJ, Olsen EA, Lee KS, et al. Inhibition of protease-resistant prion protein formation in a transformed deer cell line infected with chronic wasting disease. J Virol. 2006;80(2):596–604.
- Bone I, Belton L, Walker AS, et al. Intraventricular pentosan polysulphate in human prion diseases: an observational study in the UK. Eur J Neurol. 2008;15(5):458–464.
- Tsuboi Y, Doh-ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology. 2009;29(5):632–636.
- Honda H, Sasaki K, Minaki H, et al. Protease-resistant PrP and PrP oligomers in the brain in human prion diseases after intraventricular pentosan polysulfate infusion. Neuropathology. 2012;32(2):124–132.
- Hyeon JW, Kim SY, Lee SM, et al. Anti-prion screening for acridine, dextran, and tannic acid using real time–quaking induced conversion: a comparison with PrPSc-infected cell screening. PLoS One. 2017;12(1):e0170266.
- Fuse T, Nakagaki T, Homma T, et al. Dextran sulphate inhibits an association of prions with plasma membrane at the early phase of infection. Neurosci Res. 2021;171:34–40.
- Ehlers B, Diringer H. Dextran sulphate 500 delays and prevents mouse scrapie by impairment of agent replication in spleen. J Gen Virol. 1984;65(8):1325–1330.
- Farquhar C, Dickinson A. Prolongation of scrapie incubation period by an injection of dextran sulphate 500 within the month before or after infection. J Gen Virol. 1986;67(3):463–473.
- Ladogana A, Casaccia P, Ingrosso L, et al. Sulphate polyanions prolong the incubation period of scrapie-infected hamsters. J Gen Virol. 1992;73(3):661–665.
- Additives EPOF, Mortensen A, Aguilar F, Crebelli R, et al. Re‐evaluation of β‐cyclodextrin (E 459) as a food additive. EFSA J. 2016;14(12):e04628.
- Prior M, Lehmann S, Sy M-S, et al. Cyclodextrins inhibit replication of scrapie prion protein in cell culture. J Virol. 2007;81(20):11195–11207.
- McEvoy K, McMahon HE. Antiprion action of new cyclodextrin analogues. Biochim Biophys Acta. 2009;1790(10):1382–1386.
- FDA, U. Celebrex (celecoxib) Information. 2015 2022 April 28]; Available from: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/celebrex-celecoxib-information.
- Villa V, Thellung S, Corsaro A, et al. Celecoxib inhibits prion protein 90-231-mediated pro-inflammatory responses in microglial cells. Mol Neurobiol. 2016;53(1):57–72.
- AR-12. Study of AR-12 (2-Amino-N-[4-[5-(2 Phenanthrenyl)-3-(Trifluoromethyl)-1H-pyrazol-1-yl] Phenyl]-Acetamide) in Adult Patients With Advanced or Recurrent Solid Tumors or Lymphoma. 2014; [cited April 28, 2022], Available from: https://clinicaltrials.gov/ct2/show/NCT00978523
- Abdulrahman BA, Abdelaziz D, Thapa S, et al. The celecoxib derivatives AR-12 and AR-14 induce autophagy and clear prion-infected cells from prions. Sci Rep. 2017;7(1):1–12.
- Booth L, Roberts JL, Cruickshanks N, et al. Regulation of osu-03012 toxicity by ER stress proteins and ER stress–inducing drugs. Mol Cancer Ther. 2014;13(10):2384–2398.
- Abdelaziz DH, Abdulrahman BA, Gilch S, et al. Autophagy pathways in the treatment of prion diseases. Curr Opin Pharmacol. 2019;44:46–52.
- FDA, U., CHLORPROMAZINE HYDROCHLORIDE. 2021.
- Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proceedings of the National Academy of Sciences (PNAS), USA, 2001. Vols.98(17): p. 9836–9841.
- Baral PK, Swayampakula M, Rout M, et al. Structural basis of prion inhibition by phenothiazine compounds. Structure. 2014;22(2):291–303.
- Barreca ML, Iraci N, Biggi S, et al. Pharmacological agents targeting the cellular prion protein. Pathogens. 2018;7(1):27.
- Stincardini C, Massignan T, Biggi S, et al. An antipsychotic drug exerts anti-prion effects by altering the localization of the cellular prion protein. PloS one. 2017;12(8):e0182589.
- Cronier S, Beringue V, Bellon A, et al. Prion Strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J Virol. 2007;81(24):13794–13800.
- Daniel JA, Chau N, Abdel-Hamid MK, et al. Phenothiazine-derived antipsychotic drugs inhibit dynamin and clathrin-mediated endocytosis. Traffic. 2015;16(6):635–654.
- Roikhel V, Fokina G, Pogodina V. Influence of aminasine on experimental scrapie in mice. Acta Virol. 1984;28(4):321–324.
- Ali T, Hannaoui S, Nemani S, et al. Oral administration of repurposed drug targeting Cyp46A1 increases survival times of prion infected mice. Acta Neuropathol Commun. 2021;9(1):1–14.
- Friedel HA, Fitton A. Flupirtine. Drugs. 1993;45(4):548–569.
- Agency, E.M.,Assessment report for flupirtine containing medicinal products. 2013.
- Schwarz M, Block F, Pergande G. N-methyl-D-aspartate (NMDA)-mediated muscle relaxant action of flupirtine in rats. Neuroreport. 1994;5(15):1981–1984.
- Khosravani H, Zhang Y, Tsutsui S, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181(3):551–565.
- Fang C, Wu B, Le NTT, et al. Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog. 2018;14(9):e1007283.
- Perovic S, Pergande G, Ushijima H, et al. Flupirtine partially prevents neuronal injury induced by prion protein fragment and lead acetate. Neurodegeneration. 1995;4(4):369–374.
- Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology. 2004;62(5):714–718.
- Patil MA, Matter B, Raol Y, et al. Brain distribution and metabolism of flupirtine, a nonopioid analgesic drug with antiseizure effects, in neonatal rats. Pharmaceutics. 2018;10(4):281.
- Schröder HC, Müller W. Neuroprotective effect of flupirtine in prion disease. Drugs Today (Barc). 2002;38(1):49–58.
- FDA, U., Guanabenz. 1998.
- Tribouillard-Tanvier D, Béringue V, Desban N, et al. Antihypertensive drug guanabenz is active in vivo against both yeast and mammalian prions. PloS one. 2008;3(4):e1981.
- Sica DA. Centrally acting antihypertensive agents: an update. J Clin Hypertens. 2007;9(5):399–405.
- Holmes B, Brogden RN, Heel RC, et al. Guanabenz A review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs. 1983;26(3):212–229.
- Nguyen PH, Hammoud H, Halliez S, et al. Structure–activity relationship study around guanabenz identifies two derivatives retaining antiprion activity but having lost α2-adrenergic receptor agonistic activity. ACS Chem Neurosci. 2014;5(10):1075–1082.
- FDA, U., Glimepride. 1995.
- Bate C, Tayebi M, Diomede L, et al. Glimepiride reduces the expression of PrPC, prevents PrPSc formation and protects against prion mediated neurotoxicity. PLoS One. 2009;4(12):e8221.
- Williams A, Van Dam AM, Ritchie D, et al. Immunocytochemical appearance of cytokines, prostaglandin E2 and lipocortin-1 in the CNS during the incubation period of murine scrapie correlates with progressive PrP accumulations. Brain Res. 1997;754(1–2):171–180.
- Farooqui AA, Ong W-Y, Horrocks LA. Inhibitors of brain phospholipase A 2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006;58(3):591–620.
- Ingham V, Williams A, Bate C. Glimepiride reduces CD14 expression and cytokine secretion from macrophages. J Neuroinflammation. 2014;11(1):1–14.
- [cited April 30, 2022]. Available from: https://www.medicines.org.uk/emc/medicine/25845#gref.
- Ertmer A, Gilch S, Yun S-W, et al. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J Biol Chem. 2004;279(40):41918–41927.
- López-Pérez Ó, Badiola JJ, Bolea R, et al. An update on autophagy in prion diseases. Front Bioeng Biotechnol. 2020;8:975.
- Ertmer A, Huber V, Gilch S, et al. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21(5):936–942.
- Advani D, Kumar P. Therapeutic Targeting of Repurposed Anticancer Drugs in Alzheimer’s Disease: using the Multiomics Approach. ACS omega. 2021;6(21):13870–13887.
- Yun S-W, Ertmer A, Flechsig E, et al. The tyrosine kinase inhibitor imatinib mesylate delays prion neuroinvasion by inhibiting prion propagation in the periphery. J Neurovirol. 2007;13(4):328–337.
- Ryou C, Legname G, Peretz D, et al. Differential inhibition of prion propagation by enantiomers of quinacrine. Lab Invest. 2003;83(6):837–843.
- Barret A, Tagliavini F, Forloni G, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003;77(15):8462–8469.
- Yung L, Huang Y, Lessard P, et al. Pharmacokinetics of quinacrine in the treatment of prion disease. BMC Infect Dis. 2004;4(1):1–7.
- Vogtherr M, Grimme S, Elshorst B, et al. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J Med Chem. 2003;46(17):3563–3564.
- Murakami-Kubo I, Doh-ura K, Ishikawa K, et al. Quinoline derivatives are therapeutic candidates for transmissible spongiform encephalopathies. J Virol. 2004;78(3):1281–1288.
- Klingenstein R, Lober S, Kujala P, et al. Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J Neurochem. 2006;98(3):748–759.
- Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8(4):334–344.
- Geschwind MD, Kuo AL, Wong KS, et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology. 2013;81(23):2015–2023.
- Haik S, Brandel JP, Salomon D, et al. Compassionate use of quinacrine in Creutzfeldt–Jakob disease fails to show significant effects. Neurology. 2004;63(12):2413–2415.
- Huang Y, Okochi H, May BCH, et al. Quinacrine is mainly metabolized to mono-desethyl quinacrine by CYP3A4/5 and its brain accumulation is limited by P-glycoprotein. Drug Metab Dispos. 2006;34(7):1136–1144.
- Kwak J-Y, Kim H, Kim JA, et al. Efficacy and safety of radotinib compared with imatinib in newly diagnosed chronic phase chronic myeloid leukemia patients: 12 months result of phase 3 clinical trial. Blood. 2015;126(23):476.
- Trials C. Safety, Tolerability, Pharmacokinetics and efficacy study of radotinib in parkinson’s disease. ClinicalTrials.gov Identifier: NCT04691661. Last updated February 15, 2022. https://clinicaltrials.gov/ct2/show/NCT04691661.
- Lee J, Han BC, Goh HG, et al. IY5511, a novel Bcr-Abl tyrosine kinase inhibitor, is highly active compound in inhibition of CrkL phosphorylation and in xenograft animal chronic myeloid leukemia model. ISH-APD. APBMT Meeting Abstract. Beijing (China). 2007.
- Cho SR, et al. Plasma cell leukemia with rouleaux formation involving neoplastic cells and RBC. The Korean Journal of Hematol. 2011;46(3):152.
- Mucke HA. Drug Repurposing Patent applications January–March 2020. Assay Drug Dev Technol. 2020;18(7):341–346.
- Perrier V, Imberdis T, Lafon P-A, et al. Plasma cholesterol level determines in vivo prion propagation [S]. J Lipid Res. 2017;58(10):1950–1961.
- Pinheiro TJ. The role of rafts in the fibrillization and aggregation of prions. Chem Phys Lipids. 2006;141(1–2):66–71.
- Kempster S, Bate C, Williams A. Simvastatin treatment prolongs the survival of scrapie-infected mice. Neuroreport. 2007;18(5):479–482.
- Mok SWF, Thelen KM, Riemer C, et al. Simvastatin prolongs survival times in prion infections of the central nervous system. Biochem Biophys Res Commun. 2006;348(2):697–702.
- Jeong A, Suazo KF, Wood WG, et al. Isoprenoids and protein prenylation: implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Crit Rev Biochem Mol Biol. 2018;53(3):279–310.
- Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proceedings of the National Academy of Sciences (PNAS), USA, 2001. Vols.98(10): p. 5856–5861.
- Hooff GP, Peters I, Wood WG, et al. Modulation of cholesterol, farnesylpyrophosphate, and geranylgeranylpyrophosphate in neuroblastoma SH-SY5Y-APP695 cells: impact on amyloid beta-protein production. Mol Neurobiol. 2010;41(2):341–350.
- Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease: i. Review of epidemiological and preclinical studies. Arch Neurol. 2011;68(10):1239–1244.
- Haviv Y, Avrahami D, Ovadia H, et al. Induced neuroprotection independently from PrPSc accumulation in a mouse model for prion disease treated with simvastatin. Arch Neurol. 2008;65(6):762–775.
- Forloni G, Iussich S, Awan T, et al. Tetracyclines affect prion infectivity. Proceedings of the National Academy of Sciences (PNAS), USA, 2002. Vols.99(16): p. 10849–10854.
- Tagliavini F, Forloni G, Colombo L, et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrPSc in vitro. J Mol Biol. 2000;300(5):1309–1322.
- Tagliavini F. S3‐01–03: prion therapy: tetracyclic compounds in animal models and patients with Creutzfeldt‐Jakob disease. Alzheimers Dement. 2008;4(4S_Part_5):T149–T150.
- Ronga L, Langella E, Palladino P, et al. Does tetracycline bind helix 2 of prion? An integrated spectroscopical and computational study of the interaction between the antibiotic and α helix 2 human prion protein fragments. Proteins: structure. Funct Bioinf. 2007;66(3):707–715.
- Haïk S, Marcon G, Mallet A, et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(2):150–158.
- Merlini G, Ascari E, Amboldi N, et al. Interaction of the anthracycline 4’-iodo-4’-deoxydoxorubicin with amyloid fibrils: inhibition of amyloidogenesis. Proceedings of the National Academy of Sciences (PNAS), USA, 1995. Vols.92(7): p. 2959–2963.
- Tagliavini F, McArthur RA, Canciani B, et al. Effectiveness of anthracycline against experimental prion disease in Syrian hamsters. Science. 1997;276(5315):1119–1121.
- Balducci C, Forloni G. Doxycycline for Alzheimer’s disease: fighting β-amyloid oligomers and neuroinflammation. Front Pharmacol. 2019;10:738.
- De Luigi A, Colombo L, Diomede L, et al. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS One. 2008;3(3):e1888.
- Varges D, Manthey H, Heinemann U, et al. Doxycycline in early CJD: a double-blinded randomised phase II and observational study. J Neurol Neurosurg. 2017;88(2):119–125.
- Mangé A, Nishida N, Milhavet O, et al. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J Virol. 2000;74(7):3135–3140.
- Carmody M, Murphy B, Byrne B, et al. Biosynthesis of amphotericin derivatives lacking exocyclic carboxyl groups. J Biol Chem. 2005;280(41):34420–34426.
- Soler L, Caffrey P, McMahon HE. Effects of new amphotericin analogues on the scrapie isoform of the prion protein. Biochim Biophys Acta. 2008;1780(10):1162–1167.
- Beringue V, Adjou KT, Lamoury F, et al. Opposite effects of dextran sulfate 500, the polyene antibiotic MS-8209, and Congo red on accumulation of the protease-resistant isoform of PrP in the spleens of mice inoculated intraperitoneally with the scrapie agent. J Virol. 2000;74(12):5432–5440.
- Arshad H, Patel Z, Mehrabian M, et al. The aminoglycoside G418 hinders de novo prion infection in cultured cells. J Biol Chem. 2021;297(3):101073.
- Freyssin A, Page G, Fauconneau B, et al. Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases. Neural Regen Res. 2018;13(6):955.
- Habtemariam S. Molecular pharmacology of rosmarinic and salvianolic acids: potential seeds for Alzheimer’s and vascular dementia drugs. Int J Mol Sci. 2018;19(2):458.
- Velander P, Wu L, Henderson F, et al. Natural product-based amyloid inhibitors. Biochem Pharmacol. 2017;139:40–55.
- Cornejo A, Aguilar Sandoval F, Caballero L, et al. Rosmarinic acid prevents fibrillization and diminishes vibrational modes associated to β sheet in tau protein linked to Alzheimer’s disease. J Enzyme Inhib Med Chem. 2017;32(1):945–953.
- Toni M, Massimino ML, De Mario A, et al. Metal dyshomeostasis and their pathological role in prion and prion-like diseases: the basis for a nutritional approach. Front Neurosci. 2017;11:3.
- Mandel SA, Amit T, Weinreb O, et al. Understanding the broad-spectrum neuroprotective action profile of green tea polyphenols in aging and neurodegenerative diseases. Journal of Alzheimer’s Disease. 2011;25(2):187–208.
- Singh M, Arseneault M, Sanderson T, et al. Challenges for research on polyphenols from foods in Alzheimer’s disease: bioavailability, metabolism, and cellular and molecular mechanisms. J Agric Food Chem. 2008;56(13):4855–4873.
- Heo HJ, Kim D-O, Choi SJ, et al. Potent Inhibitory Effect of Flavonoids in Scutellaria baicalensis on Amyloid β Protein-Induced Neurotoxicity. J Agric Food Chem. 2004;52(13):4128–4132.
- Moon J-H, Park S-Y. Baicalein prevents human prion protein-induced neuronal cell death by regulating JNK activation. Int J Mol Med. 2015;35(2):439–445.
- Shen H-M, Liu Z-G. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40(6):928–939.
- Eiden M, Leidel F, Strohmeier B, et al. A medicinal herb Scutellaria lateriflora inhibits PrP replication in vitro and delays the onset of prion disease in mice. Front Psychiatry. 2012;3:9.
- Cortez LM, Campeau J, Norman G, et al. Bile acids reduce prion conversion, reduce neuronal loss, and prolong male survival in models of prion disease. J Virol. 2015;89(15):7660–7672.
- Cortez L, Sim V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion. 2014;8(2):197–202.
- Ahn T-K, Kim K-T, Joshi HP, et al. Therapeutic potential of tauroursodeoxycholic acid for the treatment of osteoporosis. Int J Mol Sci. 2020;21(12):4274.
- Dirikoc S, Priola SA, Marella M, et al. Nonpsychoactive cannabidiol prevents prion accumulation and protects neurons against prion toxicity. J Neurosci. 2007;27(36):9537–9544.
- Andrade JM, Faustino C, Garcia C, et al. Rosmarinus officinalis L.: an update review of its phytochemistry and biological activity. Future Sci OA. 2018;4(4):FSO283.
- Karagianni K, Pettas S, Kanata E, et al. Carnosic acid and carnosol display antioxidant and anti-prion properties in in vitro and cell-free models of prion diseases. Antioxidants. 2022;11(4):726.
- Satoh T, Kosaka K, Itoh K, et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S- alkylation of targeted cysteines on Keap1. J Neurochem. 2008;104(4):1116–1131.
- Shaker B, Yu M-S, Song JS, et al. LightBBB: computational prediction model of blood–brain-barrier penetration based on LightGBM. Bioinformatics. 2021;37(8):1135–1139.
- Caughey B, Raymond LD, Raymond GJ, et al. Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J Virol. 2003;77(9):5499–5502.
- Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892–5901.
- Garcea G, Jones DJL, Singh R, et al. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br J Cancer. 2004;90(5):1011–1015.
- Hafner-Bratkovič I, Gašperšič J, Šmid LM, et al. Curcumin binds to the α-helical intermediate and to the amyloid form of prion protein - a new mechanism for the inhibition of PrPSc accumulation. J Neurochem. 2008;104(6):1553–1564.
- Lin C-F, Yu K-H, Jheng C-P, et al. Curcumin reduces amyloid fibrillation of prion protein and decreases reactive oxidative stress. Pathogens. 2013;2(3):506–519.
- Kurien BT, Singh A, Matsumoto H, et al. Improving the solubility and pharmacological efficacy of curcumin by heat treatment. Assay Drug Dev Technol. 2007;5(4):567–576.
- Lee J-H, Moon J-H, Kim S-W, et al. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget. 2015;6(12):9701.
- Jeong J-K, Moon M-H, Lee Y-J, et al. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging. 2013;34(1):146–156.
- Qin W, Yang T, Ho L, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281(31):21745–21754.
- Parker JA, Arango M, Abderrahmane S, et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37(4):349–350.
- Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26(13):3169–3179.
- Liu JW, Tian SJ, de Barry J, et al. Panaxadiol glycosides that induce neuronal differentiation in neurosphere stem cells. J Nat Prod. 2007;70(8):1329–1334.
- Wu T, Kwaku OR, Li H-Z, et al. Sense ginsenosides from ginsengs: structure-activity relationship in autophagy. Nat Prod Commun. 2019;14(6):1934578X19858223.
- Moon J-H, Lee J-H, Lee Y-J, et al. Autophagy flux induced by ginsenoside-Rg3 attenuates human prion protein-mediated neurotoxicity and mitochondrial dysfunction. Oncotarget. 2016;7(52):85697.
- El Hachlafi N, Lakhdar F, Khouchlaa A, et al. Health benefits and pharmacological properties of hinokitiol. Processes. 2021;9(9):1680.
- Jayakumar T, Hsu WH, Yen TL, et al. Hinokitiol, a natural tropolone derivative, offers neuroprotection from thromboembolic stroke in vivo. Evid Based Complement Alternat Med. 2013;2013:1–8.
- Moon J-H, Lee J-H, Lee Y-J, et al. Hinokitiol protects primary neuron cells against prion peptide-induced toxicity via autophagy flux regulated by hypoxia inducing factor-1. Oncotarget. 2016;7(21):29944.
- Jeong J-K, Seo JS, Moon MH, et al. Hypoxia-inducible factor-1 alpha regulates prion protein expression to protect against neuron cell damage. Neurobiol Aging. 2012;33(5):1006. e1–1006. e10.
- Galano A, Tan DX, Reiter RJ. Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res. 2011;51(1):1–16.
- Singhal NK, Srivastava G, Patel DK, et al. Melatonin or silymarin reduces maneb- and paraquat-induced Parkinson’s disease phenotype in the mouse. J Pineal Res. 2011;50(2):97–109.
- Jeong J-K, Lee J-H, Moon J-H, et al. Melatonin-mediated β -catenin activation protects neuron cells against prion protein-induced neurotoxicity. J Pineal Res. 2014;57(4):427–434.
- Jeong J-K, Moon M-H, Lee Y-J, et al. Melatonin-induced autophagy protects against human prion protein-mediated neurotoxicity. J Pineal Res. 2012;53(2):138–146.
- Bleazard W, McCaffery JM, King EJ, et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1(5):298–304.
- Olichon A, Emorine LJ, Descoins E, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523(1–3):171–176.
- Zhang X, Zhao D, Wu W, et al. Melatonin regulates mitochondrial dynamics and alleviates neuron damage in prion diseases. Aging (Albany NY). 2020;12(11):11139.
- Loh D, Reiter RJ. Melatonin: regulation of prion protein phase separation in cancer multidrug resistance. Molecules. 2022;27(3):705.
- Wu X, Li Q, Feng Y, et al. Antitumor research of the active ingredients from traditional Chinese medical plant Polygonum cuspidatum. Evid Based Complement Alternat Med. 2018; 2018:10. https://doi.org/10.1155/2018/2313021
- Şöhretoğlu D, Baran MY, Arroo R, et al. Recent advances in chemistry, therapeutic properties and sources of polydatin. Phytochem Rev. 2018;17(5):973–1005.
- Rivière C, Papastamoulis Y, Fortin P-Y, et al. New stilbene dimers against amyloid fibril formation. Bioorg Med Chem Lett. 2010;20(11):3441–3443.
- Rivière C, Richard T, Quentin L, et al. Inhibitory activity of stilbenes on Alzheimer’s β-amyloid fibrils in vitro. Bioorg Med Chem. 2007;15(2):1160–1167.
- Sirohi PR, Kumari A, Admane N, et al. The polyphenolic phytoalexin polydatin inhibits amyloid aggregation of recombinant human prion protein. RSC Adv. 2021;11(42):25901–25911.
- Albani D, Polito L, Signorini A, et al. Neuroprotective properties of resveratrol in different neurodegenerative disorders. Biofactors. 2010;36(5):370–376.
- Wang J, Zhang B-Y, Zhang J, et al. Treatment of SMB-S15 cells with resveratrol efficiently removes the PrPSc accumulation in vitro and prion infectivity in vivo. Mol Neurobiol. 2016;53(8):5367–5376.
- Wang J, Zhang J, Shi Q, et al. Scrapie infection in experimental rodents and SMB-S15 cells decreased the brain endogenous levels and activities of Sirt1. J Mol Neurosci. 2015;55(4):1022–1030.
- Li L, Zhu Y, Zhou S, et al. Experimental and theoretical insights into the inhibition mechanism of prion fibrillation by resveratrol and its derivatives. ACS Chem Neurosci. 2017;8(12):2698–2707.
- Jeong JK, Moon MH, Bae BC, et al. Autophagy induced by resveratrol prevents human prion protein-mediated neurotoxicity. Neurosci Res. 2012;73(2):99–105.
- Xie R, Nguyen S, McKeehan WL, et al. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010;11(1):1–12.
- Phadwal K, Kurian D, Salamat MKF, et al. Spermine increases acetylation of tubulins and facilitates autophagic degradation of prion aggregates. Sci Rep. 2018;8(1):1–17.
- Chen Q, Haddad GG. Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol. 2004;207(18):3125–3129.
- Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J Biol Chem. 2007;282(8):5641–5652.
- Aguib Y, Heiseke A, Gilch S, et al. Autophagy induction by trehalose counter-acts cellular prion-infection. Autophagy. 2009;5(3):361–369.
- Béranger F, Crozet C, Goldsborough A, et al. Trehalose impairs aggregation of PrPSc molecules and protects prion-infected cells against oxidative damage. Biochem Biophys Res Commun. 2008;374(1):44–48.
- Lee H-J, Yoon Y-S, Lee S-J. Mechanism of neuroprotection by trehalose: controversy surrounding autophagy induction. Cell Death Dis. 2018;9(7):1–12.
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu. Rev Neurosci. 2008;31(1):175–193.
- Souan L, Tal Y, Felling Y, et al. Modulation of proteinase-K resistant prion protein by prion peptide immunization. Eur J Immunol. 2001;31(8):2338–2346.
- Aguzzi A, Lakkaraju AK, Frontzek K. Toward therapy of human prion diseases. Annu Rev Pharmacol Toxicol. 2018;58(1):331–351.
- Sassa Y, Kataoka N, Inoshima Y, et al. Anti-PrP antibodies detected at terminal stage of prion-affected mouse. Cell Immunol. 2010;263(2):212–218.
- Taschuk R, Van der Merwe J, Marciniuk K, et al. In vitro neutralization of prions with PrP Sc -specific antibodies. Prion. 2015;9(4):292–303.
- Marciniuk K, Määttänen P, Taschuk R, et al. Development of a multivalent, PrPSc-specific prion vaccine through rational optimization of three disease-specific epitopes. Vaccine. 2014;32(17):1988–1997.
- Taschuk R, Marciniuk K, Määttänen P, et al. Safety, specificity and immunogenicity of a PrP Sc -specific prion vaccine based on the YYR disease specific epitope. Prion. 2014;8(1):51–59.
- Wood ME, Griebel P, Huizenga ML, et al. Accelerated onset of chronic wasting disease in elk (Cervus canadensis) vaccinated with a PrPSc-specific vaccine and housed in a prion contaminated environment. Vaccine. 2018;36(50):7737–7743.
- Frontzek K, Pfammatter M, Sorce S, et al. Neurotoxic antibodies against the prion protein do not trigger prion replication. PLoS One. 2016;11(9):e0163601.
- Frontzek K, Bardelli M, Senatore A, et al. A conformational switch controlling the toxicity of the prion protein. Nat Struct Mol Biol. 2022;1–10. https://doi.org/10.1101/2021.09.20.460912
- Ilie IM, Caflisch A. Antibody binding increases the flexibility of the prion protein. Biochim Biophys Acta (Bba)-proteins and Proteomics. 2022;1870(11–12):140827.
- Bardelli M, Frontzek K, Simonelli L, et al. A bispecific immunotweezer prevents soluble PrP oligomers and abolishes prion toxicity. PLoS Pathog. 2018;14(10):e1007335.
- Ma Y, Ma J. Immunotherapy against prion disease. Pathogens. 2020;9(3):216.
- Enari M, Flechsig E, Weissmann C, Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proceedings of the National Academy of Sciences (PNAS), USA, 2001. Vols.98(16): p. 9295–9299.
- Féraudet C, Morel N, Simon S, et al. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280(12):11247–11258.
- Pankiewicz J, Prelli F, Sy M-S, et al. Clearance and prevention of prion infection in cell culture by anti-PrP antibodies. Eur J Neurosci. 2006;23(10):2635–2647.
- Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412(6848):739–743.
- Müller-Schiffmann A, Korth C. Vaccine approaches to prevent and treat prion infection. BioDrugs. 2008;22(1):45–52.
- Sigurdsson EM, Sy M-S, Li R, et al. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett. 2003;336(3):185–187.
- Sadowski MJ, Pankiewicz J, Prelli F, et al. Anti-PrP Mab 6D11 suppresses PrPSc replication in prion infected myeloid precursor line FDC-P1/22L and in the lymphoreticular system in vivo. Neurobiol Dis. 2009;34(2):267–278.
- Pankiewicz JE, Sanchez S, Kirshenbaum K, et al. Anti-prion protein antibody 6D11 restores cellular proteostasis of prion protein through disrupting recycling propagation of PrPSc and targeting PrPSc for lysosomal degradation. Mol Neurobiol. 2019;56(3):2073–2091.
- White AR, Enever P, Tayebi M. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature, 2003;422(6927):80–83. https://doi.org/10.1038/nature01457
- Khalili-Shirazi A, Kaisar M, Mallinson G, et al. β-PrP form of human prion protein stimulates production of monoclonal antibodies to epitope 91–110 that recognise native PrPSc. Biochimica Et Biophysica Acta (BBA) - Proteins and Proteomics. 2007;1774(11):1438–1450.
- Biro L, Leone N. Aplastic anemia induced by quinacrine. Arch Dermatol. 1965;92(5):574–576.
- Terada T, Tsuboi Y, Obi T, et al. Less protease-resistant PrP in a patient with sporadic CJD treated with intraventricular pentosan polysulphate. Acta Neurol Scand. 2010;121(2):127–130.
- Zerr I. Investigating new treatments for Creutzfeldt–Jakob disease. Lancet Neurol. 2022;21(4):299–300.
- Colini Baldeschi A, Zattoni M, Vanni S, et al. Innovative Non-PrP-targeted drug strategy designed to enhance prion clearance. J Med Chem. 2022;65(13):8998–9010.
- Zattoni M, Mearelli M, Vanni S, et al. Serpin signatures in prion and alzheimer’s diseases. Mol Neurobiol. 2022;59:3778–3799. https://doi.org/10.1007/s12035-022-02817-3
- Kristiansen M, Deriziotis P, Dimcheff DE, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26(2):175–188.
- Lee FK, Wong AKY, Lee YW, et al. The role of ubiquitin linkages on α-synuclein induced-toxicity in a Drosophila model of Parkinson’s disease. J Neurochem. 2009;110(1):208–219.
- Al-Ramahi I, Lam YC, Chen H-K, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281(36):26714–26724.
- Leestemaker Y, de Jong A, Witting KF, et al. Proteasome activation by small molecules. Cell Chem Biol. 2017;24(6):725–736. e7