
ABSTRACT
Cell motility is an essential cellular process for a variety of biological events. It requires cross-talk between the signaling and the cytoskeletal systems. Despite the recognized importance of aPKCζ for cell motility, there is little understanding of the mechanism by which aPKCζ mediates extracellular signals to the cytoskeleton. In the present study, we report that aPKCζ is required for the cellular organization of acto-non-muscle myosin II (NMII) cytoskeleton, for proper cell adhesion and directed cell migration. We show that aPKCζ mediates EGF-dependent RhoA activation and recruitment to the cell membrane. We also show that aPKCζ mediates EGF-dependent myosin light chain (MRLC) phosphorylation that is carried out by Rho-associated protein kinase (ROCK), and that aPKCζ is required for EGF-dependent phosphorylation and inhibition of the myosin phosphatase targeting subunit (MYPT). Finally, we show that aPKCζ mediates the spatial organization of the acto-NMII cytoskeleton in response to EGF stimulation. Our data suggest that aPKCζ is an essential component regulator of acto-NMII cytoskeleton organization leading to directed cell migration, and is a mediator of the EGF signal to the cytoskeleton.
Introduction
Cell migration is a vital phenomenon in most biological processes. It plays an important role in many physiological and pathological processes, including cellular development, tissue repair, and cancer metastasis.Citation1,2 Non-muscle myosin II (NMII) plays a key role in cell migration by moving the body of the cell forward, by retracting the rear of the cell, and by the initiation and maturation of adhesion sites.Citation3-7 NMII is a hexamer composed of 2 heavy chains (MHC) of ∼200 kDa and 2 pairs of essential (MELC) and regulatory light chains (MRLC). The MHCs include an α-helical coiled-coil rod domain responsible for the assembly of NMII monomers into filaments, which is the functional structure required for NMII activity.Citation7 NMII is in constant equilibrium between monomeric and filamentous forms. During migration, the cell achieves spatio-temporal control of NMII assembly and activation primarily through phosphorylation events. Phosphorylation of MRLC at Thr-18 and Ser-19 promotes activation of NMII ATPase and filament assembly.Citation8,9 MRLC phosphorylation is regulated by myosin light chain kinase (MLCK) and Rho-associated protein kinase (ROCK). ROCK phosphorylates MRLC directly,Citation10 but it primarily acts to inhibit the major myosin light chain phosphatase (MLCP) through phosphorylation of the myosin phosphatase-targeting (MYPT) subunit.Citation10-13 ROCK is activated by RhoA, and in migrating cells RhoA activation at the rear of the cell promotes the organization and contraction of acto-NMII filaments by phosphorylating MRLC and activating NMII to form large, thick acto-NMII bundles.Citation7 Activation of ROCK in living cells strongly induces stress fiber and focal adhesion organization in cultured fibroblasts.Citation14-17 Atypical protein kinase C subtype ζ (aPKCζ) is involved in the regulation of cell polarization, directional sensing, formation of filopodia, and cell motility.Citation18 aPKCζ is essential for the migration and invasion of multiple cancer cell typesCitation19-21 because it regulates cytoskeleton rearrangement and cell adhesion.Citation22,23 aPKCζ is required for EGF-induced chemotaxis of several cancer cell lines.Citation19,20,23 EGF-elicited translocation and phosphorylation of aPKCζ indicates that EGF could activate aPKCζ.Citation20 In vivo aPKCζ, is part of the Par complex that is involved in the polarity of migrating cells.Citation24 For example, it was demonstrated that Par6 and aPKCζ regulate cell polarity in wound-induced directed migration of astrocytes and fibroblasts, and that aPKCζ inhibition induces random cell migration.Citation25 Recently we showed that aPKCζ is important for establishing front-rear polarization of migrating cells by regulating the tumor suppressor lethal giant larvae 1 (Lgl1).Citation26 Lgl1 regulates the polarity of migrating cells by controlling the assembly state of NMII isoform A (NMIIA), its cellular localization, and focal adhesion assembly.Citation27 Phosphorylation of Lgl1 by aPKCζ affects its cellular localization and prevents its interaction with NMIIA, thus affecting the cellular organization of the acto-NMIIA cytoskeleton.Citation26 Together, these results strongly indicate that aPKCζ plays an important role in cell migration. Nevertheless, little is known about the mechanism by which aPKCζ affects cell migration and how it mediates extracellular signals to the cytoskeleton. In the present study, we report that aPKCζ is required for the proper cellular organization of the acto-NMII cytoskeleton, cell adhesion, and migration. Furthermore, we show that aPKCζ mediates EGF signaling to the cytoskeleton by activation of the RhoA-ROCK pathway that leads to MRLC phosphorylation and spatial organization of active acto-NMII.
Results
aPKCζ is important for proper cellular organization of the acto-NMII cytoskeleton
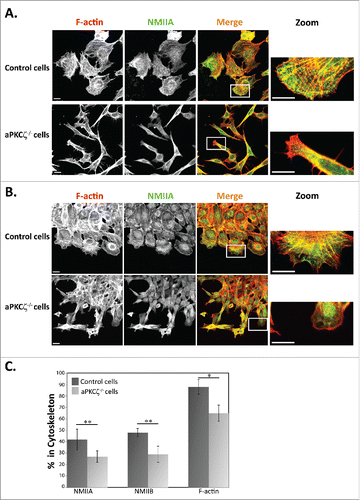
The dynamic organization of the acto-NMII cytoskeleton provides the driving force for cell movement, which directs the protrusion of the cell membrane at the front of the cell and retraction at the rear.Citation7 Therefore, the spatial regulation of the acto-NMII cytoskeleton is a critical component in the regulation of cell migration. To begin exploring the role of aPKCζ in the organization of the acto-NMII cytoskeleton, we characterized the cellular localization properties of NMIIA, NMIIB, and F-actin in aPKCζ−/− dispersed cells and in cells subjected to wound scratch assay in order to achieve cell polarization. Dispersed control cells exhibited well-defined, typical acto-NMIIA and acto-NMIIB cytoskeletons containing stress fibers ( and S1). In control cells subjected to wound scratch assay, the F-actin was localized to the lamellipodia; by contrast, NMIIA and NMIIB were missing from this region and presented in the lamella ( and S1), consistent with previous reports.Citation5,28,29 Furthermore, these cells formed one sheet with the same cell polarity as determined by the orientation of F-actin (). By contrast, dispersed aPKCζ−/− cells and cells subjected to wound scratch assay demonstrated disrupted acto-NMIIA and acto–NMIIB cytoskeletons, with a few stress fibers that were missing the typical cellular localization of NMIIA, NMIIB, and F-actin, which was observed in control cells (). Furthermore, aPKCζ−/− cells that were subjected to wound scratch assay migrated in different directions, thus exhibiting different cell polarities, with some cells detached from the main sheet (). Hence, the absence of aPKCζ may result in a loss of cell-cell contact and in independent migration of detached cells into the wound space. Collectively, these results indicate that aPKCζ plays a role in the assembly of acto-NMII that is required for cell polarity and migration. To further study the role of aPKCζ in the cellular organization of acto-NMII, we used the Triton X-100 solubility assay to determine the amount of endogenous NMIIA, NMIIB, and F-actin associated with the cytoskeleton in aPKCζ−/− and control cells. Lower levels of NMIIA, NMIIB, and F-actin were associated with the cytoskeleton in aPKCζ−/− cells than in control cells (41%, 48%, and 88% vs. 26%, 28%, and 64%, respectively, ). These results further indicate that NMIIA, NMIIB, and F-actin polymerized less in aPKCζ−/− cells than in control cells, and that aPKCζ is important for acto-NMII filament assembly.
Figure 1. aPKCζ affected the acto-NMII cytoskeleton. aPKCζ−/− and control cells were seeded on coverslips (i.e., dispersed cells) (A) or subjected to wound scratch assay (B), and stained for F-actin, using Rhodamine-Phalloidin, and for NMIIA, using C-terminal specific antibody and secondary antibody conjugated to Cy2. Bars are 20μm. (C) aPKCζ−/− and control cells were subjected to Triton X-100 solubility assay, and the percentages of total NMIIA, NMIIB, and actin in the soluble and insoluble fractions were determined, as described in Materials and Methods. Values represent the mean ± SEM for 4 independent experiments, * p < 0.05, ** p < 0.01, values are aPKCζ−/− cells compared with control cells.
aPKCζ is required for proper cell adhesion
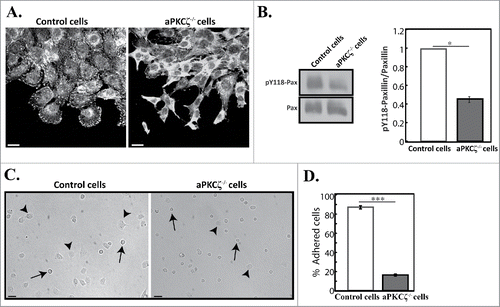
In motile cells, focal adhesions are constantly assembled and disassembled as the cell establishes new contacts at its leading edge and breaks old contacts at its trailing edge.Citation30 NMIIA and NMIIB mediate the initiation, maturation, and stabilization of adhesion sites.Citation4,5,31,32 Next, we tested how the absence of aPKCζ affected the formation of focal adhesion. Because paxillin phosphorylation is critical to cell migration and maturation of adhesions,Citation33-35 and NMII activity promotes phosphorylation of paxillin on tyrosines 31 and 118,Citation36 we characterized paxillin and paxillin phosphorylated on residue Y118 (pY118-paxillin) in aPKCζ−/− and control cells. Control cells subjected to wound scratch assay exhibited well-defined adhesions in protrusions, as well as some small adhesions near the leading edge, as viewed by paxillin () and pY118-paxillin (Fig. S2), consistent with previous reports.Citation5 Elimination of aPKCζ led to abnormal appearance of adhesions and a marked decrease in the number of focal adhesions compared to control cells ( and S2). To quantify these results, we measured the amounts of paxillin and pY118-paxillin in these cells. Whereas control and aPKCζ−/− cells demonstrated similar levels of paxillin, the ratio of pY118-paxillin to total paxillin levels in aPKCζ−/− cells was reduced by 50% compared to control cells (). These results indicate that aPKCζ is necessary for paxillin phosphorylation, which is critical for the maturation of focal adhesions. Next, we sought to determine how the aberrant focal adhesion and the reduced pY118-paxillin in aPKCζ−/− cells affected cell adhesion. shows that most aPKCζ−/− cells were round in shape, and only a few cells adhered to the substrate. This contrasted with the control cells, most of which adhered to the substrate, leaving only a few round cells detached. Quantification of the number of adherent cells indicated that only 18% of aPKCζ−/− cells adhered to the dish surface, compared to 88% of control cells (). Together, these results indicate that aPKCζ is required for the formation and maturation of focal adhesion and cell adhesion.
Figure 2. aPKCζ is required for proper cell adhesion. (A) aPKCζ−/− and control cells were subjected to wound scratch assay and immunostained with anti-paxillin antibody. Bars are 20 µm. (B) Quantification of the amount of pY118-paxillin. Lysates from aPKCζ−/− and control cells were subjected to Western blot analysis using anti-paxillin and anti-pY118-paxillin antibodies. The amounts of paxillin were normalized relative to actin. Bands were analyzed by densitometry using ImageJ. Paxillin phosphorylation was calculated as the ratio of phosphorylated paxillin to total paxillin. Values represent the mean ± SEM for 4 independent experiments, * p < 0.05, values are aPKCζ−/− cells compared with control cells. Pax, paxillin and pY118-pax, pY118-paxillin. (C) aPKCζ−/− and control cells were seeded on dishes, and after 2.5 h phase-contrast images of the cells were taken by confocal microscopy. Arrows indicate rounded detached cells and arrowheads indicate adhered cells. Bars are 50 µm. (D) Quantification of adhered cells. Adhered and rounded detached cells in 15 randomly-chosen fields were counted and the percentage of adhered cells relative to the total number of cells in each field was calculated. Values represent the mean ± SEM for 4 independent experiments subjected to 2-tailed, 2-sampled unequal variance Student's t test, *** p < 0.001, values are aPKCζ−/− cells compared with control cells.
aPKCζ is important for directed cell migration
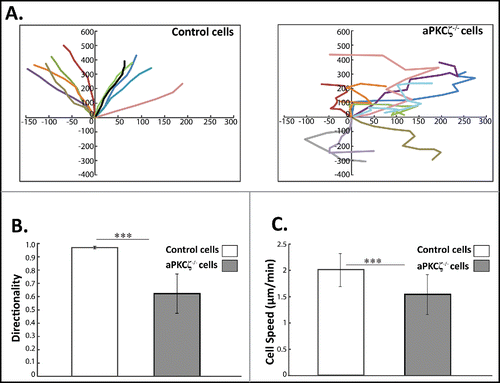
The cellular organization of F-actin and NMII plays a critical role in directed cell migration.Citation7 Migratory front-back polarity emerges from the cooperative effects of NMIIA and NMIIB on adhesive signaling. The results described above indicate that aPKCζ plays a crucial role in the cellular organization of acto-NMII and focal adhesion assembly. We therefore investigated how the absence of aPKCζ affected directed cell migration. To this end, we subjected aPKCζ−/− cells to a wound healing assay, and tracked single cell migration using time-lapse microscopy. The persistence of migration (the ratio of the vectorial distance traveled to the total path length of the cell) was extracted from the track plots. Whereas control cells moved in relatively straight paths, with high persistence of migration, aPKCζ−/− cells moved randomly, with several changes in direction, showing both a significant decrease in persistence () and significantly lower directionality (). Furthermore, the cell migration speed of control cells was higher than that of aPKCζ−/− cells (), possibly because of their high persistence of migration. To further characterize the effect of aPKCζ on cell migration, we assessed the ability of aPKCζ−/− cells to initiate migration in a wound scratch assay. Control cells exhibited broad lamellipodia, whereas aPKCζ−/− cells appeared elongated, with long filopodia ( and S3). Moreover, control cells formed one sheet in which the cells showed the same cell polarity, whereas aPKCζ−/− cells exhibited different cell polarities, with some cells detached from the main sheet ( and S3). These results indicate that aPKCζ, is important for directional migration, persistence of movement, and the regulation of cell-cell contact.
Figure 3. aPKCζ affects persistence and speed of cell movement. (A) aPKCζ−/− and control cells were seeded on 2-well chamber coated with Collagen I. After wounding, the cells were tracked using time-lapse confocal microscopy at 30 min intervals. The paths of 10 randomly chosen cells were plotted for each experimental group. Paths are oriented in such a way that the start point is normalized to the origin. (B) the persistence of migration and (C) the speed of migration were extracted from the track plots. Persistence is defined as the ratio of the vectorial distance traveled to the total path length described by the cell. Values represent the mean ± SEM for n > 30 track plots. Persistence and speed, *** p < 0.001, values are aPKCζ−/− cells compared with control cells.
aPKCζ mediates EGF-dependent MRLC phosphorylation
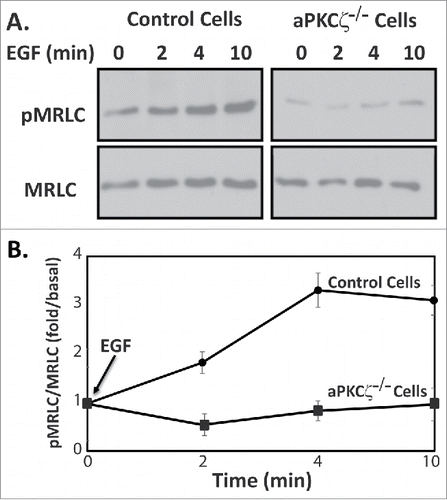
To begin understanding the role of aPKCζ in directed cell migration, we studied its role in the activation of NMII. Phosphorylation of the MRLC on Thr-18 and Ser-19 residues is known to promote contraction of the actin-based cytoskeleton and generate the force required for cell motility.Citation7 We speculated that aPKCζ affects the phosphorylation state of MRLC, hence affecting NMII activity and directional cell motility. Extracellular cues, such as growth factors, are known to elicit various intracellular responses in the organization of acto-NMII cytoskeletons.Citation1,37,38 Because aPKCζ is activated by EGF,Citation39,40 we tested this hypothesis using EGF as a directional cue. We stimulated aPKCζ−/− and control cells with EGF and measured the levels of phosphorylated MRLC (pMRLC). shows that EGF stimulation of control cells, but not of aPKCζ−/− cells, resulted in an increase in pMRLC, with a maximum increase taking place 4 min after EGF stimulation. These results indicate that aPKCζ is involved in EGF-dependent pMRLC and activation of NMII.
Figure 4. aPKCζ is required for EGF-dependent MRLC phosphorylation. (A) aPKCζ−/− and control cells were stimulated with EGF, and at the indicated time point the cells were lysed in SDS-PAGE sample buffer divided into 2 samples that were subjected to Western blot analysis. One sample was probed with anti-MRLC antibodies and the other anti-pMRLC antibodies. Shown are representative Western blots. (B) Densitometry analysis of Western blots. Bands were analyzed by densitometry using ImageJ and the relative amounts of pMRLC to total MRLC are shown. Values represent the mean ± SEM for 5 independent experiments.
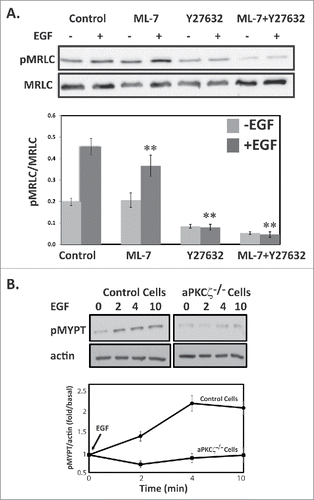
ROCK but not MLCK affects the level of pMRLC after EGF stimulation
MLCK and ROCK appear to be the 2 important kinases that phosphorylate MRLC in vitro as well as in vivo; they play distinct roles in membrane protrusions and focal adhesion dynamics during cell migration.Citation10,12,13 ROCK also phosphorylates the MYPT subunit, inhibiting MLCP and indirectly promoting MRLC phosphorylation.Citation41 To dissect the kinases involved in the EGF-dependent MRLC phosphorylation, we used the ROCK inhibitor, Y-27632, and the MLCK inhibitor, ML-7. Control cells treated with Y-27632 or ML-7 were stimulated with EGF for 4 min and subjected to Western blots using antibodies specific to pMRLC. Inhibition of ROCK but not of MLCK led to a significant decrease in EGF-dependent pMRLC (). ML-7 resulted in ∼20% and Y-27632 in ∼83% decrease in EGF-dependent pMRLC. These results suggest that the ROCK-MRLC phosphorylation cascade plays a key role in mediating the effects of aPKCζ upon EGF stimulation. Addition of ML-7 and Y-27632 further decreased the EGF-dependent pMRLC by 90%. Cells treated with Y-27632 but not with ML-7 showed decreased basal levels of pMRLC, indicating that ROCK is also involved in MRLC phosphorylation in unstimulated cells as well. ROCK inactivates MLCP through phosphorylation of MYPT at Thr-696 and Thr-850,Citation11,41 thereby increasing MRLC phosphorylation. To test whether aPKCζ is also involved in EGF-dependent MYPT phosphorylation through the activation of ROCK, we investigated the phosphorylation levels of MYPT (pMYPT) upon EGF stimulation in aPKCζ−/− cells. pMYPT in control cells but not in aPKCζ−/− cells was phosphorylated upon EGF stimulation, further supporting the involvement of aPKCζ in EGF-dependent MRLC phosphorylation (). Together, these results indicate that EGF stimulation of control but not of aPKCζ−/− cells activates ROCK, leading to direct phosphorylation of MRLC and to phosphorylation and inhibition of MYPT, thus increasing MRLC phosphorylation, highlighting the role of aPKCζ in the activation of NMII upon EGF stimulation.
Figure 5. ROCK increase MRLC phosphorylation upon EGF stimulation. (A) ROCK but not MLCK phosphorylates MRLC upon EGF stimulation. aPKCζ−/− and control cells were preincubated with ML-7, Y-27632, or both, stimulated with EGF for 4 min, lysed in SDS-PAGE sample buffer, divided into 2 samples that were subjected to Western blot analysis, One sample was probed with anti-MRLC antibodies and the other with anti-pMRLC antibodies. Shown are representative Western blots. The relative amounts of pMRLC to total MRLC are shown. Values represent the mean ± SEM for 5 independent experiments, ** p < 0.01, values are compared with control cells. (B) aPKCζ is required for EGF-dependent phosphorylation of MYPT. aPKCζ−/− and control cells were stimulated with EGF, and at the indicated time point cells were lysed in SDS-PAGE sample buffer and subjected to Western blot analysis with anti-pMYPT and anti-actin antibodies. Shown are representative Western blots. (B) Densitometry analysis of Western blots. The relative amounts of pMYPT to total actin are shown. Values represent the mean ± SEM for n = 3.
aPKCζ mediates EGF-dependent RhoA activation
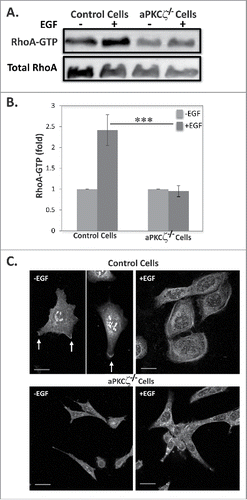
The finding that ROCK is activated upon EGF stimulation led us to investigate whether the ROCK effector, RhoA, is activated upon EGF stimulation and whether aPKCζ is required for this activation. For this purpose, we stimulated control and aPKCζ−/− cells with EGF and measured the amounts of GTP-bound RhoA, using the Rhotekin binding assay.Citation42 shows that EGF stimulation of control cells resulted in ∼2.5-fold increase in active RhoA; by contrast, aPKCζ−/− cells did not present such increase (). Because RhoA must be targeted to the plasma membrane for its activation,Citation43 we examined the effect of EGF on the cellular localization of RhoA in aPKCζ−/− cells. In untreated control cells, RhoA localized to the cell extensions (), consistent with previous report.Citation44 Upon EGF stimulation, RhoA was confined to the cell cortex, forming a shell-like structure around the cell. By contrast, RhoA was completely diffused in aPKCζ−/− cells, regardless of EGF stimulation. These results indicate that aPKCζ is required for EGF-dependent RhoA activation.
Figure 6. aPKCζ is required for EGF-dependent RhoA activation. (A) Lysates aPKCζ−/− and control cells that were stimulated with EGF for 4 min, were subjected to affinity precipitation with GST-Rhotekin beads. Proteins bound to GST-Rhotekin beads (RhoA-GTP) and cell lysate (total RhoA) were separated on SDS-PAGE and immunoblotted for RhoA. (B) Relative amounts of RhoA-GTP to RhoA are shown. Values represent the mean ± SEM for 4 independent experiments, *** p < 0.001, values are aPKCζ−/− cells compared with control cells. (C) aPKCζ−/− and control cells were placed on cover slips, stimulated with EGF for 4 min, stained with anti-RhoA antibodies, and secondary antibody conjugated to Cy2. Bars are 10 μm.
aPKCζ affects the spatial organization of pMRLC
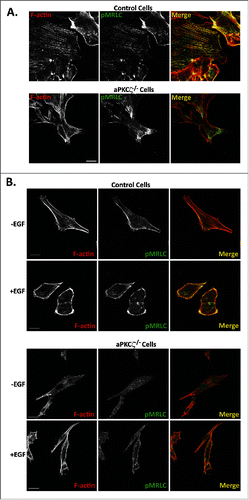
Phosphorylation of MRLC is required for proper assembly of stress fibers and focal adhesion, and for NMII dynamics during cell migration.Citation12,45,46 Next, we studied how the absence of aPKCζ affects the spatial organization of pMRLC in polarized migrating cells. aPKCζ−/− and control cell lines were subjected to a wound scratch assay and stained for pMRLC and F-actin. In control cells, pMRLC colocalized with F-actin, forming the stress fibers, and with branched F-actin in the lamellipodia and in the cell cortex, but it was missing from cell protrusions (). aPKCζ−/− cells subjected to a wound scratch assay, had substantially reduced amounts of stress fibers, as well as very little pMRLC and F-actin colocalization (). To study the effect of aPKCζ on pMRLC cellular localization upon EGF stimulation, we stimulated dispersed control and aPKCζ−/− cells with EGF and monitored the localization properties of pMRLC and F-actin. In unstimulated control cells, pMRLC and F-actin colocalized to form stress fibers (). Upon EGF stimulation, there was enrichment in pMRLC-F-actin in the cell cortex (), consistent with a previous report.Citation47 By contrast, unstimulated and EGF-stimulated aPKCζ−/− cells showed fewer stress fibers than did control cells, and the distributions of pMRLC and F-actin were not affected by EGF stimulation (). These results indicate that aPKCζ plays a role in the cellular organization of pMRLC-F-actin upon EGF stimulation. Together, the results presented above point to the EGF-aPKCζ-RhoA-ROCK-pMRLC pathway as affecting the organization of the acto-NMII cytoskeleton.
Figure 7. aPKCζ affects the spatial organization of pMRLC. aPKCζ−/− and control cells were subjected to wound scratch assay (A) or seeded on coverslips and stimulated with EGF for 4 min (B). Cells were stained for F-actin, using Rhodamine-Phalloidin, and with anti-pMRLC antibody and secondary antibody conjugated to Alexa flour 488. Bars are 20 μm.
Discussion
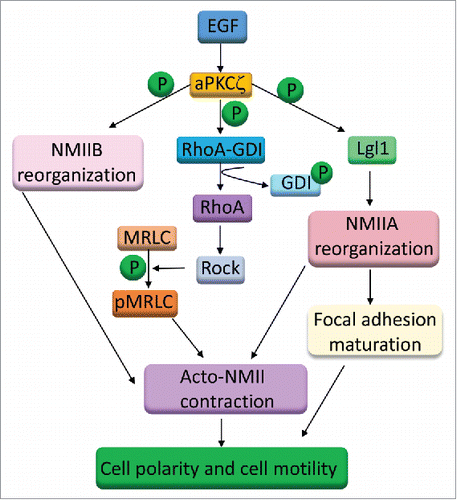
Directional sensing and polarization are crucial steps for cell migration. Despite the recognized importance of aPKCζ for these processes, there is little understanding of the molecular mechanism by which aPKCζ regulates these functions. The studies presented here reveal an important role of aPKCζ in directed cell migration, possibly through multiple pathways. We showed that aPKCζ is a key player in acto-NMII cytoskeleton regulation and reorganization during cell migration. The cellular organization of NMII is a dynamic process that involves the assembly-disassembly of NMII. It is regulated by phosphorylation of both MHC and MRLC, as well as by proteins such as Lgl1 and mts1.Citation7,27,48 We have shown previously that NMIIB resides in a complex with aPKCζ and p21-activated kinase 1 (PAK1), and that the interaction between these proteins is EGF-dependent.Citation49 We have also shown that aPKCζ phosphorylates the MHC of NMIIB directly in an EGF-dependent manner, and leads to slower filament assembly of NMIIB. Furthermore, a decrease in aPKCζ expression in the cells alters NMIIB cellular organization.Citation49 The present study indicates that aPKCζ is involved in EGF-dependent MRLC phosphorylation. Together, these results indicate that aPKCζ affects both MHC and MRLC phosphorylation, thereby affecting the cellular organization of NMII ().
Figure 8. A model for the role of aPKCζ in cell migration. aPKCζ may affect cell migration by multiple mechanisms: through the phosphorylation of GDI and activation of RhoA, through the phosphorylation of NMIIB heavy chains, and through the phosphorylation of Lgl1.
In the process of cell migration, focal adhesions are constantly being assembled and disassembled as the cell establishes new contacts at the leading edge, and breaks old ones at the trailing edge of the cell.Citation30 NMIIA mediates the maturation of adhesion sites.Citation4,5,31 We showed here that the absence of aPKCζ significantly decreased the number of focal adhesions among cells as well as actual cell adhesion. We propose that this effect may be due to the absence of Lgl1 phosphorylation in aPKCζ−/− cells (). We have recently shown that Lgl1 binds NMIIA, inhibiting its ability to form filaments, and that phosphorylation of Lgl1 by aPKCζ prevents its interaction with NMIIA both in vitro and in vivo, thus removing the inhibition of NMIIA filament assembly.Citation26,27 Furthermore, phosphorylation of Lgl1 by aPKCζ affects its cellular localization and is important for the cellular organization of the acto-NMII cytoskeleton.Citation26 We therefore propose that in the absence of aPKCζ, Lgl1 is not phosphorylated and binds with NMIIA constitutively, inhibiting its activity. Because NMIIA is required for focal adhesion maturation, its inhibition leads to the small number of focal adhesions observed in aPKCζ−/− cells. We further propose that the permanent inhibition of NMIIA filament assembly by unphosphorylated Lgl1 also prevents the formation of acto-NMII stress fibers, thus negatively affecting cell migration. The present study complements others showing that inhibition of aPKCζ activity leads to down-regulation of actin polymerization and cell adhesion.Citation20,23
Several lines of evidence point to the involvement of aPKCζ in cell migration induced by EGF. In motile cells aPKCζ is constitutively associated with the plasma membrane, whereas in non-motile cells aPKCζ is excluded from the plasma membrane, suggesting that the membrane-associated intracellular distribution of aPKCζ plays a critical role in maintaining cell motility.Citation50 Wounding induces strong activation of aPKCζ in rat astrocytes and becomes associated with the plasma membrane at the leading edge of migrating cells.Citation25 It has been shown that EGF induces the phosphorylation of aPKCζ and its translocation from the cytosol to the plasma membrane, and that it is an essential component of the EGF-stimulated chemotactic signaling pathway.Citation19,23,51 These results indicate that aPKCζ membrane association is required for directed cell migration. We propose that the membrane-associated aPKCζ recruits and activates other signaling proteins. We showed here that aPKCζ is required for EGF-dependent RhoA membrane association and activation. In addition, it has been shown that aPKCζ associates with activated RhoA and that this interaction is strictly stimulus-dependent.Citation52 Furthermore, aPKCζ forms a complex with the RhoA inhibitor, GDP dissociation inhibitor (Rho-GDI).Citation53 Phosphorylation of Rho-GDI by aPKCζ leads to Rho-GDI dissociation from RhoA and subsequent activation of RhoA.Citation53 We propose that EGF stimulation leads to aPKCζ activation and association with cell membrane, after which active aPKCζ forms a complex with RhoA and the inhibitor Rho-GDI. Phosphorylation of Rho-GDI removes its inhibition from RhoA, allowing RhoA to bind ROCK and activate it (). It is well established that MRLC phosphorylation triggers a key regulatory event that controls NMII assembly and activation, as well as cellular organization, and plays a crucial role in cell migration.Citation54-56 We showed here that EGF-dependent MRLC phosphorylation is carried out by RhoA activating ROCK, and that aPKCζ is required for execution of this pathway. Our study complement the studies indicating that ROCK mediates signals from RhoA to induce the formation of stress fibers and focal adhesions in fibroblasts stimulated by extracellular signals.Citation10,57,58
In sum, we propose that aPKCζ may regulate the EGF-dependent directed cell migration through multiple phosphorylation events (). aPKCζ phosphorylates NMIIB heavy chains, leading to its cellular organization, which is required for the contraction of the rear part of the cell. aPKCζ phosphorylates Lgl1, removing the inhibitory effect from NMIIA, thereby affecting its cellular localization and inducing stress fiber formation and focal adhesion maturation. Finally, aPKCζ phosphorylates Rho-GDI, activating RhoA and ROCK, which increase MRLC phosphorylation and activation of NMII. These events are required for stress fiber formation, NMII contraction, and focal adhesion assembly and maturation, which are essential for cell motility and directed cell migration.
Materials and methods
Cell lines and culture conditions
Mouse embryonic fibroblasts obtained from wild type (control) and aPKCζ−/− day 13.5 embryos.Citation59 These cell lines were kindly received from Dr. Jorge Moscat (Sanford-Burnham Medical Research Institute, La Jolla). Cell lines were maintained in high-glucose DMEM, supplemented with 2 mM l-glutamine, 10% FCS, and antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin, and 1:100 Biomyc3 anti-mycoplasma antibiotic solution) (Biological Industries, Beit HaEmek, Israel). Cells were grown at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Antibodies and reagents
Antibodies specific for the C-terminal region of mouse NMIIA were a kind gift from Dr. R. S. Adelstein. Antibodies specific for the C-terminal region of NMIIB were generated in rabbits, as described.Citation60 Recombinant GFP antibodies were prepared in rabbits, as described.Citation61 Anti-mouse monoclonal MRLC, pMRLC (Thr-18/Ser-19) and pMYPT (Thr-853) antibodies were purchased from Cell Signaling Technology. Anti-mouse monoclonal β-actin and vinculin antibodies, Collagen type I, EGF, ML-7, and Y-27632 were purchased from Sigma-Aldrich. Anti-mouse monoclonal paxillin antibodies were purchased from BD Transduction Laboratories, and anti-rabbit pY118-paxillin antibodies were purchased from Invitrogen. Anti-mouse monoclonal aPKCζ and anti-RhoA antibodies were purchased from Santa Cruz Biotechnology. Horseradish peroxidase-conjugated secondary antibodies, Cy2 rabbit anti-mouse, Cy2 goat anti-rabbit, and Alexa flour 488 goat anti-mouse secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (Pike West Grove, PA). Rhodamine-Phalloidin was obtained from Molecular Probes (Eugene, OR).
Wound scratch assay and confocal microscopy
6 × 105 control and 6 × 105 aPKCζ−/− cells were seeded on cover slips coated with 27 μg/ml Collagen type I. After 16 h, cells were washed twice with 1 ml of starvation medium (high glucose Dulbecco's Modified Eagle's Medium supplemented with 2 mM L-glutamine, 100 units/ml penicillin, 100 g/ml streptomycin, and 0.1% fatty acid-free bovine serum albumin (Sigma)), and were serum-starved for 3 h in 2 ml of starvation medium. Three parallel scratches were performed using a small pipette tip, and the cells were washed twice in PBS to remove cell debris. Next, high-glucose DMEM medium was added, and cells were incubated for 7 h. Cells were washed 3 times with PBS and fixed for 20 min in 1.5 ml of 3.7% formaldehyde in PBS. Cells were immunostained with anti-NMIIA or anti-NMIIB antibodies and secondary antibody conjugated to Cy2. F-Actin was stained with rhodamine-phalloidin, as described.Citation62 For focal adhesion analysis, aPKCζ−/− and control cells were immunostained with paxillin and pY118-paxillin antibodies and secondary antibody conjugated to Cy2 and Cy3, respectively. For RhoA analysis, cells were immunostained with anti-mouse RhoA antibodies and secondary antibody conjugated to Cy2. To test the effect of EGF on the cellular localization of RhoA and pMRLC, cells were seeded on collagen-coated glass coverslips, 10 ng/ml of EGF was added, and after 4 min the cells were fixed. Cells were visualized using a 60× magnification, numeric aperture 1.4 objective, under a TE2000 inverted confocal laser scanning system (Nikon, Tokyo, Japan).
Triton solubility assay
6 × 105 control and 8 × 105 of aPKCζ−/− cells were seeded on 30 mm dishes, one day before the experiment. Cells were washed twice with 1 ml PBS and lysed by the addition of 200 μl PEM buffer (100 mM PIPES pH 6.9, 1 mM MgCl2, 1 mM EGTA) + 1% Triton-X-100 + protease inhibitors mix (Sigma) to the plates, followed by incubation for 5 min at 4°C. The Triton-soluble fractions were collected from the plates and centrifuged for 5 min at 16,000 g to remove the remnants of the insoluble fraction. 100 μl of supernatant was removed to fresh tubes, 25 μl of 5x SDS-PAGE sample buffer was added, and the tubes were boiled for 5 min at 100°C. Insoluble fraction was washed once with 300 ml PEM buffer, 120 μl SDS-PAGE sample buffer was added to the plates, and the insoluble fractions were collected and boiled for 5 min at 100°C. After separation on 8% SDS-PAGE, Western blot analysis was performed using anti-NMIIA, anti-NMIIB, and anti-Actin antibodies. The Western blots were developed using the EZ-ECL Chemiluminescence Detection Kit (Biological Industries), and the intensity of the bands was analyzed using Fujifilm ImageGauge V software. In the final calculations of the percentage of the proteins in the soluble fractions, the amount of the proteins in the Triton-soluble fraction was corrected by a factor of 2, and the intensity of proteins in the Triton-soluble fraction was divided by the sum of the intensities of the proteins in the Triton-soluble and Triton-insoluble fractions.
Adhesion assay
1.5 × 105 aPKCζ−/− and control cells were seeded on 30 mm plates after removal of trypsin by centrifugation. 2.5 h post-seeding, the cells were washed gently with PBS, and high-glucose DMEM medium was added to the dishes. Phase-contrast images of 15 randomly-chosen fields were taken using an Olympus DP70 camera attached to the confocal microscope. Quantification of the percent of adhered cells relative to total cells in each field was performed.
Live-cell imaging and persistence measurement
aPKCζ−/− and control cells were seeded on a Lab-Tek (Nalge Nunc International, Rochester, NY) 2-well chamber, coated with 27 μg/ml Collagen type I. After wounding, the cells were tracked by time-lapse confocal microscopy at 30 min intervals for 20 h, using a 40× objective. Images were processed and converted to QuickTime (Apple, Cupertino, CA) movies, using ZEN2009 and ImageJ software. The paths of 10 randomly-chosen cells were plotted for each experimental group, and the speed and persistence of migration were extracted from the track plots using ImageJ software. The results were subjected to a 2-tailed, 2-sampled, unequal-variance Student's t test, using at least 30 cells for each group. Directionality represents the ratio of the vectorial distance traveled to the total path length described by the cell.
Determination of pMRLC and pMYPT
3 × 105 control and 5 × 105 aPKCζ−/− cells were seeded on 30 mm plates and starved for 3 h as described above. Cells were stimulated with 10 ng/ml EGF and lysed in SDS-PAGE sample buffer, separated on SDS-PAGE. Western blot analysis was performed using anti-MRLC, anti-pMRLC, or anti-pMYPT and anti-Actin antibodies. To detemine the effects of ROCK and MLCK on pMRLC, cells were treated with 10 μM Y-27632 or 50 μM ML-7 for 30 min prior to being stimulated with EGF. For control, cells were preincubated with DMSO (the ML-7 solvent).
RhoA activity assay
RhoA activity was measured using a previously described method.Citation42 Briefly, 5 × 105 control and 8 × 105 aPKCζ−/− cells were seeded on 60 mm plates. After 16 h, cells were washed twice with 1 ml of starvation medium and serum-starved for 3 h, as described above. Cells were stimulated with 10 ng/ml EGF for 4 min and lysed in Rho buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% NP-40, 10 mM MgCl2, 1 mM EDTA and 10% glycerol, 10 μg/ml leupeptin, 10 μg/ ml pepstatin, and 10 μg/ml aprotinin), and GTP-RhoA was affinity-precipitated using GST-Rhotekin beads. The affinity-precipitated GTP-RhoA was quantified in parallel with total cellular RhoA from cell lysates by Western blot analysis, with an antibody against RhoA.
Statistical analysis
Results are expressed as means ± SEM of the mean and were analyzed by using 2-tailed, 2-sampled unequal variance Student's t test. Student's t-test was performed to check statistical significance in experimental data. The results were considered significant when p values were less than < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_materials.zip
Download Zip (6.8 MB)Acknowledgments
We thank Jorge Moscat (Sanford-Burnham Medical Research Institute, La Jolla) for aPKCζ wild type (control) and aPKCζ−/− cell lines.
Funding
This study was supported by the Israel Science Foundation (Grant No. 1174/12), Israel Cancer Research Foundation, and Israel Cancer Association (Grant No. 20140082). S.R. holds the Dr. Daniel G. Miller Chair in Cancer Research. I.D. was supported by the Canadian Friends of Hebrew University. D.P. was supported by the Luxemburg Foundation.
Related Research Data
References
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science 2003; 302:1704-9; PMID:14657486; https://doi.org/10.1126/science.1092053
- Devreotes P, Horwitz AR. Signaling networks that regulate cell migration. Cold Spring Harb Perspect Biol 2015; 7:a005959; PMID:26238352; https://doi.org/10.1101/cshperspect.a005959
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 2010; 11:633-43; PMID:20729930; https://doi.org/10.1038/nrm2957
- Giannone G, Dubin-Thaler BJ, Rossier O, Cai Y, Chaga O, Jiang G, Beaver W, Döbereiner HG, Freund Y, Borisy G, et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 2007; 128:561-75; PMID:17289574; https://doi.org/10.1016/j.cell.2006.12.039
- Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol 2007; 176:573-80; PMID:17312025; https://doi.org/10.1083/jcb.200612043
- Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol 2008; 183:543-54; PMID:18955554; https://doi.org/10.1083/jcb.200806030
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 2009; 10:778-90; PMID:19851336; https://doi.org/10.1038/nrm2786
- Ikebe M, Hartshorne DJ. Proteolysis of smooth muscle myosin by Staphylococcus aureus protease: Preparation of heavy meromyosin and subfragment 1 with intact 20000-dalton light chains. Biochemistry 1985; 24:2380-7; PMID:3158349; https://doi.org/10.1021/bi00330a038
- Ikebe M, Hartshorne DJ, Elzinga M. Identification, phosphorylation, and dephosphorylation of a second site for myosin light chain kinase on the 20,000-dalton light chain of smooth muscle myosin. J Biol Chem 1986; 261:36-9; PMID:3079756
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 1996; 271:20246-9; PMID:8702756; https://doi.org/10.1074/jbc.271.34.20246
- Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol 1999; 147:1023-38; PMID:10579722; https://doi.org/10.1083/jcb.147.5.1023
- Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol 2000; 150:797-806; PMID:10953004; https://doi.org/10.1083/jcb.150.4.797
- Totsukawa G, Wu Y, Sasaki Y, Hartshorne DJ, Yamakita Y, Yamashiro S, Matsumura F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J Cell Biol 2004; 164:427-39; PMID:14757754; https://doi.org/10.1083/jcb.200306172
- Katoh K, Kano Y, Amano M, Kaibuchi K, Fujiwara K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am J Physiol Cell Physiol 2001; 280:C1669-79; PMID:11350763
- Katoh K, Kano Y, Amano M, Onishi H, Kaibuchi K, Fujiwara K. Rho-kinase–mediated contraction of isolated stress fibers. J Cell Biol 2001; 153:569-84; PMID:11331307; https://doi.org/10.1083/jcb.153.3.569
- Katoh K, Kano Y, Noda Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J R Soc Interface 2011; 8:305-11; PMID:20826475; https://doi.org/10.1098/rsif.2010.0419
- Katoh K, Kano Y, Ookawara S. Rho-kinase dependent organization of stress fibers and focal adhesions in cultured fibroblasts. Genes Cells 2007; 12:623-38; PMID:17535253; https://doi.org/10.1111/j.1365-2443.2007.01073.x
- Xiao H, Liu M. Atypical protein kinase C in cell motility. Cell Mol Life Sci 2013; 70:3057-66; PMID:23096778; https://doi.org/10.1007/s00018-012-1192-1
- Sun R, Gao P, Chen L, Ma D, Wang J, Oppenheim JJ, Zhang N. Protein kinase C zeta is required for epidermal growth factor-induced chemotaxis of human breast cancer cells. Cancer Res 2005; 65:1433-41; PMID:15735031; https://doi.org/10.1158/0008-5472.CAN-04-1163
- Liu Y, Wang B, Wang J, Wan W, Sun R, Zhao Y, Zhang N. Down-regulation of PKCzeta expression inhibits chemotaxis signal transduction in human lung cancer cells. Lung Cancer 2009; 63:210-8; PMID:18701187; https://doi.org/10.1016/j.lungcan.2008.05.010
- Lee H, Park M, Shin N, Kim G, Kim YG, Shin JS, Kim H. High mobility group box-1 is phosphorylated by protein kinase C zeta and secreted in colon cancer cells. Biochem Biophys Res Commun 2012; 424:321-6; PMID:22750245; https://doi.org/10.1016/j.bbrc.2012.06.116
- Keenan C, Kelleher D. Protein kinase C and the cytoskeleton. Cell Signal 1998; 10:225-32; PMID:9617479; https://doi.org/10.1016/S0898-6568(97)00121-6
- Guo H, Gu F, Li W, Zhang B, Niu R, Fu L, Zhang N, Ma Y. Reduction of protein kinase C zeta inhibits migration and invasion of human glioblastoma cells. J Neurochem 2009; 109:203-13; PMID:19187446; https://doi.org/10.1111/j.1471-4159.2009.05946.x
- Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene 2008; 27:6970-80; PMID:19029938; https://doi.org/10.1038/onc.2008.347
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 2001; 106:489-98; PMID:11525734; https://doi.org/10.1016/S0092-8674(01)00471-8
- Dahan I, Petrov D, Cohen-Kfir E, Ravid S. The tumor suppressor Lgl1 forms discrete complexes with NMII-A and Par6alpha-aPKCzeta that are affected by Lgl1 phosphorylation. J Cell Sci 2014; 127:295-304; PMID:24213535; https://doi.org/10.1242/jcs.127357
- Dahan I, Yearim A, Touboul Y, Ravid S. The tumor suppressor Lgl1 regulates NMII-A cellular distribution and focal adhesion morphology to optimize cell migration. Mol Biol Cell 2012; 23:591-601; PMID:22219375; https://doi.org/10.1091/mbc.E11-01-0015
- Kolega J. Cytoplasmic dynamics of myosin IIA and IIB: spatial “sorting” of isoforms in locomoting cells. J Cell Sci 1998; 111:2085-95; PMID:9664030
- Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 2006; 125:1361-74; PMID:16814721; https://doi.org/10.1016/j.cell.2006.05.029
- Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Curr Opin Cell Biol 1995; 7:697-706; PMID:8573345; https://doi.org/10.1016/0955-0674(95)80112-X
- Owen KA, Pixley FJ, Thomas KS, Vicente-Manzanares M, Ray BJ, Horwitz AF, Parsons JT, Beggs HE, Stanley ER, Bouton AH. Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. J Cell Biol 2007; 179:1275-87; PMID:18070912; https://doi.org/10.1083/jcb.200708093
- Vicente-Manzanares M, Newell-Litwa K, Bachir AI, Whitmore LA, Horwitz AR. Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J Cell Biol 2011; 193:381-96; PMID:21482721; https://doi.org/10.1083/jcb.201012159
- Petit V, Boyer B, Lentz D, Turner CE, Thiery JP, Valles AM. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J Cell Biol 2000; 148:957-70; PMID:10704446; https://doi.org/10.1083/jcb.148.5.957
- Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci 2007; 120:137-48; PMID:17164291; https://doi.org/10.1242/jcs.03314
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol 2006; 173:587-9; PMID:16717130; https://doi.org/10.1083/jcb.200509075
- Pasapera AM, Schneider IC, Rericha E, Schlaepfer DD, Waterman CM. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J Cell Biol 2010; 188:877-90; PMID:20308429; https://doi.org/10.1083/jcb.200906012
- Lauffenburger DA, Horwitz AF. Cell migration: A physically integrated molecular process. Cell 1996; 84:359-69; PMID:8608589; https://doi.org/10.1016/S0092-8674(00)81280-5
- Vicente-Manzanares M, Horwitz AR. Cell migration: an overview. Methods Mol Biol 2011; 769:1-24; PMID:21748665; https://doi.org/10.1007/978-1-61779-207-6_1
- Corbit KC, Soh JW, Yoshida K, Eves EM, Weinstein IB, Rosner MR. Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol Cell Biol 2000; 20:5392-403; PMID:10891480; https://doi.org/10.1128/MCB.20.15.5392-5403.2000
- Cohen EE, Lingen MW, Zhu B, Zhu H, Straza MW, Pierce C, Martin LE, Rosner MR. Protein kinase C zeta mediates epidermal growth factor-induced growth of head and neck tumor cells by regulating mitogen-activated protein kinase. Cancer Res 2006; 66:6296-303; PMID:16778206; https://doi.org/10.1158/0008-5472.CAN-05-3139
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996; 273:245-8; PMID:8662509; https://doi.org/10.1126/science.273.5272.245
- Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol 2000; 325:264-72; PMID:11036609; https://doi.org/10.1016/S0076-6879(00)25448-7
- O'Connor K, Chen M. Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases 2013; 4:141-7; PMID:24025634; https://doi.org/10.4161/sgtp.25131
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 2006; 440:1069-72; PMID:16547516; https://doi.org/10.1038/nature04665
- Fumoto K, Uchimura T, Iwasaki T, Ueda K, Hosoya H. Phosphorylation of myosin II regulatory light chain is necessary for migration of HeLa cells but not for localization of myosin II at the leading edge. Biochem J 2003; 370:551-6; PMID:12429016; https://doi.org/10.1042/bj20021559
- Watanabe T, Hosoya H, Yonemura S. Regulation of myosin II dynamics by phosphorylation and dephosphorylation of its light chain in epithelial cells. Mol Biol Cell 2007; 18:605-16; PMID:17151359; https://doi.org/10.1091/mbc.E06-07-0590
- Ben-Ya'acov A, Ravid S. Epidermal growth factor-mediated transient phosphorylation and membrane localization of myosin II-B are required for efficient chemotaxis. J Biol Chem 2003; 278:40032-40; PMID:12874274; https://doi.org/10.1074/jbc.M306948200
- Dulyaninova NG, Malashkevich VN, Almo SC, Bresnick AR. Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry 2005; 44:6867-76; PMID:15865432; https://doi.org/10.1021/bi0500776
- Even-Faitelson L, Ravid S. PAK1 and aPKCzeta regulate myosin II-B phosphorylation: a novel signaling pathway regulating filament assembly. Mol Biol Cell 2006; 17:2869-81; PMID:16611744; https://doi.org/10.1091/mbc.E05-11-1001
- Laudanna C, Sorio C, Tecchio C, Butcher EC, Bonora A, Bassi C, Scarpa A. Motility analysis of pancreatic adenocarcinoma cells reveals a role for the atypical zeta isoform of protein kinase C in cancer cell movement. Lab Invest 2003; 83:1155-63; PMID:12920244; https://doi.org/10.1097/01.LAB.0000081390.92179.F3
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science 1995; 268:247-51; PMID:7716516; https://doi.org/10.1126/science.7716516
- Teusch N, Lombardo E, Eddleston J, Knaus UG. The low molecular weight GTPase RhoA and atypical protein kinase Czeta are required for TLR2-mediated gene transcription. J Immunol 2004; 173:507-14; PMID:15210811; https://doi.org/10.4049/jimmunol.173.1.507
- Kuribayashi K, Nakamura K, Tanaka M, Sato T, Kato J, Sasaki K, Takimoto R, Kogawa K, Terui T, Takayama T, et al. Essential role of protein kinase C zeta in transducing a motility signal induced by superoxide and a chemotactic peptide, fMLP. J Cell Biol 2007; 176:1049-60; PMID:17389234; https://doi.org/10.1083/jcb.200607019
- Post PL, DeBiasio RL, Taylor DL. A fluorescent protein biosensor of myosin II regulatory light chain phosphorylation reports a gradient of phosphorylated myosin II in migrating cells. Mol Biol Cell 1995; 6:1755-68; PMID:8590803; https://doi.org/10.1091/mbc.6.12.1755
- Saitoh T, Takemura S, Ueda K, Hosoya H, Nagayama M, Haga H, Kawabata K, Yamagishi A, Takahashi M. Differential localization of non-muscle myosin II isoforms and phosphorylated regulatory light chains in human MRC-5 fibroblasts. FEBS Lett 2001; 509:365-9; PMID:11749957; https://doi.org/10.1016/S0014-5793(01)03186-6
- Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol 2001; 11:471-7; PMID:11719051; https://doi.org/10.1016/S0962-8924(01)02153-5
- Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 1997; 275:1308-11; PMID:9036856; https://doi.org/10.1126/science.275.5304.1308
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992; 70:389-99; PMID:1643657; https://doi.org/10.1016/0092-8674(92)90163-7
- Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell 2001; 8:771-80; PMID:11684013; https://doi.org/10.1016/S1097-2765(01)00361-6
- Phillips CL, Yamakawa K, Adelstein RS. Cloning of the cDNA encoding human nonmuscle myosin heavy chain-B and analysis of human tissues with isoform-specific antibodies. J Muscle Res Cell Motil 1995; 16:379-89; PMID:7499478; https://doi.org/10.1007/BF00114503
- Rosenberg M, Ravid S. Protein kinase Cg regulates myosin IIB phosphorylation, cellular localization, and filament assembly. Mol Biol Cell 2006; 17:1364-74; PMID:16394101; https://doi.org/10.1091/mbc.E05-07-0597
- Ronen D, Ravid S. Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. J Biol Chem 2009; 284:24948-57; PMID:19553683; https://doi.org/10.1074/jbc.M109.023754