
ABSTRACT
Myeloid cell leukemia-1 (MCL-1), closely related to B-cell lymphoma 2 (BCL-2), has a well-established role in cell survival and has emerged as an important target for cancer therapeutics. We have demonstrated that inhibiting MCL-1 is efficacious in suppressing tumour progression in pre-clinical models of breast cancer and revealed that in addition to its role in cell survival, MCL-1 modulated cellular invasion. Utilizing a MCL-1-specific genetic antagonist, we found two possible mechanisms; firstly MCL-1 directly binds to and alters the phosphorylation of the cytoskeletal remodeling protein, Cofilin, a protein important for cytoskeletal remodeling during invasion, and secondly MCL-1 modulates the levels SRC family kinases (SFKs) and their targets. These data provide evidence that MCL-1 activities are not limited to endpoints of extracellular and intracellular signaling culminating in cell survival as previously thought, but can directly modulate the output of SRC family kinases signaling during cellular invasion. Here we review the pleotropic roles of MCL-1 and discuss the implications of this newly discovered effect on protein kinase signaling for the development of cancer therapeutics.
Myeloid cell leukemia-1 (MCL-1), a unique pro-survival member or the BCL-2 family of proteins
Recently, work performed in our laboratory demonstrated that Myeloid cell leukemia-1 (MCL-1) antagonism suppressed tumour progression in pre-clinical models of breast cancer and revealed that in addition to its role in cell survival, MCL-1 modulated cellular invasion .Citation1 MCL-1 was first described as an immediate-early response gene in human myeloid leukaemia cells induced to differentiate with phorbol ester.Citation2 MCL-1 is best known as a pro-survival member of the BCL-2 family of proteins that regulates the intrinsic (mitochondrial) apoptotic cascade.Citation3 MCL-1 is important for the survival of most normal and malignant tissues (reviewed in.Citation4) The C-terminal region of MCL-1 shares homology with the BCL-2 family of proteins, which contain four BCL-2-homology (BH1–4)-domains that form a binding pocket for interaction with the pro-apoptotic BH3 only proteins and by doing so protect normal and malignant cells from cell death. The BCL-2 family members include two pro-apoptotic subgroups: the BH3-only sensor proteins (e.g. BIM, PUMA, NOXA, tBID etc), which trigger the intrinsic apoptotic cascade in response to cytotoxic insults or cellular stresses and BAX and BAK, the apoptotic effectors (reviewed elsewhereCitation3). The BH domain hydrophobic pocket dictates MCL-1 binding specificity for BIM, tBID, PUMA, NOXA and BAK thereby restraining cellular apoptotic activity.
MCL-1 is important for cancer cell survival and therapeutic resistance
MCL1 is one of the most common somatic copy number amplifications, observed in 11% of cancers across multiple tissue types, with the highest rates observed in breast (36% of cases) and lung (54% of cases).Citation5 MCL-1 protein levels correlate with outcome, tumor grade and therapeutic resistance in many cancers including those of the hematopoietic system, breast, lung and pancreas.Citation6–Citation12 MCL-1 has been validated to participate in neoplastic progression of B-cell lymphomasCitation13 as well as haematopoietic progenitor/stem cell tumoursCitation14 and accelerate Myc induced myeloid leukaemia.Citation15 Several studies have also shown that MCL-1 is a barrier to therapeutic sensitivity, including those that target BCL-2 (reviewed inCitation16). MCL-1 is responsible for therapeutic resistance in a range of cancers including oral cancers,Citation17 lung cancer,Citation18 pancreatic cancer,Citation19 ovarian cancer, colon cancerCitation20 and triple negative breast cancer.Citation21 MCL-1 also confers the survival of breast cancer cells in vitroCitation22 and protects HER-2 positive breast cancers from hypoxia-induced death.Citation23 It is for the above reasons that there have been significant advances in the development of small molecule inhibitors that bind to and inhibit MCL-1, including the most recent and promising compounds A1210477 and S63845,Citation24,Citation25 with the later inhibitor having high affinity, efficacy at low doses, and low toxicity. Recent work has shown that the MCL-1 inhibitor S63845 could increase the sensitivity of patient derived xenografts to docetaxel and traztuzumab.Citation26 More work is to determine the genetic or proteomic biomarkers that would stratify patients to this type of therapy as a single agent or in combination with other therapies. Thus MCL-1 is a potent survival factor in hematopoietic and solid tumors and can be targeted with small molecule inhibitors to treat a wide range of cancers.
Protein kinase signalling control of MCL-1 activity
Unlike other members of the BCL-2 family, MCL-1 contains a unique 150 amino acid N-terminus consisting of PEST-like sequences (rich in proline (P), glutamic acid (E), serine (S), and threonine (T)) implicated in the control of protein stability and activity. Degradation of MCL-1 is thought to be primarily controlled primarily by ubiquitylation via 13 different lysine residues, and modulated by phosphorylation, targeting it to the proteasome.Citation27 E3 ubiquitin-protein ligases known to regulate MCL-1 stability include HUWE1/MULE,Citation28,Citation29 SCF F-box containing proteins F-box/WD repeat-containing protein 7 (Fbw7,Citation20,Citation30) and F-box/WD repeat-containing protein 1A (βTrCP) and the deubuitinase Ubiquitin Specific Peptidase 9 (USPX,Citation31). As a result MCL-1 has a short half-life of approximately 3 hours.Citation32 MCL-1 expression and activity is also controlled by a variety of stress, growth factor, hormone, cytokine and signals culminating in receptor tyrosine kinase signalling involved in the stimulation of differentiation of myeloid lineage cells or in response to stress to enhance cell survival.Citation7,Citation33 A summary of the various signalling pathways and kinases responsible for the regulation of MCL-1 is provided in and illustrated in . Best known in the regulation of MCL-1, is output of the PI3K/AKT, MAPK/ERK and JAK/STAT pathways that can be activated by a variety of receptor tyrosine kinases that including EGFR. The interleukin (IL) family of cytokines, which have roles in differentiation, growth and survival,Citation34 also induce MCL-1 transcription. These include IL3,Citation35,Citation36 IL5,Citation37 IL6,Citation38,Citation39 IL7Citation40 and IL15.Citation41 Signalling downstream of the interleukin family predominantly occurs via the JAK/STAT pathway,Citation37,Citation39,Citation41 with a STAT binding site present in the MCL-1 promoter.Citation38 Consequently STAT5 has been shown to induce MCL-1 transcription.Citation42 MCL-1 transcription is also activated after endoplasmic reticulum stressCitation43 and hypoxiaCitation44 resulting in the death of diseased cells. Intracellular kinases (eg GSK3β, CDK1 and CDK2) downstream of these signalling pathways also control MCL-1 activity resulting via phosphorylation on nine potential phosphosites influencing the stability, activity, degradation and even localisation (summarised in ). Thus MCL-1 is a critical cell survival factor in normal and malignant tissues that is induced and activated in response to a variety of extracellular and intracellular cues via protein kinase signalling.
Table 1. Extracellular and intracellular regulation of MCL-1 function. The effects of extracellular and intracellular signalling (signalling pathways) on MCL-1 transcription in various cellular contexts.
Figure 1. Schematic diagram of MCL-1 regulation and function. MCL-1 is induced by a variety of extracellular receptor tyrosine kinases (RTKs) (purple), growth factors, stress induced cytokines and hormones resulting in the activation of intracellular signals (yellow) and inducing the transcription of MCL-1 mRNA by target transcription factors (TF). MCL-1 activity and stability is regulated by E3 ubiquitin-ligase and protein kinase induced phosphorylation (P) during post-translational processing. The best recognized function of MCL-1 is its role in maintaining cell survival via interaction with the intrinsic apoptotic machinery at the mitochondria. MCL-1 can also participate in the regulation of mitochondrial structure and function, cell cycle (CellC) and DNA damage mechanisms. In diseased and damaged cells, MCL-1 can be ubiquitinated (U) and targeted for degradation at the proteasome resulting in cell death.
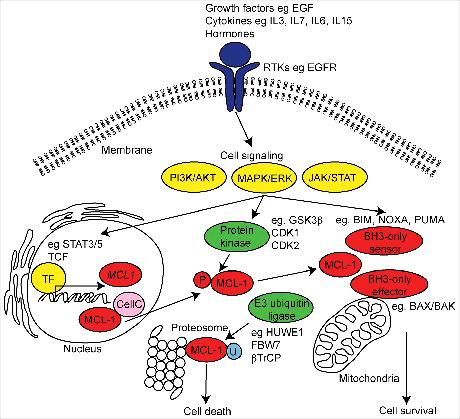
Table 2. Protein kinase regulation of MCL-1. The effects of protein kinase phosphorylation on target residues of MCL-1 by protein kinases in various cellular contexts.
MCL-1 is not just a mediator of cell survival
In addition to its role in cell survival, MCL-1 has been shown to possess multiple functions in different cellular compartments. Deletion of MCL-1 in mice resulted in peri-implantation lethality an effect that was independent of apoptosis.Citation45 Additionally, MCL-1 and STAT3 have been shown to interact during embryonic implantation, which resulted in the expression of epithelial to mesenchymal markers, increased apoptosis and decreased invasion.Citation46 MCL-1 is highly expressed in both human and mouse embryonic stem cells (ESCs), with the loss of MCL-1 through siRNA or up-regulation of NOXA by CDK1 inhibitor treatment leading to significant induction of cell death, pointing to MCL-1 playing an active role in ESC homeostasis.Citation47
The divergent roles of MCL-1 are dependent on its post-translational modification and protein-protein interactions. Proteolytically cleaved amino-truncated MCL-1 can localise to the inner mitochondrial membrane and is important for mitochondrial structure and physiology.Citation48,Citation49 Furthermore, cell cycle progression by MCL-1 is also mediated through the direct recruitment and inhibition of CDK1, due a reduced capacity of CDK1 to bind to Cyclin 1B.Citation50 In this manner MCL-1 binding may subvert the interaction of other target proteins thereby restricting kinase activity eg by binding to CDK1 and preventing cell cycle progression. MCL-1 has also been shown to directly interact with other proteins such as CDK1, PCNA and CHK1 in the nucleus, where it similarly regulates cell cycle progression and DNA damage.Citation51 In the nervous system, MCL-1 is important for neural precursor cell survivalCitation52 but also for cell cycle progression in embryonic neural precursor cells. Furthermore, MCL-1 expression correlates with the levels of VEGF (vascular endothelial growth factor),Citation53 and although this expression pattern is important for the survival of endothelial cells, it is also important for vessel sprouting and invasion.Citation54,Citation55
MCL-1 has also been implicated in the migration and invasion of normal and malignant tissues, for example MCL-1 has shown to be important for neuronal progenitor cell migration from the ventricular zone into the cortical plate during cortical neurogenesis.Citation56 MCL-1 has further been demonstrated to play a role in the migration and invasion of colorectalCitation57 and gastricCitation58 cancer cell lines, whereby siRNA knockdown led to a loss of motility in wound healing and trans-well assays. Furthermore forced expression of MIR26a, which targets MCL-1 in breast cancer cell lines led to the loss of migration in wound healing assays.Citation59 MIR26a does have other targets such as MTDH and EZH2 therefore it is still unclear whether this effect is solely dependent on the activities of MCL-1. These data suggest that MCL-1 possesses functions beyond merely its role in cell survival, including roles in mitochondrial physiology, cell cycle progression, DNA damage and possibly invasion but it has been difficult to discriminate these cellular functions from apoptosis as they are intrinsically linked to cellular viability.
MCL-1 is a new regulator of protein kinase signalling during invasion
We investigated the consequences of inhibiting MCL-1 in triple negative breast cancer cells,Citation1 a subtype of breast cancer with limited treatment options and some of the poorest outcomes.Citation60 We inhibited MCL-1 using a genetic approach via inducible expression of a modified form (L62A/F69A double mutant) of the short isoform of BIM (BIMs2A).Citation61 This approach mimics that of small molecules (BH3-mimetics) targeting MCL-1 and was chosen as it was highly specific for MCL-1 and previously validated permitting an investigation of MCL-1 in a tumor cell autonomous manner.Citation62 Expression of BIMs2A increased cell death in basal-like MDA-MB-468 cells but did not induce apoptosis in highly invasive claudin-low MDA-MB-231 cells when cultured on plastic. When seeded on contracted collagen I matrices that more accurately recapitulated key aspects of the in vivo microenvironment,Citation63 MCL-1 antagonism suppressed invasion, an effect that was independent on its effect on apoptosis.Citation1 Furthermore inhibition of MCL-1 significantly suppressed both the size and number of lung metastases in the lungs of mice bearing both MDA-MB-468 and MDA-MB-231 mammary intraductal xenografts, indicating that MCL-1 was essential for metastatic progression in both models. This specific model of antagonism provided definitive proof that MCL-1 controls invasive capacity of cancer cells, as what had been suggested previously using wound healing assays.Citation59
To invade, cancer cells form specialised membrane protrusions termed invadopodiaCitation64 rich in filamentous (F)-actin filaments initiated by a kinase signalling cascade (often involving the SRC family kinases cSRC, FYN and YES and others).Citation65 This signalling cascade results in the phosphorylation and activation of cytoskeletal remodelling proteins that include Dynamin, Cortactin, Cofilin, Talin, N-Wasp and ARP2/3 complex, augmented by the activity of GTPases CDC42 and RACCitation66 and the co-ordinated assembly of adhesion proteins (eg FAK) and those that promote F-Actin stabilisation (eg Paxillin, Vimentin).Citation67
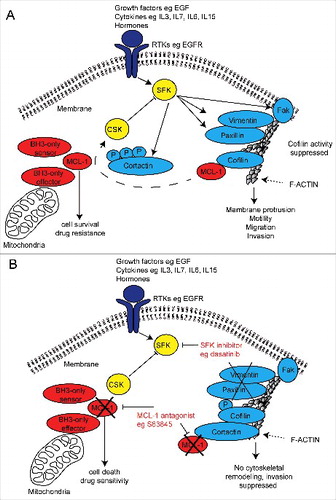
Additional work in our laboratory provided a potential mechanism for the effects of MCL-1 during cancer cell invasion and suggested that MCL-1 may modulate cytoskeletal remodelling during invasion. Kinomic profiling data revealed that MCL-1 inhibition altered a large number of proteins important for invasion and regulated by SRC family kinases.Citation1 These included increased CSK levels (cSRC tyrosine kinase), a negative regulator of SRC family kinases,Citation68 decreased total levels of the SRC family kinase, FYN, and the cSRC target, ABL. The cSRC target and adhesion protein FAK was also decreased as was the phosphorylation of Paxillin and Vimentin. Serine 3 phosphorylation of Cofilin 1 and 2 was increased, an effect that suppresses actin remodelling during invasion. All of these targets are regulated by SRC family kinase activity.Citation69,Citation70 Western blotting confirmed the observed decrease in total FAK and an increase in E-Cadherin (data not shown), suggestive of a more epithelial and less invasive state.Citation71 MCL-1 antagonism also decreased the auto-phosphorylation site Y1148 in EGFR indicating suppression of invasion activity, perhaps due to loss of cSRC activation.Citation72 Mass spectrometry and proximity ligation assays showed for the first time that MCL-1 was in direct contact with Cofilin, which perhaps is similar to what has been observed for MCL-1 and CDK1,Citation51 may be important for restricting activity,Citation1 via preventing the binding of its inhibitory partner Cortactin.Citation73,Citation74 Our data possibly places MCL-1 in complexes in direct contact with F-Actin signalling apparatus important for dynamic cytoskeletal changes during invasion (). Although additional experimentation is required to confirm this hypothesis, these results suggest that MCL-1 may play a critical role in invasion via the modulation of SRC family kinase signalling.
Figure 2. Schematic diagram of a putative mechanism for MCL-1 regulation of SRC family kinase signalling in invasive cancer cells and dual therapeutic strategy. (A) Receptor tyrosine kinase (RTK) activation induces SRC family kinase (SFK) signalling and its targets important for cytoskeletal invasion by cancer cells. MCL-1 binds and prevents serine 3 phosphorylation of Cofilin, which may prevent Cortactin inhibition of Cofilin, permitting cytoskeletal (F-actin) remodelling and cellular invasion. MCL-1 modulates the output of the SRC family kinases (eg Vimentin, Paxillin, FAK and CSK) via an unknown mechanism promotes cellular invasion. (B) MCL-1 antagonism using pharmaceutical inhibition (eg S63845) may allow Cortactin inhibition of Cofilin activity thereby preventing its cytoskeletal remodelling function and also alters the output of the SRC family kinases. When combined with SRC family kinase inhibitors (eg dasatinib, saracatinib and bosutinib), MCL-1 inhibition suppresses invasion while simultaneously induce cell death and increasing drug sensitivity.
MCL-1 regulation of SRC family kinase signalling; implications for cancer therapeutics
Our findings have significant implications for the treatment of cancers that rely on MCL-1 and SRC family kinase signalling for survival and metastatic progression. SRC family kinases are important in the development, maintenance, progression and the metastatic spread of several malignancies leading to extensive research into the development of agents that target this family. Examples of these agents include dasatinib (targeting ABL, cSRC and c-KIT), saracatinib (targeting cYES, FYN, LYN, BLK, FGR and LCK) and bosutinib (targeting SRC and ABR).Citation65 Dasatinib has shown profound improvements in tumor and metastatic outcomes in pancreatic xenograft pre-clinical models in miceCitation75,Citation76 but despite this Phase II clinical trials of SFK inhibitors alone and/or in combination with gemcitabine have failed to show any improvement in progression free or overall survival in patients with advanced pancreatic adenocarcinoma.Citation77–Citation79 In breast cancer, FYN activation plays an important role in breast cancer cell motility and drug resistance in vitro,Citation80 but so far trials of saracatinib targeting FYN have not succeeded.Citation81
Conversely, Phase II clinical trials of single agent dasatinib have shown durable and objective clinical responses in a small proportion (5%) of patients with locally advanced and metastatic triple negative breast cancer.Citation82 Combination trials have shown improved outcomes in patients with breast cancer, for example Phase I clinical trials of dasatinib with Capecitabine show clinical response rates of 56% in unselected patients.Citation83 Other trials combining dasatinib with paclitaxelCitation84 and bosutinib with exemestaneCitation85 are currently underway in patients with advanced metastatic breast cancer and are showing improved responses compared to single agents alone. These preliminary results suggest that the efficacy of SFK inhibitors will likely be improved by combining these drugs with others that increase potency or have parallel cytotoxic activity.
As MCL-1 modulated the output of SFK signalling, we then examined whether MCL-1 antagonism could be one way to increase the efficacy of these SFK inhibitors. Encouragingly, MCL-1 inhibition greatly enhanced the anti-invasive potential of dasatinib in 3D organotypic assays in vitro and suppressed tumour progression in pre-clinical models of breast cancer.Citation1 The next logical step is to investigate whether this effect extends to other inhibitors of SRC family and their targets (eg saracatinib, bosutinib and others), as recently achieved for other highly metastatic cancers,Citation86 and examine efficacy of this new dual targeting therapeutic strategy in clinical trials for metastatic disease (). Importantly the advantage of using an MCL-1 antagonist to improve potency of anti-metastatic agents is the simultaneous suppression of cell survival and increased therapeutic sensitivity that may result in a substantial improvement in patient survival.
Concluding remarks
MCL-1 is an important regulator of normal and cancer cell viability but there is increasing evidence that MCL-1 has additional roles in mitochondrial structure and function, cell cycle regulation, DNA damage response and cellular invasion. Receptor tyrosine kinase signalling upstream of MCL-1 is important for its effects but our new evidence suggests that MCL-1 can also feed back to directly modulate protein kinase invasion signalling during metastasis (). Although more work is needed to understand to fully understand the mechanisms underlying these effects, the regulation of the cytoskeletal machinery by MCL-1 regulation via modulation of protein kinase signalling provides a valuable opportunity to increase the potency of drugs that antagonise these networks. For those SRC family kinase inhibitors that have largely disappointed in clinical trials, it may now be prudent to consider pharmaceutical inhibitors of MCL-1 (eg S63845Citation24,Citation25) in combination with these to improve clinical response.
Abbreviations
| ABL | = | Abelson murine leukaemia viral oncogene homolog 1 |
| AKT/PKB | = | Protein kinase B |
| BAX/BCL2L4 | = | Bcl-2 associated X protein/Bcl-2-like protein 4 |
| BAK/BCL2L7 | = | Bcl-2 homologous antagonist killer/Bcl-2-like protein 7 |
| BCL-2 | = | B-cell lymphoma 2 |
| BIM/BCL2L11 | = | Bcl-2-like protein 11 |
| BH | = | BCL-2 homology |
| BLK | = | Tyrosine-protein kinase Blk |
| βTrCP | = | F-box/WD repeat-containing protein 1A |
| CDC42 | = | Cell division control protein 42 homolog |
| CDK1 | = | Cyclin dependent kinase 1 |
| CDK2 | = | Cyclin dependent kinase 2 |
| CHK1/CHEK1 | = | Checkpoint kinase 1 |
| CSK | = | cSRC tyrosine kinase |
| cKIT/CD117 | = | Tyrosine-protein kinase Kit |
| cSRC | = | Tyrosine kinase Src |
| cYES | = | Tyrosine-protein kinase Yes |
| EGFR/HER1 | = | Epidermal growth factor receptor |
| ERK | = | Extracellular signal-related kinases |
| ESC | = | Embryonic stem cell |
| EZH2 | = | Enhancer of zeste homolog 2 |
| FAK/PTK2 | = | Focal adhesion kinase/protein tyrosine kinase 2 |
| FBW7 | = | SCF F-box containing proteins F-box/WD repeat-containing protein 7 |
| FGR/SRC2 | = | Gardner-Rasheed feline sarcoma viral |
| FYN | = | Tyrosine-protein kinase Fyn |
| GSK3β | = | Glycogen synthase kinase-3 beta |
| HUWE1/MULE | = | E3 ubiquitin ligase HUWE1/MCL-1 ubiquitin ligase E3 |
| JAK | = | Janus kinase |
| JNK | = | c-Jun N-terminal kinases |
| LCK | = | Tyrosine-protein kinase Lck |
| LYN | = | Tyrosine protein kinaseLyn |
| MAPK | = | Mitogen-activated protein kinases |
| MCL-1 | = | Myeloid cell leukemia-1 |
| MIR26a | = | microRNA 26a |
| MTDH/AEG1 | = | Metadherin/Astrocytevelevated gene-1 |
| MYC | = | Avian myelocytomatosis viral oncogene homolog |
| NOXA/PMAIP1 | = | Phorbol-12-myristate-13-acetate-induced protein 1 |
| N-Wasp | = | Neural Wiskott-Aldrich syndrome protein |
| PCNA | = | Proliferating cell nuclear antigen |
| PI3K | = | Phosphoinositide 3-kinase |
| PUMA/BBC3 | = | p53 upregulated modulator of apoptosis/BCL-2 binding component 3 |
| RAC | = | Ras-related C3 botulinum toxin substrate 1 |
| SFK | = | SRC family kinase |
| STAT | = | Signal transducer and activator of transcription |
| STAT3 | = | Signal transducer and activator of transcription 3 |
| STAT5 | = | Signal transducer and activator of transcription 5 |
| tBID | = | truncated BH3- interacting-domain death agonist |
| USPX | = | Ubiquitin Specific Peptidase 9 |
| YES | = | Tyrosine-protein kinase Yes |
Disclosure statement
The authors declare no competing or financial interests.
Additional information
Funding
References
- Young AI, Law AM, Castillo L, et al. MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast cancer research: BCR. 2016;18(1):125. doi:10.1186/s13058-016-0781-6.
- Kozopas KM, Yang T, Buchan HL, et al. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proceedings of the National Academy of Sciences of the United States of America 1993;90(8):3516–3520. doi:10.1073/pnas.90.8.3516.
- Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. Embo J. 2011;30(18):3667–3683. doi:10.1038/emboj.2011.307.
- Perciavalle RM, Opferman JT. Delving deeper: MCL-1's contributions to normal and cancer biology. Trends Cell Biol. 2013;23(1):22–29. doi:10.1016/j.tcb.2012.08.011.
- Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi:10.1038/nature08822.
- Abulwerdi F, Liao C, Liu M, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13(3):565–575. doi:10.1158/1535-7163.MCT-12-0767.
- Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cellular and molecular life sciences: CMLS. 2009;66(8):1326–1336. doi:10.1007/s00018-008-8637-6.
- Chowdry RP, Sica GL, Kim S, et al. Phosphorylated Bcl-2 and Mcl-1 as prognostic markers in small cell lung cancer. Oncotarget. 2016; Feb 18. doi:10.18632/oncotarget.7485.
- Goodwin CM, Rossanese OW, Olejniczak ET, et al. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015;22(12):2098–2106. doi:10.1038/cdd.2015.73.
- Leverson JD, Zhang H, Chen J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015;6:e1590. doi:10.1038/cddis.2014.561.
- Modugno M, Banfi P, Gasparri F, et al. Mcl-1 antagonism is a potential therapeutic strategy in a subset of solid cancers. Experimental cell research. 2015;332(2):267–277. doi:10.1016/j.yexcr.2014.11.022.
- Quinn BA, Dash R, Azab B, et al. Targeting Mcl-1 for the therapy of cancer. Expert opinion on investigational drugs. 2011;20(10):1397–1411. doi:10.1517/13543784.2011.609167.
- Zhou P, Levy NB, Xie H, et al. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood. 2001;97(12):3902–3909. doi:10.1182/blood.V97.12.3902.
- Campbell KJ, Bath ML, Turner ML, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116(17):3197–3207. doi:10.1182/blood-2010-04-281071.
- Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28(9):1274–1279. doi:10.1038/onc.2008.466.
- Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015;6(6):3519–3530. doi:10.18632/oncotarget.2792.
- Palve V, Mallick S, Ghaisas G, et al. Overexpression of Mcl-1L splice variant is associated with poor prognosis and chemoresistance in oral cancers. PLoS One. 2014;9(11):e111927. doi:10.1371/journal.pone.0111927.
- Toge M, Yokoyama S, Kato S, et al. Critical contribution of MCL-1 in EMT-associated chemo-resistance in A549 non-small cell lung cancer. Int J Oncol. 2015;46(4):1844–1848. doi:10.3892/ijo.2015.2861.
- Wei D, Zhang Q, Schreiber JS, et al. Targeting mcl-1 for radiosensitization of pancreatic cancers. Translational oncology. 2015;8(1):47–54. doi:10.1016/j.tranon.2014.12.004.
- Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–114. doi:10.1038/nature09779.
- Balko JM, Giltnane JM, Wang K, et al. Molecular Profiling of the Residual Disease of Triple-Negative Breast Cancers after Neoadjuvant Chemotherapy Identifies Actionable Therapeutic Targets. Cancer discovery. 2014;4(2):232–45. doi:10.1158/2159-8290.CD-13-0286.
- Xiao Y, Nimmer P, Sheppard GS, et al. MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor. Molecular cancer therapeutics. 2015;14(8):1837–1847. doi:10.1158/1535-7163.MCT-14-0928.
- Bashari MH, Fan F, Vallet S, et al. Mcl-1 confers protection of Her2-positive breast cancer cells to hypoxia: therapeutic implications. Breast cancer research: BCR. 2016;18(1):26. doi:10.1186/s13058-016-0686-4.
- Bruncko M, Wang L, Sheppard GS, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem. 2015;58(5):2180–2194. doi:10.1021/jm501258m.
- Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482. doi:10.1038/nature19830.
- Merino D, Whittle JR, Vaillant F, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med. 2017;9(401). (pii):eaam7049. doi: 10.1126/scitranslmed.aam7049.
- Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–1095. doi:10.1016/j.cell.2005.06.009.
- Czabotar PE, Lee EF, van Delft MF, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(15):6217–6222. doi:10.1073/pnas.0701297104.
- Gomez-Bougie P, Menoret E, Juin P, et al. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem Biophys Res Commun. 2011;413(3):460–464. doi:10.1016/j.bbrc.2011.1008.1118. Epub 2011 Sep 1011. doi:10.1016/j.bbrc.2011.08.118.
- Inuzuka H, Shaik S, Onoyama I, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471(7336):104–109. doi:10.1038/nature09732.
- Ding Q, He X, Xia W, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67(10):4564–4571. doi:10.1158/0008-5472.CAN-06-1788.
- De Biasio A, Vrana JA, Zhou P, et al. N-terminal truncation of antiapoptotic MCL1, but not G2/M-induced phosphorylation, is associated with stabilization and abundant expression in tumor cells. J Biol Chem. 2007;282(33):23919–23936. doi:10.1074/jbc.M700938200.
- Warr MR, Shore GC. Unique biology of Mcl-1: therapeutic opportunities in cancer. Curr Mol Med. 2008;8(2):138–147. doi:10.2174/156652408783769580.
- Arai KI, Lee F, Miyajima A, Miyatake S, et al. Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem. 1990;59:783–836. doi:10.1146/annurev.bi.59.070190.004031.
- Wang JM, Chao JR, Chen W, et al. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19(9):6195–6206. doi:10.1128/MCB.19.9.6195.
- Wang JM, Lai MZ, Yang-Yen HF. Interleukin-3 stimulation of mcl-1 gene transcription involves activation of the PU.1 transcription factor through a p38 mitogen-activated protein kinase-dependent pathway. Mol Cell Biol. 2003;23(6):1896–1909. doi:10.1128/MCB.23.6.1896-1909.2003.
- Huang HM, Huang CJ, Yen JJ. Mcl-1 is a common target of stem cell factor and interleukin-5 for apoptosis prevention activity via MEK/MAPK and PI-3K/Akt pathways. Blood. 2000;96(5):1764–1771.
- Isomoto H, Kobayashi S, Werneburg NW, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42(6):1329–1338. doi:10.1002/hep.20966.
- Liu H, Ma Y, Cole SM, et al. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood. 2003;102(1):344–352. doi:10.1182/blood-2002-11-3396.
- Opferman JT, Letai A, Beard C, et al. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–676. doi:10.1038/nature02067.
- Shenoy AR, Kirschnek S, Hacker G. IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur J Immunol. 2014;44(8):2500–2507. doi:10.1002/eji.201344238.
- Aichberger KJ, Mayerhofer M, Krauth MT, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105(8):3303–3311. doi:10.1182/blood-2004-02-0749.
- Jiang CC, Lucas K, Avery-Kiejda KA, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 2008;68(16):6708–6717. doi:10.1158/0008-5472.CAN-08-0349.
- Piret JP, Minet E, Cosse JP, Ninane N, et al. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J Biol Chem. 2005;280(10):9336–9344. doi:10.1074/jbc.M411858200.
- Rinkenberger JL, Horning S, Klocke B, et al. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14(1):23–27.
- Renjini AP, Titus S, Narayan N, Murali M, et al. STAT3 and Mcl-1 unite to cause mesenchymal epithelial transition. J Cell Sci. 2014;127(Pt 8):1738–50. doi:10.1242/jcs.138214.
- Huskey NE, Guo T, Evason KJ, et al. CDK1 inhibition targets the p53-NOXA-MCL1 axis, selectively kills embryonic stem cells, and prevents teratoma formation. Stem Cell Reports. 2015;4(3):374–389. doi:10.1016/j.stemcr.2015.01.019.
- Morciano G, Giorgi C, Balestra D, et al. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell. 2016;27(1):20–34. doi:10.1091/mbc.E15-01-0028.
- Perciavalle RM, Stewart DP, Koss B, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14(6):575–583. doi:10.1038/ncb2488.
- Jamil S, Sobouti R, Hojabrpour P, et al. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. The Biochemical journal. 2005;387(Pt 3):659–667. doi:10.1042/BJ20041596.
- Jamil S, Stoica C, Hackett TL, et al. MCL-1 localizes to sites of DNA damage and regulates DNA damage response. Cell cycle (Georgetown, Tex). 2010;9(14):2843–2855. doi:10.4161/cc.9.14.12354.
- Malone CD, Hasan SM, Roome RB, et al. Mcl-1 regulates the survival of adult neural precursor cells. Mol Cell Neurosci. 2012;49(4):439–447. doi:10.1016/j.mcn.2012.02.003.
- Kuramoto K, Sakai A, Shigemasa K, et al. High expression of MCL1 gene related to vascular endothelial growth factor is associated with poor outcome in non-Hodgkin's lymphoma. Br J Haematol. 2002;116(1):158–161. doi:10.1046/j.1365-2141.2002.03253.x.
- Watson EC, Whitehead L, Adams RH, et al. Endothelial cell survival during angiogenesis requires the pro-survival protein MCL1. Cell Death Differ. 2016;23(8):1371–1379. doi:10.1038/cdd.2016.20.
- Lee WS, Park YL, Kim N, et al. Myeloid cell leukemia-1 is associated with tumor progression by inhibiting apoptosis and enhancing angiogenesis in colorectal cancer. Am J Cancer Res. 2015;5(1):101–113.
- Hasan SM, Sheen AD, Power AM, et al. Mcl1 regulates the terminal mitosis of neural precursor cells in the mammalian brain through p27Kip1. Development. 2013;140(15):3118–3127. doi:10.1242/dev.090910.
- Koehler BC, Scherr AL, Lorenz S, et al. Beyond cell death – antiapoptotic Bcl-2 proteins regulate migration and invasion of colorectal cancer cells in vitro. PLoS One. 2013;8(10):e76446. doi:10.1371/journal.pone.0076446.
- Lee WS, Kim N, Park YR, et al. Myeloid cell leukemia-1 promotes epithelial-mesenchymal transition of human gastric cancer cells. Oncol Rep. 2015;34(2):1011–1016. doi:10.3892/or.2015.4040.
- Gao J, Li L, Wu M, Liu M, et al. MiR-26a Inhibits Proliferation and Migration of Breast Cancer through Repression of MCL-1. PLoS One. 2013;8(6):e65138. doi:10.1371/journal.pone.0065138.
- O'Toole SA, Beith JM, Millar EKA, et al. Therapeutic targets in triple negative breast cancer. J Clin Pathol. 2013;66(6):530–542. doi:10.1136/jclinpath-2012-201361.
- Lee EF, Czabotar PE, van Delft MF, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180(2):341–355. doi:10.1083/jcb.200708096.
- Glaser SP, Lee EF, Trounson E, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26(2):120–125. doi:10.1101/gad.182980.111.
- Timpson P, McGhee EJ, Erami Z, et al. Organotypic collagen I assay: a malleable platform to assess cell behaviour in a 3-dimensional context. Journal of visualized experiments: JoVE. 2011;(56):e3089.
- Yamaguchi H, Pixley F, Condeelis J. Invadopodia and podosomes in tumor invasion. European journal of cell biology. 2006;85(3–4):213–218. doi:10.1016/j.ejcb.2005.10.004.
- Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. The oncologist. 2009;14(7):667–678. doi:10.1634/theoncologist.2009-0009.
- Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5(8):647–657. doi:10.1038/nrm1436.
- Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005;17(5):559–564. doi:10.1016/j.ceb.2005.08.002.
- Okada M. Regulation of the SRC family kinases by Csk. International journal of biological sciences. 2012;8(10):1385–1397. doi:10.7150/ijbs.5141.
- Reynolds AB, Kanner SB, Bouton AH, et al. SRChing for the substrates of Src. Oncogene. 2014;33(37):4537–4547. doi:10.1038/onc.2013.416.
- Roy S, Ruest PJ, Hanks SK. FAK regulates tyrosine phosphorylation of CAS, paxillin, and PYK2 in cells expressing v-Src, but is not a critical determinant of v-Src transformation. J Cell Biochem. 2002;84(2):377–388. doi:10.1002/jcb.10025.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013.
- Jorissen RN, Walker F, Pouliot N, et al. Epidermal growth factor receptor: mechanisms of activation and signalling. Experimental cell research. 2003;284(1):31–53. doi:10.1016/S0014-4827(02)00098-8.
- Oser M, Condeelis J. The cofilin activity cycle in lamellipodia and invadopodia. J Cell Biochem. 2009;108(6):1252–1262. doi:10.1002/jcb.22372.
- Oser M, Yamaguchi H, Mader CC, et al. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol. 2009;186(4):571–587. doi:10.1083/jcb.200812176.
- Morton JP, Karim SA, Graham K, et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139(1):292–303. doi:10.1053/j.gastro.2010.03.034.
- Nobis M, McGhee EJ, Morton JP, et al. Intravital FLIM-FRET imaging reveals dasatinib-induced spatial control of src in pancreatic cancer. Cancer Res. 2013;73(15):4674–4686. doi:10.1158/0008-5472.CAN-12-4545.
- Chee CE, Krishnamurthi S, Nock CJ, et al. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. The oncologist. 2013;18(10):1091–1092. doi:10.1634/theoncologist.2013-0255.
- Evans TR, Van Cutsem E, Moore MJ, et al. Phase 2 Placebo-Controlled, Double-Blind Trial of Dasatinib Added to Gemcitabine for Patients with Locally-Advanced Pancreatic Cancer. Ann Oncol. 2017;28(2):354–361. doi:10.1093/annonc/mdw607.
- Renouf DJ, Moore MJ, Hedley D, et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2012;30(2):779–786. doi:10.1007/s10637-010-9611-3.
- Elias D, Ditzel HJ. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol Res. 2015;100:250–254. doi:10.1016/j.phrs.2015.08.010.
- Gucalp A, Sparano JA, Caravelli J, et al. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer. 2011;11(5):306–311. doi:10.1016/j.clbc.2011.03.021.
- Finn RS, Bengala C, Ibrahim N, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(21):6905–6913. doi:10.1158/1078-0432.CCR-11-0288.
- Somlo G, Atzori F, Strauss LC, et al. Dasatinib plus capecitabine for advanced breast cancer: safety and efficacy in phase I study CA180004. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(7):1884–1893. doi:10.1158/1078-0432.CCR-12-0652.
- Fornier MN, Morris PG, Abbruzzi A, et al. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann Oncol. 2011;22(12):2575–2581. doi:10.1093/annonc/mdr018.
- Moy B, Neven P, Lebrun F, et al. Bosutinib in combination with the aromatase inhibitor exemestane: a phase II trial in postmenopausal women with previously treated locally advanced or metastatic hormone receptor-positive/HER2-negative breast cancer. The Oncologist. 2014;19(4):346–347. doi:10.1634/theoncologist.2014-0022.
- Erami Z, Herrmann D, Warren SC, et al. Intravital FRAP Imaging using an E-cadherin-GFP Mouse Reveals Disease- and Drug-Dependent Dynamic Regulation of Cell-Cell Junctions in Live Tissue. Cell Rep. 2016;14(1):152–167. doi:10.1016/j.celrep.2015.1012.1020. Epub 2015 Dec 1024. doi:10.1016/j.celrep.2015.12.020.
- Vickers ER, Kasza A, Kurnaz IA, et al. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol. 2004;24(23):10340–10351. doi:10.1128/MCB.24.23.10340-10351.2004.
- Townsend KJ, Trusty JL, Traupman MA, et al. Expression of the antiapoptotic MCL1 gene product is regulated by a mitogen activated protein kinase-mediated pathway triggered through microtubule disruption and protein kinase C. Oncogene 1998;17(10):1223–1234. doi:10.1038/sj.onc.1202035.
- Fritsch RM, Schneider G, Saur D, et al. Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem. 2007;282(31):22551–22562. doi:10.1074/jbc.M702673200.
- Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11(6):427–439. doi:10.1038/nrm2900.
- Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. Embo J. 2010;29(14):2407–2420. doi:10.1038/emboj.2010.112.
- Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW, Craig RW, Kaufmann SH, Gores GJ. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem. 2007;282(25):18407–18417. doi:10.1074/jbc.M610010200.
- Nakajima W, Hicks MA, Tanaka N, Krystal GW, Harada H. Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer. Cell Death Dis. 2014;5:e1052. doi:10.1038/cddis.2014.6.
- Inoshita S, Takeda K, Hatai T, et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem. 2002;277(46):43730–43734. Epub 42002 Sep 43739. doi:10.1074/jbc.M207951200.
- Kodama T, Hikita H, Kawaguchi T, et al. Mcl-1 and Bcl-xL regulate Bak/Bax-dependent apoptosis of the megakaryocytic lineage at multistages. Cell Death Differ. 2012;19(11):1856–1869. doi:10.1038/cdd.2012.88.
- Ding Q, Huo L, Yang JY, et al. et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008;68(15):6109–6117. doi:10.1158/0008-5472.CAN-08-0579.
- Domina AM, Vrana JA, Gregory MA, et al. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23(31):5301–5315. doi:10.1038/sj.onc.1207692.
- Nifoussi SK, Vrana JA, Domina AM, et al. Thr 163 phosphorylation causes Mcl-1 stabilization when degradation is independent of the adjacent GSK3-targeted phosphodegron, promoting drug resistance in cancer. PLoS One. 2012;7(10):e47060. doi:10.41371/journal.pone.0047060. Epub 0042012 Oct 0047069. doi:10.1371/journal.pone.0047060.
- Chu R, Terrano DT, Chambers TC. Cdk1/cyclin B plays a key role in mitotic arrest-induced apoptosis by phosphorylation of Mcl-1, promoting its degradation and freeing Bak from sequestration. Biochem Pharmacol. 2012;83(2):199–206. doi:10.1016/j.bcp.2011.10.008.
- Thomas E, Lee-Pullen T, Rigby P, et al. Receptor activator of NF-kappaB ligand promotes proliferation of a putative mammary stem cell unique to the lactating epithelium. Stem Cells (Dayton, Ohio). 2012;30(6):1255–1264. doi:10.1002/stem.1092.
- Iqbal S, Howard S, LoGrasso PV. Serum- and Glucocorticoid-Inducible Kinase 1 Confers Protection in Cell-Based and in In Vivo Neurotoxin Models via the c-Jun N-Terminal Kinase Signaling Pathway. Mol Cell Biol. 2015;35(11):1992–2006. doi:10.1128/MCB.01510-01514. Epub 02015 Mar 01530. doi:10.1128/MCB.01510-14.
- Maurer U, Charvet C, Wagman AS, et al. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–760. doi:10.1016/j.molcel.2006.02.009.
- Morel C, Carlson SM, White FM, et al. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol. 2009;29(14):3845–3852. doi:10.1128/MCB.00279-00209. Epub 02009 May 00211. doi:10.1128/MCB.00279-09.