
ABSTRACT
MARCKS is an actin and PIP2-binding protein that plays an essential role in neutrophil migration and adhesion; however, the molecular details regarding MARCKS function in these processes remains unclear. Neutrophil adhesion and migration also require the cell surface receptors β2-integrins. We hypothesized that MARCKS inhibition would alter neutrophil β2-integrin activation and signaling. We utilized a MARCKS-targeting peptide to inhibit MARCKS in inside-out and outside-in β2-integrin activation in neutrophils. MANS-mediated MARCKS inhibition had no significant effect on inside-out β2-integrin activation. MANS treatment significantly attenuated ICAM-1/Mn2+-stimulated static adhesion, cell spreading and β2-integrin clustering, suggesting a role for MARCKS function in outside-in β2-integrin activation. Additional work is needed to better understand the molecular mechanisms of MARCKS role in outside-in β2-integrin activation and signaling.
Introduction
Neutrophils play an essential role during the innate immune response. To transmigrate from the vasculature to sites of inflammation and/or microbial infection, neutrophils must be recruited from the bloodstream using specialized cell surface receptors, known as beta2 (β2)-integrins, that interact with adhesion molecules expressed on the surface of endothelial cells (e.g., intercellular adhesion molecule 1 (ICAM-1)). The details of neutrophil capture, rolling, endothelial adhesion, post-adhesion strengthening, crawling, and transmigration have largely been determined; however, there are still gaps in knowledge regarding specific cell signaling mechanisms [Citation1,Citation2], including β2-integrins. It is clear that β2-integrins play an important role in adhesion strengthening, crawling, and transmigration [Citation3], all of which are required processes for normal biological functions and host defense mechanisms. This is evidenced by the severe consequences of lack of functional β2-integrins seen in patients with leukocyte adhesion deficiency (LAD), which severely weakens the host immune system, resulting in recurrent and/or chronic bacterial infections leading to significant morbidity and mortality [Citation4].
Although critical for survival, neutrophils are considered a double-edged sword. When their destructive powers designed to combat pathogens become dysregulated, excessive neutrophil infiltration or neutrophil activation can cause significant damage to host tissues [Citation5]. In fact, neutrophil derived tissue injury is known to play a role in numerous acute and chronic inflammatory diseases, including myocardial infarction [Citation6], stroke [Citation7], ischemia-reperfusion injury [Citation8,Citation9], rheumatoid arthritis [Citation10,Citation11], severe SARS-CoV-2 [Citation12,Citation13], and sepsis-associated organ damage (i.e., acute lung injury) [Citation14,Citation15]. Several animal models have highlighted the potential and/or ability to target neutrophils by inhibiting β2-integrins [Citation14,Citation16]; however, the use of monoclonal antibodies targeting β2-integrins in clinical settings have not been successful [Citation17,Citation18]. Despite this, β2-integrins remain a desirable target for modulating neutrophil responses [Citation19]. A more detailed understanding of how β2-integrins are regulated may help identify novel targets for modulating dysregulated neutrophil responses.
B2-integrins are transmembrane heterodimers that consist of a common β-subunit (CD18), which is non-covalently associated with one of the four known α-subunits (CD11a, CD11b, CD11c, and CD11d) [Citation20]. The two most prominent integrins on neutrophils are CD11a (LFA-1 or αLβ2) and CD11b (Mac-1 or αLβ2). Within circulating, inactive neutrophils, CD11b integrins are primarily contained within the cytoplasm, secondary and tertiary granules, and secretory vesicles. The CD11a integrins that are present on the surface of resting neutrophils exist in an inactive/low affinity state with a ‘bent’ conformation. This combination of low surface expression and inactive conformation are control measures to help prevent nonspecific neutrophil binding and activation, as this could have damaging effects on host tissue and vasculature. When neutrophils receive an activation signal through intracellular signaling pathways, β2-integrin surface expression is increased (upregulation), and conformational changes take place (‘affinity’). This type of integrin stimulation is termed ‘inside-out’ activation and is driven by intracellular signals received by G protein coupled receptors (GPCRs) and Fc receptors, among others [Citation21]. B2-integrins can also become activated when the extracellular domain interacts directly with extracellular matrix proteins or other cell surface ligands (ICAM-1, fibrinogen, etc.) and initiates its own signaling, termed ‘outside-in’ activation [Citation1,Citation20,Citation22,Citation23]. Details regarding the downstream signaling pathway from outside-in β2-integrin activation are incomplete; however, multiple lines of evidence show that this type of activation is linked to neutrophil firm adhesion and cell spreading [Citation23].
Myristoylated alanine-rich C kinase substrate (MARCKS) is a well-known protein kinase C (PKC) substrate and actin-binding protein. It is essential for several integrin-dependent cellular functions, such as adhesion, cell spreading, and migration in numerous cell types [Citation24–26]. In most resting cells, MARCKS protein is associated with the inner leaflet of the plasma membrane due to hydrophobic insertion of an N-terminal myristoyl-moiety into the lipid bilayer, and electrostatic interactions between the MARCKS effector domain (ED) and 3–4 PIP2 molecules [Citation27–29]. The ED of MARCKS is subject to reversible regulation by PKC phosphorylation or Ca++/calmodulin binding. Either of these events displace MARCKS from the membrane to the cytosol and releases PIP2. Once dephosphorylated, MARCKS reassociates with the plasma membrane [Citation30–33]. This reversible association with the plasma membrane is known as the ‘myristoyl-electrostatic switch’ and has been shown to play a key role in adhesion and cell spreading in multiple cell types [Citation34,Citation35].
To date, in vitro research has demonstrated a role for MARCKS protein in exo-, endo- and phagocytosis, embryogenesis, growth-cone formation, myoblast spreading and adhesion, mast cell and neutrophil degranulation, tumor cell motility, and directed migration of vascular endothelial cells, fibroblast and neutrophils [Citation24,Citation25,Citation33,Citation36–42]. MARCKS function has been examined in various cell types by a variety of methods including transgenic expression of mutant proteins, RNAi cellular knock-down, knock-out mice (embryonic lethal) and inhibitor peptides designed to mimic either the effector domain or N-terminus amino acid sequence. Derivatives of the N-terminus mimetic peptide known as MANS (myristoylated N-terminal sequence) have entered Phase II clinical trials as potential airway therapeutics [Citation43–45]. Additional mechanistic understanding regarding the effect of MARCKS-targeting peptides on neutrophil function could further inform development of novel anti-inflammatory therapies.
Previous results from our lab and others have shown that inhibition of MARCKS with an N-terminal mimetic peptide known as MANS significantly attenuates functional responses of primary neutrophils, including migration and adhesion [Citation24,Citation26,Citation36,Citation43,Citation46]. Studies by Eckert et al. determined that MANS peptide displaces MARCKS from the membrane to the cytosol in resting neutrophils, but does not alter fMLP-induced MARCKS phosphorylation [Citation26]; however, additional insight into the mechanisms of MANS effect on neutrophil functions have not been elucidated. In other cell types, such as myoblasts and fibroblasts, MARCKS has been implicated in integrin-dependent cellular responses [Citation25,Citation47]. In the current study, we hypothesized that inhibition of MARCKS with the MANS peptide would alter β2-integrin activation and signaling in primary human neutrophils. We examined the impact of MANS-mediated MARCKS inhibition on β2-integrins using methods to target both ‘inside-out’ and ‘outside-in’ β2-integrin activation pathways. Our results show that MANS peptide does not impact fMLP-stimulated inside-out β2-integrin upregulation or affinity change but does inhibit outside-in β2-integrin-dependent adhesion, cell spreading and avidity, as well as β2-integrin-dependent respiratory burst. This study further informs our understanding of the cellular mechanism of MARCKS inhibition with MANS and is the first study to suggest a role for MARCKS protein in outside-in β2-integrin activation and signaling in primary human neutrophils.
Materials and methods
Isolation of human PMNs
Human blood collection protocols were reviewed and approved by Institutional Research Ethics Committee of NCSU (IRB approval #616). For all neutrophil experiments, 10–30 ml of whole blood was collected using heparinized syringes from healthy human volunteers that provided informed consent for participation. Neutrophils were isolated from whole blood using Ficoll-Paque Plus (GE Healthcare) density gradient centrifugation. Briefly, heparinized whole blood was mixed with 0.6% Dextran with 0.9% NaCl in a 15 mL polypropylene conical and allowed to settle at room temperature for 45–60 minutes. Up to 10 ml of leukocyte-rich plasma was aspirated using a bulb syringe and layered on 5 mL of Ficoll in a separate 15 mL conical tube. Cells were then centrifuged at 1800 rpm for 20 minutes with the brake off. The supernatant was discarded and remaining blood cells within the cell pellet were removed by 60 seconds of hypotonic lysis. Isolated neutrophils were resuspended/washed in sterile HBSS (Life Technologies) without additives. Cell number and viability was quantified using trypan blue exclusion (1:1) and a manual hemocytometer count. Final suspension of cells was in HBSS++ chemotaxis buffer [1× HBSS, 1 mM Ca2+, 1 mM Mg2+, 5% fetal bovine serum (Gibco)] at the indicated concentration for each experiment. All experiments were completed within 4 to 6 hours of blood collection. Neutrophils from individual human donors were used for all time points and treatment conditions for each experiment (i.e., ‘n’ represents a separate human donor).
Peptide treatment
The MyristoylAted N-terminal Sequence (MANS) and Random Nucleotide Sequence (RNS) peptides were synthesized by Genemed Synthesis, Inc. The sequence of MANS is identical to the first 24 amino acids of the human MARCKS protein: myristic acid-GAQFSKTAAKGEAAAERPGEAAVA. The RNS peptide is a randomly scrambled control: myristic acid-GTPAPAAEGAGAEVKRASAEAKQAF. Peptide working solutions were resuspended in sterile PBS. Where indicated, pretreatment of cell suspensions with indicated peptide concentrations occurred at 37°C for 30 minutes.
Fluorescence labeling of neutrophils
For adhesion experiments, isolated neutrophils (1×107/mL in HBSS) were incubated with the fluorescent dye calcein AM (Corning) at 2 μg/mL for 30 minutes at room temperature, protected from light. Cells were then centrifuged at 1200 rpm for 10 minutes and resuspended in HBSS++ with 2% heat inactivated FBS to the appropriate final experimental concentration.
Beta2-integrin inhibition
As a positive control β2-integrin inhibition, isolated human neutrophils were pretreated with 30 μg/mL anti-human CD18 F(ab’)2 (Ancell Corp) at 37°C for 30 minutes. Anti-human IgG2a F(ab’)2 (Ancell Corp) was used as an isotype control.
Flow cytometry
To avoid unintended stimulation of isolated neutrophils, hypotonic lysis of red blood cells was not performed prior to flow cytometry. Surface expression assay: Cells were pretreated as specified, stimulated with 100 nM fMLP (Sigma) or vehicle control (DMSO) for 5 minutes, diluted with 1 volume ice cold PBS and placed on ice for 5 minutes, spun at 1200 rpm for 5 minutes, resuspended in sterile PBS with 5% FBS and labeled with PE-conjugated anti-CD11b antibody clone ICFR44 (Novus Biologicals), PE-conjugated anti-CD11b CBRM1/5 antibody (Biolegend), or PE-conjugated IgG1 and IgG2a isotype controls (Biolegend). Flow cytometry was performed using Calibur FACScan. ICAM-1 binding assay: Cells were treated with 50 µM MANS, 50 µM RNS, or PBS for 30 minutes then washed with PBS and resuspended in HBSS++ +2% FBS buffer. Cells were incubated with 40 µg/mL ICAM-1/Fc and/or 100 nM fMLP for 5 minutes. Cells were fixed with 2% PFA, washed, resuspended with PE-conjugated anti-Fc F(ab’)2 fragment (1:100 dilution) for 20 minutes at 4°C, and washed again with PBS prior to acquisition. Flow cytometry was performed using BD LSRII. Flow cytometry experiments were performed in the Flow Cytometry and Cell Sorting facility at North Carolina State University, College of Veterinary Medicine.
Static adhesion assay
Individual wells of Immulon2HB flat bottom 96-well plates were coated with 10 μg/mL Recombinant ICAM-1 (R&D Systems) or 5% FBS in PBS (for controls). Isolated human neutrophils at 1 × 107/mL in HBSS were labeled with calcein AM (Corning) at a concentration of 2 μg/mL and primed with human GM-CSF (EMD Millipore) at 5 ng/mL for 30 minutes, protected from light. Cells were then centrifuged at 1000 rpm for 8 minutes and then resuspended in HBSS++ +2% FBS buffer at 7.0 × 105 cells/mL. Appropriate blocking antibodies and/or peptides were added to cells and incubated for 30 minutes at 37°C. The ICAM-1 coated plate was washed once with 1X PBS before cells were added to individual wells. Cells were added to wells and allowed to settle for 10 minutes at 37°C. 0.5 mM Mn2+ was added to individual wells. Plates were incubated for 10 minutes prior to initial fluorescence reading. Cells were gently dumped and washed with PBS, reading fluorescence (485 nm excitation, 535 nm emission) at each wash step. Fluorescence washing was divided by initial fluorescence and multiplied by 100 to calculate percent adhesion. The first wash that demonstrated less than 10% adhesion in the non-stimulated cells (plated on 5% FBS coating) was considered the result. Treatment groups were tested in triplicate.
Immunofluorescence microscopy
Chambered coverslips were either coated with 5% FBS in PBS (unstimulated control) or 10 µg/mL ICAM-1/Fc (R&D Systems) overnight. Isolated primary neutrophils were primed with 5 ng/mL human GM-CSF for 30 minutes at room temperature protected from light. Cells were then centrifuged at 1000 rpm for 8 minutes and then resuspended in the chemotaxis buffer HBSS++ +2% FBS buffer at 1.0 × 106 cells/mL. Appropriate concentrations of peptides were added to cells and incubated for 30 minutes at 37°C. The ICAM-1 (or ICAM-1/Fc) coated chamber coverslip was washed once with 1X PBS before cells were added to individual wells. 1.3 × 105 cells were added to the ICAM-1 or ICAM-1/Fc-coated chamber coverslips for 10 minutes. For fMLP stimulated wells, cells were allowed to settle for 10 minutes prior to addition of 100 nM fMLP or vehicle control for 5 minutes. For Mn2+ treated cells, 0.5 mM Mn2+ was added to individual wells after neutrophils were plated for 10 minutes on ICAM-1. Cells were then fixed to the coverslip, washed with PBS, blocked with rabbit serum, and labeled with AF549-conjugated anti-CD11b antibody clone M1/70 (Biolegend) before imaging with an Olympus I×83 Inverted Microscope. Cells that displayed morphology indicative of spreading were counted and averaged for each stimulation group.
Respiratory burst
Isolated neutrophils were resuspended in HBSS++ +2% FBS buffer to a final concentration of 3.0 × 106 cells/mL. Cells were then incubated with indicated treatments at 37°C for 30 minutes prior to each experiment and 100 μL of cells from indicated treatment groups were placed in individual wells of 5% FBS PBS or insoluble immune complex (IIC) coated Immulon2HB plates. Plates were coated overnight at 4°C with BSA (100 μg/mL). To generate IIC substrate, BSA (Sigma)-coated wells were washed three times and recoated with anti-BSA antibody (50 μg/well) and incubated for 2 hours at 37°C. Prior to the addition of cells, all wells were washed three times with sterile PBS. For PMA-stimulated respiratory burst, cells were allowed to settle for 10 minutes prior to the addition of dihydrorhodamine-123 (DHR-123) (Sigma) (10 μM final concentration) and PMA (100 ng/mL final concentration) (Sigma). In the case of IIC-mediated respiratory burst, DHR-123 was added immediately following addition of cells to the well. An fMax fluorescence plate reader (Molecular Devices) was used to measure initial fluorescence (485 nm excitation, 530 nm emission) followed by a fluorescence reading every 15 minutes for 120 minutes. Results are reported as nm fluorescence. Treatment groups were tested in triplicate. Individual time points were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test. *p < .05 MANS compared to IIC. #p < .05 anti-CD18 F(ab)’2 compared to IIC. Staurosporine p < .05 for all time points beginning at 30 minutes.
Cell Lysate stimulation
Prior to the experiment, low density insoluble immune complex was prepared as follows. Wells of a 24 well tissue culture plate was coated with 100 µg/mL bovine serum albumin overnight at 4°C. The wells were washed three times with PBS before the addition of coating with 33.35 µg/mL rabbit anti-bovine albumin antibody (Sigma) for 2 hours at 37°C. The wells were washed three times with PBS prior to the addition of cells. Isolated neutrophils (7×106 were resuspended in HBSS++ with 2% FBS and pretreated with PBS, 50 µM MANS, 50 µM RNS, 30 µM 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-D] pyrimidine (PP2) (Sigma), or 30 µg/mL anti-CD18 F(ab’)2 prior to stimulation with immune complexes for 15 minutes at 37°C. An Eppendorf tube of cells remained unstimulated for a suspension control. Plates were centrifuged at 1200 rpms for 10 minutes then immediately transferred onto ice and lysed with ice cold RIPA buffer [1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 5 mM sodium pyrophosphate and 50 mM sodium fluoride] containing protease inhibitors and phosphatase inhibitors for 30 minutes shaking on ice. After lysis, cell supernatants were centrifuged for at 1300 rpm for 10 minutes at 4°C. Supernatants were collected and frozen at −80°C until immunoblotting.
Immunoblotting
Frozen samples were thawed on ice and protein concentrations were measured using BCA Protein Assay Reagent (Pierce). Cell lysate was mixed with 5X sample buffer [25% glycerol, 2% SDS, 60 mM Tris-HCL (pH 6.8), 5% β-mercaptoethanol, 0.1% bromophenol blue in diH20] and boiled for 10 minutes. Equal amount of protein was analyzed in 4–12% SDS-PAGE. Resolved samples were transferred to Immobilon-P PVDF membrane (Millipore). Total protein was determined using No-Stain Protein Labeling Reagent (Invitrogen) followed by blocking 1 hour with 5% nonfat dry milk with Tween-20 (TBS/T; 136 μM NaCl, 20 μM Tris-base (pH: 7.6) and 0.1% Tween-20 v/v). Primary antibody (1:1000) was incubated overnight in 5% BSA TBS/T at 4°C. Membranes were washed with TBS/T and incubated with anti-rabbit horseradish peroxidase (HRP) secondary antibody (1:2000) (Cell Signaling Technology) in 5% BSA TBS/T for one hour, then washed three times for 5 minutes, developed using Bio-Rad Clarity Western ECL Substrate, and imaged using a Bio-Rad ChemiDoc. Western blot images were analyzed in Image Lab and results were normalized to total protein detected by the No-Stain Protein Labeling Reagent [Citation48–50].
Statistical analysis
Data are reported as mean ± SD. All statistical tests were performed using GraphPad Prism software (San Diego, CA) with the appropriate test details in the figure legend. P values < 0.05 were considered statistically significant. *Indicates p < .05, ** p < .01, *** p < .001, **** p < .0001.
Results
MARCKS inhibition with MANS peptide does not alter inside-out activation of neutrophil β2-integrins.
Previous findings in our lab demonstrated that inhibition of MARCKS protein function significantly decreased chemoattractant-induced (e.g., fMLP, IL-8, and LTB4) neutrophil adhesion and migration [Citation24,Citation26]. While these studies did not determine the mechanism for MARCKS in neutrophils stimulated with chemoattractants, Sheats et al. determined that MARCKS may play a role in neutrophil β2-integrins [Citation24]. Neutrophils within the vasculature are non-adherent and express low numbers of surface β2-integrins. When neutrophils are stimulated by chemoattractants, there is a significant increase in surface expression of β2-integrins, as determined by the expression of CD18 and CD11b. To determine whether inhibition of MARCKS with the MANS peptide altered inside-out activation of β2-integrins, neutrophils were pretreated with MANS or RNS and stimulated with fMLP, and CD11b surface expression was measured by flow cytometry. As shown in , fMLP stimulation induced a significant increase in total CD11b on the surface of neutrophils compared to control. MANS peptide treatment had no effect on fMLP-induced surface expression of CD11b. Calcium chelation with EDTA was used as a positive control for inhibition of CD11b upregulation. MARCKS inhibition with MANS also had no effect on fMLP-induced surface expression of CD18 or MHC class I expression (Supplemental Figure 1), consistent with prior observations [Citation26].
Figure 1. MANS peptide treatment does not inhibit fMLP-induced β2-integrin affinity conformation change of neutrophils. Neutrophils were pretreated with 50 µM MANS, 50 µM RNS, 10 mM EDTA, or PBS (untreated) for 30 minutes then stimulated with 100 nM fMLP or vehicle control (DMSO) for 5 minutes. Flow cytometry was used to measure total CD11b (ICRF44) (a-b) and high affinity CD11b (CBRM1/5) (c-d) as described. Histograms (b & d) show unstimulated (UTC) cells stained with IgG1 isotype control in red, DMSO (vehicle control/unstimulated) treated in blue, fMLP in green, MANS treated cells in orange, RNS treated cells in cyan, and EDTA (positive control for inhibition) in magenta. Data are represented as mean ± SD, n = 4–9. Mixed-effects analysis with Dunnett’s multiple comparisons test. *p < .05, ** p < .01, *** p < .001, **** p < .0001 indicates when compared to fMLP stimulation.
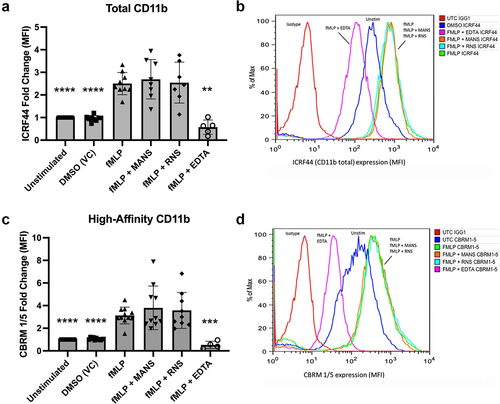
In addition to increasing the number of surface expressed β2-integrins, stimulation by chemoattractants also cause integrin conformation to shift from low to high affinity conformation, which facilitates ligand binding. Stimulation with fMLP resulted in a significant increase in the surface expression of high affinity CD11b compared to controls, as detected by CBRM1/5 antibody binding (). MANS peptide treatment had no effect on fMLP-induced β2-integrin high affinity conformation surface expression. Calcium chelation with EDTA was used as a positive control for inhibition of high affinity conformation expression. The high affinity conformation is required for β2-integrin binding to ICAM-1 [Citation51,Citation52]; therefore, we evaluated whether MARCKS inhibition had an effect on neutrophil binding to ICAM-1 upon activation with fMLP. Pretreatment of neutrophils with MANS or RNS had no effect on neutrophil binding to ICAM-1 (Supplemental Figure 2). This evidence demonstrates that inhibition of MARCKS with the MANS peptide does not alter fMLP-induced inside-out activation of neutrophil β2-integrins.
MARCKS inhibition with MANS peptide attenuates outside-in β2-integrin mediated neutrophil adhesion.
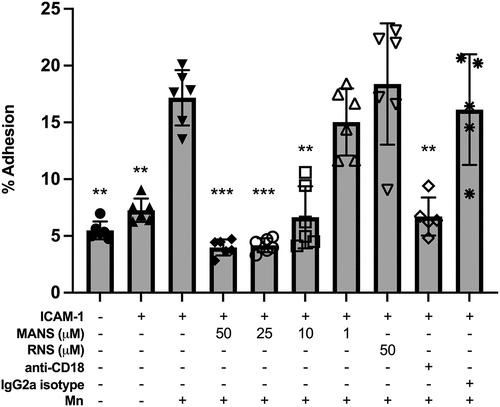
When neutrophils become activated by chemoattractants through inside-out stimulation, the high affinity conformation promotes β2-integrin binding to cellular adhesion molecules expressed on endothelial cells, such as intercellular adhesion molecule 1 (ICAM-1) [Citation51,Citation52]. This binding results in ‘outside-in’ activation and signaling. Outside-in activation and signaling controls firm adhesion, which is a necessary step for several neutrophil effector functions [Citation53,Citation54]. Although inside-out and outside-in activation happen together in vivo, we utilized an in vitro model of outside-in adhesion to determine if MANS-mediated MARCKS inhibition altered outside-in activation of β2-integrins. Static neutrophil adhesion was induced on ICAM-1 coated wells and enhanced with the application of manganese (Mn2+) [Citation21]. Stimulation of neutrophils using the divalent cation Mn2+ is used to induce the high affinity conformation of β2-integrins without increasing total β2-integrin integrin expression or inducing β2-integrin inside-out signaling [Citation21,Citation55]. This model induced significant static neutrophil adhesion (). MARCKS inhibition with MANS peptide significantly attenuated ICAM-1 + Mn2+-induced adhesion in a concentration-dependent manner. The addition of anti-CD18 F(ab’)2 confirmed that ICAM-1 + Mn2+ adhesion is β2-integrin dependent. The RNS control peptide and isotype controls had no effect on neutrophil adhesion. These results indicate that MARCKS protein plays an essential role in outside-in β2-integrin mediated static adhesion.
Figure 2. MANS peptide treatment attenuates outside-in β2-integrin-mediated neutrophil adhesion. Neutrophils were pretreated with indicated concentrations of MANS and RNS peptides, 30 µg/mL anti-CD18 F(ab)’2, or 30 µg/mL IgG2a control F(ab)’2 for 30 minutes prior to application to 96 well plate coated with 10 μg/mL ICAM-1. Cells were allowed to settle for 10 minutes before application of 0.5 mM Mn2+ for 10 minutes. Adhesion was quantified as described in the materials and methods section. MANS inhibition of MARCKS demonstrated concentration-dependent inhibition of ICAM-1+Mn-induced adhesion. Data represented as mean ± SD, n = 5–6. Mixed-effects analysis with Dunnett’s multiple comparisons test. * p < .05, ** p < .01, *** p < .001, **** p < .0001 indicates when compared to ICAM-1 + Mn group.
MARCKS inhibition with MANS peptide alters neutrophil cell spreading and β2-integrin clustering.
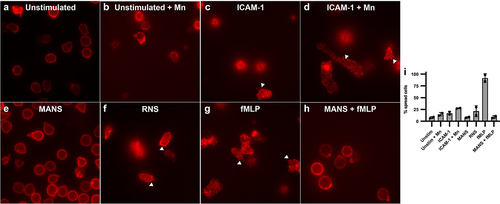
Having demonstrated a quantitative effect of MANS-mediated MARCKS inhibition on β2-integrin outside-in adhesion, we next sought qualitative information regarding the effect of MANS peptide treatment on changes in neutrophil morphology, including cell spreading, and β2-integrin clustering or avidity using immunofluorescence microscopy staining of CD11b [Citation56]. Unstimulated neutrophils appeared rounded with CD11b staining in the periphery of the cell and relatively homogenous and faint staining in the cell body, indicating no β2-integrin clustering (). Neutrophils stimulated with Mn2+ appeared similar to unstimulated cells with some increased staining of CD11b at the periphery of the cell (). Neutrophils adhered to ICAM-1 exhibited cell spreading and intense staining of CD11b within the body of the cell that is punctate and bright throughout, indicating clustered β2-integrins (). This response was more robust in neutrophils stimulated with ICAM-1 and Mn2+ (). MANS treatment significantly inhibited both cell spreading and β2-integrin clustering () to a level that resembled the unstimulated control (). The control peptide RNS had no effect on ICAM-1 + Mn2+ neutrophil spreading or β2-integrin clustering (). We further stimulated neutrophils plated on ICAM-1 + Mn2+ with fMLP () and demonstrated the addition of the inside-out stimulus fMLP did not restore the clustering and spreading defects caused by MANS peptide (). Similar results were observed in neutrophils plated on ICAM-1/Fc (Supplemental Figure 3). We also performed a semi-quantitative analysis of cell spreading to determine the percent spread neutrophils in each condition (), with the white arrows in displaying examples of cells with spreading and β2-integrin clustering morphology. As the microscopy indicates, MANS peptide decreased neutrophil cell spreading induced by ICAM-1 + Mn2+ and the spreading induced by ICAM-1 + Mn2+ with fMLP. These qualitative and semi-quantitative data indicate that inhibition of MARCKS with the MANS peptide alters the outside-in β2-integrin mediated events that follow initial ligand adhesion, including cell spreading and integrin clustering.
Figure 3. MANS peptide treatment alters neutrophil spreading and β2-integrin clustering on ICAM-1 enhanced with Mn2+. fMLP does not restore defects. Immunofluorescence microscopy of CD11b: Neutrophils were pretreated with 50 µM MANS, 50 µM RNS, or PBS for 30 minutes prior to application to ICAM-1 coated chambered coverslip. Cells were incubated for 10 minutes at 37°C. Mn2+ was added following the 10 minute incubation and allowed to adhere for another 10 minutes at 37°C. fMLP was applied 5 minutes into stimulation. (a) Neutrophils plated on 5% FBS in PBS. (b) Neutrophils plated on 5% FBS in PBS followed by Mn2+. (c) Neutrophils plated on ICAM-1. (d) Neutrophils plated on ICAM-1 and enhanced with Mn2+. (e) MANS pretreated neutrophils on ICAM-1 with Mn2+. (f) RNS pretreated neutrophils plated on ICAM-1 with Mn2+. (g) Neutrophils plated on ICAM-1/Mn2+ with fMLP applied 5 minutes into stimulation. (H) MANS pretreated neutrophils plated on ICAM-1/Mn2+ with fMLP applied 5 minutes into stimulation. (i) Percent spread cells in each condition. Data are represented as mean ± SD, n = 2.
MARCKS inhibition with MANS peptide attenuates β2-integrin-dependent, but not β2-integrin independent, neutrophil respiratory burst
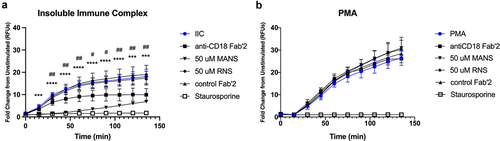
The data presented thus far have shown that inhibition of MARCKS with the MANS peptide alters outside-in activation of β2-integrins in primary human neutrophils. In addition to being responsible for the physical interactions of neutrophils with ligands, β2-integrins are also responsible for transmitting intracellular signals to elicit appropriate cellular responses. Therefore, we next evaluated whether MANS-mediated MARCKS inhibition alters signaling of neutrophil β2-integrins. Reactive oxygen species (ROS) are essential mediators of cellular signaling, so we first examined whether MANS peptide treatment altered respiratory burst using two-different β2-integrin-dependent and β2-integrin independent respiratory burst models. Neutrophil respiratory burst was stimulated with either low density insoluble immune complexes (IIC) or phorbol myristate acetate (PMA) and measured using DHR-123, which emits fluorescence once it is oxidized by reactive oxygen species.
As shown in , both IIC and PMA stimulated robust and sustained neutrophil respiratory burst. MANS peptide treatment significantly inhibited IIC-induced respiratory burst (), while RNS peptide had no effect. IIC-induced respiratory burst was β2-integrin dependent, as shown by significant inhibition with anti-CD18 F(ab’)2 treatment and was PKC dependent, as indicated by staurosporine inhibition of respiratory burst (). MANS peptide treatment had no effect on PMA-induced respiratory burst of neutrophils. PMA-induced respiratory burst was independent of β2-integrins, as indicated by lack of inhibition seen with anti-CD18 F(ab’)2 treatment, but was dependent on PKC, as indicated by staurosporine inhibition of respiratory burst (). These results demonstrate that inhibition of MARCKS with the MANS protein attenuates β2-integrin-dependent respiratory burst, but not β2-integrin independent respiratory burst. These data led us to hypothesize that MARCKS protein functions upstream of assembly of the NADPH oxidase complex.
Figure 4. MANS peptide inhibits β2-integrin-dependent neutrophil respiratory burst stimulated by insoluble immune complexes but not PMA-stimulated β2-integrin independent neutrophil respiratory burst. Neutrophils were pretreated for 30 minutes with 30 µg/mL anti-CD18 F(ab)’2, 30 µg/mL IgG2a control F(ab)’2, 50 µM MANS, 50 µM RNS, 100 nM staurosporine, or media alone (control). Respiratory burst was quantified by DHR-123. Cells were stimulated by insoluble immune complexes (IIC) (a) or PMA (b). Data are represented as mean ± SD, n = 2–4. Individual time points were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test. *p < .05, ** p < .01, *** p < .001, **** p < .0001 indicates MANS compared to IIC. #p < .05, # p < .01 indicates anti-CD18 F(ab)’2 compared to IIC. Staurosporine p < .05 for all time points beginning at 30 minutes.
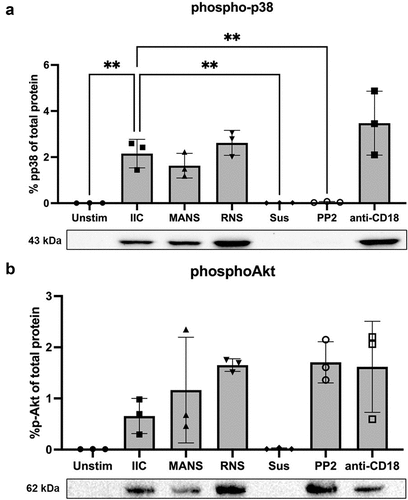
MARCKS inhibition with MANS peptide does not alter IIC-induced activation of p38 MAPK nor Akt
The p38 MAPK is an important cell signaling protein known to play an essential role in neutrophil functions [Citation57–59]. Previous studies have identified a role for p38 MAPK in β2-integrin-dependent neutrophil adhesion and ROS production [Citation60,Citation61]. Neutrophil stimulation with immune complexes induces activation (phosphorylation) of p38 MAPK, and p38 MAPK inhibition decreases neutrophil ROS production on IIC [Citation62]. Akt is also an important signaling molecule in neutrophils. Akt phosphorylation is observed in neutrophils stimulated by immune complexes, iC3b, ICAM-1, and by CBR LFA-1/2 antibodies [Citation62–64], indicating that Akt plays an important role in β2-integrin outside-in signaling.
We investigated whether attenuation of IIC-stimulated respiratory burst with the MANS peptide was associated with altered phosphorylation of p38 MAPK and Akt in MANS treated neutrophils stimulated by low density IIC. Isolated neutrophils were treated with either MANS, RNS, PP2, or anti-CD18 F(ab’)2 prior to stimulation with IICs (). The Src kinase inhibitor PP2 was included as a positive control for inhibition of p38 MAPK and Akt phosphorylation in IIC-stimulated neutrophils [Citation62]. MANS and RNS peptides had no effect on p38 MAPK phosphorylation (). Treatment with anti-CD18 F(ab’)2 also did not inhibit p38 MAPK phosphorylation (). MANS and RNS peptides and anti-CD18 F(ab’)2 had no significant effect on Akt phosphorylation (). These results indicate that inhibition of MARCKS with the MANS peptide does not alter IIC-induced p38 MAPK or Akt activation in neutrophils.
Figure 5. MANS peptide treatment does not alter IIC-induced p38 nor Akt activation in neutrophils. Neutrophils (7x106) were pretreated with either PBS, 50 µM MANS, 50 µM RNS, 30 µM PP2 (positive control for inhibition), or 30 µg/mL anti-CD18 F(ab)’2 then stimulated on low density immune complexes for 15 minutes or left in suspension (Sus). Phosphorylation of p38 or Akt was analyzed by Western blotting of whole-cell lysates. Equal loading was confirmed following transfer with Invitrogen No-Stain Protein Labeling Reagent. The percentage of phosphop38 or phosphoAkt was determined based on the total protein in the respective lane and the signal of the phosphop38 or phosphoAkt band. Blot shown is representative of three independent experiments. Data are represented as mean ± SD, n = 3. One-way ANOVA with Dunnett’s multiple comparisons test. *p < .05, *p < .01 indicates when compared to IIC.
Discussion
MARCKS is a natively unfolded, ubiquitously expressed protein that demonstrates reversible actin and PIP2 binding. Interaction of MARCKS with its binding partners is regulated by competing signals from reversible PKC-phosphorylation, and calcium/calmodulin binding, of the effector domain [Citation35,Citation65–68]. MARCKS is known to play a key role in neutrophil adhesion, migration, and respiratory burst. Previous results from our lab and others have shown that inhibition of MARCKS with an N-terminal mimetic peptide known as MANS significantly attenuates functional responses of primary neutrophils [Citation24,Citation26,Citation36,Citation46]; however, details regarding the cellular mechanism of MANS-mediated inhibition of neutrophil functions has not been fully elucidated. In the current study, we hypothesized that inhibition of MARCKS with the MANS peptide would alter β2-integrin activation and signaling in primary human neutrophils. We examined the impact of MANS-mediated MARCKS inhibition on β2-integrins using methods to target both ‘inside-out’ and ‘outside-in’ β2-integrin activation pathways. Our results show that MANS peptide does not impact fMLP-stimulated inside-out β2-integrin upregulation or affinity change but does inhibit outside-in β2-integrin-dependent adhesion, cell spreading and avidity, as well as β2-integrin-dependent respiratory burst. This is the first study to suggest that inhibition of MARCKS with the MANS peptide alters β2-integrin-dependent neutrophil functions, such as adhesion and respiratory burst, by inhibiting outside-in β2-integrin activation.
In this study we used a MARCKS-targeting peptide known as MANS, which stands for MyristoylAted N-terminus Sequence, as a tool for MARCKS inhibition. This peptide is identical to the first 24 amino acids of MARCKS sequence and includes an N-terminal myristoyl-moiety. In vitro and in vivo studies demonstrating the effects of MANS-mediated MARCKS inhibition on cellular migration, adhesion, secretion, degranulation and respiratory burst are numerous [Citation24,Citation25,Citation36,Citation41–43,Citation46,Citation69–72]. A derivative of MANS known as BIO-11006, identical to the first 10 amino acids of MARCKS, has been shown to be suitable for therapeutic use as an aerosolized peptide and has been evaluated in Phase II clinical trials in patients with chronic obstructive pulmonary disease, late-stage non-small cell lung cancer and acute lung injury [Citation43–45,Citation73]. Although these MARCKS inhibitor peptides have been reported on extensively, the cellular mechanisms of inhibition have not been fully elucidated in neutrophils. Given the clinical treatment potential of MARCKS-targeting therapies and the need for novel targets in neutrophil-mediated diseases such as severe neutrophilic asthma and acute lung injury, we would like to fill the knowledge gap regarding how MARCKS inhibition with the MANS peptide alters neutrophil activation and signaling in the hopes that this understanding will inform novel therapeutic approaches to neutrophil-mediated diseases.
MARCKS inhibition with MANS peptide disrupted neutrophil spreading and β2-integrin clustering on both ICAM-1 enhanced with Mn2+ and ICAM-1/Fc substrates ( and Supplemental Figure 3c). These findings further inform our understanding of the effect of MANS peptide on neutrophil adhesion. Interestingly, the addition of fMLP as an inside-out stimulus did not restore the defects caused by MANS peptide. While these results are consistent with previous studies that showed that MARCKS plays an essential role in cell spreading in several cell types [Citation47,Citation74–77], they are in contrast to the findings by Eckert et al., whose results showed that MANS peptide treatment did not impact fMLP-stimulated cell spreading and polarization [Citation26]. The reason for this discrepancy is not clear. Similar to our results, Mac-1 deficient neutrophils also display a rounded morphology and lack spreading on immune complexes [Citation78]. Additionally, clustering of β2-integrins is considered to be an essential part of post-adhesion strengthening and sustained adhesion to ligand [Citation79,Citation80]. Taken together, our results show that inhibition of MARCKS with the MANS peptide disrupts key steps of neutrophil β2-integrin outside-in activation, including cell spreading and integrin clustering.
Respiratory burst is an important neutrophil effector function and window into neutrophil intracellular signaling. In the current study, we evaluated MARCKS role in both β2-integrin-dependent (insoluble immune complex – IIC) and independent (PMA) respiratory burst. MANS peptide treatment significantly inhibited IIC-induced respiratory burst but not PMA-stimulated respiratory burst (). From these findings, we conclude that MANS treatment does not hinder the assembly and function of the NADPH oxidase complex. Furthermore, because the pan-PKC inhibitor staurosporine blocked both types of respiratory burst, we confirm that PKC is required for neutrophils to undergo successful respiratory burst. Therefore, we suggest that at least one aspect of MARCKS function, which is inhibited by the MANS peptide, must be essential for IIC-stimulated respiratory burst between the level of FcγR-signaling and PKC activation.
Fc receptors and β2-integrins cooperate in many neutrophil functional responses. Zhou and Brown demonstrated that β2-integrin and FcγRIII cooperation generates a respiratory burst that is unproductive when each receptor is stimulated separately [Citation81]. The respiratory burst response was also inhibited when either receptor was individually blocked [Citation81]. Other studies have also shown that β2-integrins are required for neutrophil respiratory burst [Citation82–84], and our results further confirm that β2-integrins are required for IIC-induced neutrophil respiratory burst (). This finding is in contrast to a previous study showing that superoxide production by immune complex adherent neutrophils was not β2-integrin dependent [Citation85]. However, superoxide production was only measured 20 minutes into the stimulation and the density of immune complex was not specified. In the current report, anti-CD18 F(ab’)2 treated cells are significantly different than untreated cells at the 30-minute time point but not at 15 minutes (). It is possible that the limited time point analysis is the reason that the study by Graham et al. did not capture any differences attributed β2-integrin inhibition [Citation85].
P38 MAPK is known to play an important role in inflammation and several neutrophil functions [Citation58,Citation60,Citation86]. P38 is also involved in neutrophil β2-integrin mediated adhesion, as either p38 inhibition or Mac-1 receptor blocking antibodies significantly diminish IL-8-induced adhesion to ICAM-1 [Citation87]. Interestingly, similar to our results with inhibition of MARCKS, inhibition of p38 MAPK did not attenuate PMA-stimulated neutrophil adhesion or ROS production [Citation60]. Based on these similarities, we hypothesized that MARCKS could play a role in a pathway involving p38 MAPK and β2-integrin-dependent adhesion. However, we found that MANS peptide has no effect on p38 MAPK activation in IIC-stimulated primary neutrophils. This is in contrast to previous work that showed MANS treatment significantly diminished both LPS-induced cytokine production and p38 phosphorylation in murine macrophages [Citation88]. P38 is activated by several different pathways, so the differences between our findings and Lee et al. may be explained by different cell types or mechanisms of stimulation (LPS vs IIC).
In our results reported here, anti-CD18 F(ab’)2 did not attenuate p38 activation. This is in contrast with previously published results showing that β2-integrin blocking antibodies did inhibit p38 activation; however, there are key differences between these previous studies and our current study. In the current report, we utilized low density insoluble immune complexes to stimulate neutrophil activation, which is mediated by both FcγRs and β2-integrins. The prior studies utilized chemoattractants (e.g., TNFα and IL-8) to stimulate inside-out β2-integrin responses on a variety of ligands [Citation60,Citation61,Citation87,Citation89]. The differences in cell signaling pathways initiated by these different stimuli could explain differences in our findings. Additionally, p38 has also been implicated in inside-out β2-integrin activation by studies that showed inhibition of p38 MAPK decreased TNFα-induced upregulation of CD11b in neutrophils [Citation61,Citation90]. We determined in that MARCKS inhibition does not impact inside-out β2-integrin activation stimulated by fMLP; therefore, MANS peptide treatment is unlikely to affect p38 activation induced by IICs if p38 MAPK activation is linked to inside-out β2-integrin activation and signaling. Finally, our finding that MANS peptide does not decrease p38 MAPK activation provides further indirect support for MARCKS role in outside-in β2-integrin activation, as opposed to FcγR signaling. Indeed, several reports have demonstrated that outside-in β2-integrin activation does not result in p38 phosphorylation [Citation64,Citation91]. Therefore, p38 activation in our IIC-stimulated neutrophils is likely the result of FcγR engagement, and it was not altered in MANS treated neutrophils. This indicates that either MARCKS function is downstream of p38 phosphorylation or that MARCKS function is involved in a separate parallel and essential pathway that leads to IIC-induced respiratory burst in neutrophils.
We also evaluated the effect of MARCKS inhibition on Akt activation due to Akt’s putative role in β2-integrin outside-in signaling [Citation64]. When localized to the membrane, MARCKS effector domain sequesters PIP2 molecules at a ratio of 1:4 [Citation68]. Displacement of MARCKS from the membrane by calcium/calmodulin binding or PKC-phosphorylation results in increased available membrane PIP2. PIP2 is then converted by phosphoinositide-3-kinase (PI3K) to PIP3, which leads to the increased translocation and subsequent phosphorylation of Akt [Citation92]. Interestingly, previous results in fibroblasts show that MANS inhibition of MARCKS did decrease Akt phosphorylation [Citation25]. This is in contrast to our findings that Akt phosphorylation was not significantly altered in MANS-treated neutrophils. Previous evaluation of Akt activation also utilized PP2 as a positive control for inhibition [Citation62], however, neutrophils treated with PP2 stimulated with IIC resulted in Akt phosphorylation in our study, weakening our conclusions regarding Akt signaling. Although Akt has been associated with outside-in signaling, the signaling events leading to Akt phosphorylation are different than those leading to spreading, which is a primary result of outside-in β2-integrin activation [Citation92]. While MANS peptide treatment did not inhibit Akt phosphorylation in our study, additional work is needed to better understand how MANS disruption of endogenous MARCKS function impacts other PH-domain containing protein regulators of neutrophil β2-integrins.
Several lines of evidence have converged to suggest that the ‘root’ function of MARCKS is to regulate PIP2 microdomain availability, upstream of and in response to, PKC and (or) Ca2±calmodulin signaling pathways and related to the myristoyl-electrostatic switch [Citation33,Citation93]. Studies by Glaser and colleagues showed that physiological concentrations of MARCKS (<10 μM) inhibit phospholipase C (PLC)-catalyzed hydrolysis of PIP2 in phospholipid vesicles by sequestering the acidic phospholipid at a 1:4 molar ratio (MARCKS:PIP2) [Citation68]. In various cell types PIP2 co-localizes with MARCKS in several morphologically active cellular locations, including focal adhesions and membrane ruffles [Citation93–97]. Most recently, studies using live cell fluorescence imaging of macrophages provide convincing evidence of a signaling loop that controls the leading edge of amoeboid cell migration that consists of Ca2+-PKC activation of MARCKS leading to release of concentrated pools of PIP2 which are phosphorylated by PI3K to PIP3 [Citation98]. Ultimately, the association between MARCKS and PIP2 influences cellular actin organization [Citation28]. Studies show that alterations in MARCKS ability to shuttle between the cell membrane and cytosol disrupts dynamic cellular events such as adhesion and spreading [Citation47,Citation99]. These data are consistent with findings by Sheats et al., which show that PKC-delta mediated MARCKS phosphorylation, which causes MARCKS to shuttle to the cytosol, is essential for human neutrophil adhesion [Citation100]. In future studies, we will investigate whether MANS disruption of β2-integrin outside-in activation in neutrophils is associated with differences in distribution or availability of membrane PIP2 microdomains, changes in actin organization, or both.
This is the first study to show that inhibition of MARCKS with the MANS peptide inhibits outside-in, but not inside-out, β2-integrin activation in neutrophils. A role for MARCKS in integrin-dependent cellular responses has been investigated previously in other cell types, but the molecular details remain incompletely understood. This report expands the understanding of MANS-mediated MARCKS inhibition and implicates a role for MARCKS function in neutrophil responses requiring outside-in neutrophil β2-integrin activation and signaling. Additional work is needed to determine whether this finding is specific for MANS-mediated peptide inhibition of MARCKS compared to other methods of MARCKS-targeting. In future studies, we will further investigate the mechanism of MANS effect on outside-in activation of neutrophil β2-integrins by investigating subcellular localization of MARCKS upstream and downstream signaling partners including PI3K, PIP2/PIP3, actin and PH-domain containing actin-binding proteins.
Supplemental Material
Download MS Word (1.5 MB)Acknowledgments
We would like to thank Dr. Lauren Schnabel at North Carolina State University for microscope access.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The original contributions presented in the study are included in the article and supplementary material. Data files can also be found at https://doi.org/10.5281/zenodo.8121775. Further inquiries can be directed to the corresponding author at [email protected].
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19336918.2023.2233204
Additional information
Funding
References
- Herter J, Zarbock A. Integrin regulation during leukocyte recruitment. J Immunol. 2013;190(9):4451–4457. doi: 10.4049/jimmunol.1203179
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi: 10.1038/nri3399
- Schmidt S, Moser M, Sperandio M. The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol. 2013; 55(1):49–58. doi: 10.1016/j.molimm.2012.11.006.
- Kuijpers TW, Verhoeven AJ, Yin Thung D, et al. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. J Clin Invest. 1997;100(7):1725–1733. doi: 10.1172/JCI119697
- Mittal M, Siddiqui MR, Tran K, et al. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20(7):1126–1167. doi: 10.1089/ars.2012.5149
- Meisel SR, Shapiro H, Radnay J, et al. Increased expression of neutrophil and monocyte adhesion molecules LFA- 1 and Mac-1 and their ligand ICAM-1 and VLA-4 throughout the acute phase of myocardial infarction: Possible implications for leukocyte aggregation and microvascular plugging. J Am Coll Cardiol. 1998;31(1):120–125. doi: 10.1016/S0735-1097(97)00424-5
- Edwards DN, Bix GJ. The Inflammatory Response After Ischemic Stroke: targeting β2 and β1 integrins. Front Neurosci. 2019 [cited 2020];13:540 doi: 10.3389/fnins.2019.00540
- Schofield ZV, Woodruff TM, Halai R, et al. Neutrophils—A Key component of ischemia-reperfusion injury. Shock. 2013;40(6):463–470. doi: 10.1097/SHK.0000000000000044
- Yago T, Petrich BG, Zhang N, et al. Blocking neutrophil integrin activation prevents ischemia–reperfusion injury. J Exp Med. 2015;212(8):1267–1281. doi: 10.1084/jem.20142358
- Lin J, He Y, Wang B, et al. Blocking of YY1 reduce neutrophil infiltration by inhibiting IL-8 production via the PI3K-Akt-Mtor signaling pathway in rheumatoid arthritis. Clin Exp Immunol. 2019;195(2):226–236. doi: 10.1111/cei.13218
- Wright HL, Makki FA, Moots RJ, et al. Low-density granulocytes: functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J Leukocyte Biol. 2017;101(2):599–611. doi: 10.1189/jlb.5A0116-022R
- Middleton E, Cody M, Rondina M. Neutrophil Extracellular Traps (NETs) Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood. 2020;136(10):1169–1179. doi: 10.1182/blood.2020007008
- Wang J, Li Q, Yin Y, et al. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front Immunol. 2020;11:1–13. doi: 10.3389/fimmu.2020.02063
- Neumann B, Zantl N, Veihelmann A, et al. Mechanisms of acute inflammatory lung injury induced by abdominal sepsis. 1999. doi: 10.1093/intimm/11.2.217
- Giacalone VD, Margaroli C, Mall MA, et al. Neutrophil adaptations upon recruitment to the lung: new concepts and implications for homeostasis and disease. Int J Mol Sci. 2020;21(3):1–21. doi: 10.3390/ijms21030851
- Wang Z, Li J, Cho J, et al. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotech. 2014;9(3):204–210. doi: 10.1038/nnano.2014.17
- Harlan JM, Winn RK. Leukocyte-endothelial interactions: clinical trials of anti-adhesion therapy. Crit Care Med. 2002;30(5 Suppl):S214–9. doi: 10.1097/00003246-200205001-00007
- Vicente-Manzanares M, Sá Nchez-Madrid F. Targeting the integrin interactome in human disease. Curr Opin Cell Biol Internet. 2018;55:17–23. doi: 10.1016/j.ceb.2018.05.010
- Zimmerman T, Blanco F. Inhibitors targeting the LFA-1/ICAM-1 cell-adhesion interaction: design and mechanism of action. Curr Pharm Des. 2008;14(22):2128–2139. doi: 10.2174/138161208785740225
- Schymeinsky J, Mocsai A, Walzog B. Neutrophil activation via β2 integrins (CD11/CD18): molecular mechanisms and clinical implications. Thromb Haemost. 2007;98(8):262–273. doi: 10.1160/TH07-02-0156
- Jones SL, Knaus UG, Bokoch GM, et al. Two Signaling Mechanisms for Activation of αMβ2 Avidity in Polymorphonuclear Neutrophils. Journal Of Biological Chemistry. 1998;273(17):10556–10566. doi: 10.1074/jbc.273.17.10556
- Williams MA, Solomkin JS. Integrin-mediated signaling in human neutrophil functioning. J Leukocyte Biol. 1999;65(6):725–736. doi: 10.1002/jlb.65.6.725
- Abram CL, Lowell CA. The Ins and outs of Leukocyte Integrin signaling. Annu Rev Immunol. 2009;339–62. doi: 10.1146/annurev.immunol.021908.132554
- Sheats MK, Pescosolido KC, Hefner EM, et al. Myristoylated Alanine Rich C Kinase Substrate (MARCKS) is essential to β2-integrin dependent responses of equine neutrophils. Vet Immunol Immunopathol. 2014;160(3–4):167–176. doi: 10.1016/j.vetimm.2014.04.009
- Ott LE, Sung EJ, Melvin AT, et al. Fibroblast Migration is Regulated by Myristoylated Alanine-Rich C-Kinase Substrate (MARCKS) Protein. PLoS One. 2013;8(6):e66512. doi: 10.1371/journal.pone.0066512
- Eckert RE, Neuder LE, Park J, et al. Myristoylated Alanine-Rich C-Kinase substrate (MARCKS) protein regulation of human neutrophil migration. Am J Respir Cell Mol Biol. 2010;42:586–94. doi: 10.1165/rcmb.2008-0394OC
- Wang J, Gambhir A, Hangyá S-Mihá Lyné G N, et al. Lateral Sequestration of Phosphatidylinositol 4,5-Bisphosphate by the Basic Effector Domain of Myristoylated Alanine-rich C Kinase Substrate is Due to Nonspecific Electrostatic Interactions. J Biol Chem. 2002;277(37):34401–12. doi: 10.1074/jbc.M203954200
- Kalwa H, Michel T. The MARCKS protein plays a critical role in Phosphatidylinositol 4,5-Bisphosphate metabolism and directed cell movement in vascular endothelial cells. J Biol Chem. 2011;286:2320–30. doi: 10.1074/jbc.M110.196022
- Wang J, Arbuzova A, Hangyas-Mihalyne G, et al. The effector domain of myristoylated alanine-rich c kinase substrate binds strongly to Phosphatidylinositol 4,5-Bisphosphate. J Biol Chem. 2001;276(7):5012–5019. doi: 10.1074/jbc.M008355200
- Aderem A. The MARCKS brothers: A family of protein kinase C substrates. Cell. 1992;71(5):713–716. doi: 10.1016/0092-8674(92)90546-O
- Blackshear PJ. The MARCKS family of cellular protein Kinase C substrates. J Biol Chem. 1993;268(3):1501–1504. doi: 10.1016/S0021-9258(18)53878-3
- Arbuzova A, Schmitz AAP, Res GV. Cross-talk unfolded: MARCKS proteins. Biochem J. 2002;362(Pt 1):1–12. doi: 10.1042/0264-6021:3620001
- Sundaram M, Cook HW, Byers DM. The MARCKS family of phospholipid binding proteins: regulation of phospholipase D and other cellular components. Biochem Cell Biol. 2004;82(1):191–200. doi: 10.1139/o03-087
- Seykora JT, Monn Myat M, Allen L-A, et al. Molecular Determinants of the Myristoyl-electrostatic Switch of MARCKS. Journal Of Biological Chemistry. 1996;271(31):18797–18802. doi: 10.1074/jbc.271.31.18797
- McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20(7):272–276. doi: 10.1016/S0968-0004(00)89042-8
- Takashi S, Park J, Fang S, et al. A peptide against the N-Terminus of Myristoylated Alanine-Rich C Kinase Substrate Inhibits Degranulation of Human Leukocytes in vitro. Am J Respir Cell Mol Biol. 2006;34:647–52. doi: 10.1165/rcmb.2006-0030RC
- Park J-A, Crews AL, Lampe WR, et al. Protein Kinase Cδ Regulates Airway Mucin Secretion via Phosphorylation of MARCKS Protein. Am J Pathol. 2007;171(6):1822–1830. doi: 10.2353/ajpath.2007.070318
- Estrada-Bernal A, Gatlin JC, Sunpaweravong S, et al. Dynamic adhesions and MARCKS in melanoma cells. J Cell Sci. 2009;122(13):2300–2310. doi: 10.1242/jcs.047860
- Chen X, Rotenberg SA. PhosphoMARCKS drives motility of mouse melanoma cells. Cell Signal. 2010;22(7):1097–1103. doi: 10.1016/j.cellsig.2010.03.003
- Ott LE, Mcdowell ZT, Turner PM, et al. Two myristoylated alanine-rich C-kinase substrate (MARCKS) paralogs are required for normal development in zebrafish. Anat Rec. 2011;294:1511–24. doi: 10.1002/ar.21453
- Green TD, Park J, Yin Q, et al. Directed migration of mouse macrophages in vitro involves myristoylated alanine-rich C-kinase substrate (MARCKS) protein. J Leukocyte Biol. 2012;92(3):633–639. doi: 10.1189/jlb.1211604
- Chen C-H, Thai P, Yoneda K, et al. A peptide that inhibits function of Myristoylated Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer metastasis. Oncogene. 2014;33:3696–706. doi: 10.1038/onc.2013.336
- Damera G, Jester WF, Jiang M, et al. Inhibition of myristoylated alanine-rich C kinase substrate (MARCKS) protein inhibits ozone-induced airway neutrophilia and inflammation. Exp Lung Res. 2010;36(2):75–84. doi: 10.3109/01902140903131200
- Adler KB, Parikh I, Crews A, et al. Effect of an anti-MARCKS peptide (BIO-11006) on metastasis of lung cancer in vivo. 2017 ASCO Annual Meeting I. 2017;35:15_suppl. doi: 10.1200/JCO.2017.35.15_suppl.e23017
- Adler K, Kraft M, Parikh I, et al. Results of a phase 2a clinical trial with a peptide inhibitor of MARCKS protein indicate improvement of indices of bronchitis and lung function in patients with COPD. Eur Respir J. 2011;38(Suppl 55):4901.
- Li J, D’Annibale-Tolhurst M, Adler K, et al. A Myristoylated Alanine-Rich C Kinase Substrate–Related Peptide Suppresses Cytokine mRNA and protein expression in LPS-Activated Canine Neutrophils. Am J Respir Cell Mol Biol. 2013;48(3):314–321. doi: 10.1165/rcmb.2012-0278OC
- Disatnik MH-H, Boutet SC, Pacio W, et al. The bi-directional translocation of MARCKS between membrane and cytosol regulates integrin-mediated muscle cell spreading. J Cell Sci. 2004;117:4469–4479. doi: 10.1242/jcs.01309
- Rivero-Gutiérrez B, Anzola A, Martínez-Augustin O, et al. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal Biochem. 2014;467:1–3. doi: 10.1016/j.ab.2014.08.027
- Moritz CP. Tubulin or not tubulin: heading toward total protein staining as loading control in western blots. Proteomics. 2017;17(20):1600189. doi: 10.1002/pmic.201600189
- Gilda JE, Gomes AV. Stain-Free total protein staining is a superior loading control to β-actin for Western blots. Anal Biochem. 2013;440(2):186–188. doi: 10.1016/j.ab.2013.05.027
- Fan Z, Mcardle S, Marki A, et al. Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nat Commun. 2016;7:12658. doi: 10.1038/ncomms12658
- Spillmann C, Osorio D, Waugh R. Integrin activation by divalent ions affects neutrophil homotypic adhesion. Ann Biomed Eng. 2002;30(8):1002–1011. doi: 10.1114/1.1511241
- Zarbock A, Ley K. Neutrophil Adhesion and Activation under Flow. Microcirc. 2009;16(1):31–42. doi: 10.1080/10739680802350104
- Giagulli C, Ottoboni L, Caveggion E, et al. The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating β2 integrin affinity and valency in neutrophils, but are required for β2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol. 2006;177(1):604–611. doi: 10.4049/jimmunol.177.1.604
- Altieri DC. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J Immunol. 1991;147(6):1891–1898. doi: 10.4049/jimmunol.147.6.1891
- Whitlock BB, Gardai S, Fadok V, et al. Differential rolesfor αMβ2 integrin clustering or activation in the control of apoptosis via regulation of Akt and ERK survival mechanisms. J Cell Bio. 2000;151(6):1305–1320. doi: 10.1083/jcb.151.6.1305
- Kim D, Haynes CL. The Role of p38 MAPK in Neutrophil functions: single cell chemotaxis and surface marker expression. Analyst. 2013;138(22):138. doi: 10.1039/c3an01076g
- Eckert RE, Sharief Y, Jones SL. P38 mitogen-activated kinase (MAPK) is essential for equine neutrophil migration. Vet Immunol Immunopathol. 2009;129(3–4):181–191. doi: 10.1016/j.vetimm.2008.11.007
- Zu YL, Qi J, Gilchrist A, et al. P38 mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-α or FMLP stimulation. J Immunol. 1998;160(4):1982–9.
- Detmers PA, Zhou D, Polizzi E, et al. Role of stress-activated mitogen-activated protein Kinase (p38) in β2-integrin-dependent neutrophil adhesion and the adhesion-dependent oxidative burst. J Immunol. 1998;161(4):1921–9.
- Forsberg M, Löfgren R, Zheng L, et al. Tumour necrosis factor-α potentiates CR3-induced respiratory burst by activating p38 MAP kinase in human neutrophils. Immunology. 2001;103(4):465–72. doi: 10.1046/j.1365-2567.2001.01270.x
- Behnen M, Leschczyk C, Möller S, et al. Immobilized Immune Complexes Induce Neutrophil Extracellular Trap Release by Human Neutrophil Granulocytes via FcγRIIIB and Mac-1. J Immunol. 2014;193:1954–65 doi: 10.4049/jimmunol.1400478.
- Gakidis MAMG, Cullere X, Olson T, et al. Vav GEFs are required for β2 integrin-dependent functions of neutrophils. J Cell Biol. 2004;166(2):273–82. doi: 10.1083/jcb.200404166
- Lefort CT, Hyun Y-M, Schultz JB, et al. Outside-In signal transmission by conformational changes in Integrin Mac-1. J Immunol. 2009;183:6460–8. doi: 10.4049/jimmunol.0900983
- Hartwig JH, Thelen M, Rosen A, et al. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium–calmodulin. Nature. 1992;356(6370):618–622. doi: 10.1038/356618a0
- Nairn AC, Aderem A. Calmodulin and protein kinase C cross-talk: the MARCKS protein is an actin filament and plasma membrane cross-linking protein regulated by protein kinase C phosphorylation and calmodulin. Ciba Found Symp. 1992;164:145–154; discussion 154–61. doi: 10.1002/9780470514207.ch10
- Ziemba BP, Swisher GH, Masson G, et al. Regulation of a Coupled MARCKS–PI3K Lipid Kinase Circuit by Calmodulin: Single-Molecule Analysis of a Membrane-Bound Signaling Module. Biochem. 2016;55(46):6395–6405. doi: 10.1021/acs.biochem.6b00908
- Glaser M, Wanaski S, Buser CA, et al. Myristoylated Alanine-rich C Kinase Substrate (MARCKS) Produces Reversible Inhibition of Phospholipase C by Sequestering Phosphatidylinositol 4,5-Bisphosphate in Lateral Domains. J Biol Chem. 1996;271(42):26187–93. doi: 10.1074/jbc.271.42.26187
- Singer M, Martin LD, Vargaftig B, et al. A MARCKS-related peptide blocks mucus hypersecretion in a mouse model of asthma. Nat Med. 2004;10(2):193–6. doi: 10.1038/nm983
- Westerman TL, Bogomolnaya L, Andrews-Polymenis HL, et al. The Salmonella type-3 secretion system-1 and flagellar motility influence the neutrophil respiratory burst. PLoS One. 2018;13(9):e0203698. doi: 10.1371/journal.pone.0203698
- Haddock BJ, Zhu Y, Doyle SP, et al. Role of MARCKS in regulated secretion from mast cells and airway goblet cells. Am J Physiol Lung Cell Mol Physiol. 2014;306(10):L925–36. doi: 10.1152/ajplung.00213.2013
- Yang DC, Li J-M, Xu J, et al. Tackling MARCKS-PIP3 circuit attenuates fibroblast activation and fibrosis progression. Faseb J. 2019;33:14354–14369. doi: 10.1096/fj.201901705R
- Yin Q, Fang S, Park J, et al. An Inhaled Inhibitor of Myristoylated Alanine-Rich C Kinase Substrate Reverses LPS-Induced Acute Lung Injury in Mice. Am J Respir Cell Mol Biol. 2016;55:617–622. doi: 10.1165/rcmb.2016-0236RC
- Yue L, Lu S, Garces J, et al. Protein Kinase C-regulated Dynamitin-Macrophage-enriched Myristoylated Alanine-Rice C Kinase Substrate Interaction is Involved in Macrophage Cell Spreading. J Biol Chem. 2000;275:23948–56. doi: 10.1074/jbc.M001845200
- Li J, Zhu Z, Bao Z. Role of MacMARCKS in Integrin-dependent Macrophage Spreading and Tyrosine Phosphorylation of Paxillin. J Biol Chem. 1996;271(22):12985–90. doi: 10.1074/jbc.271.22.12985
- Myat MM, Anderson S, Allen LAH, et al. MARCKS regulates membrane ruffling and cell spreading. Curr Biol. 1997;7(8):611–614. doi: 10.1016/S0960-9822(06)00262-4
- Manenti S, Malecaze F, Darbon JM. The major myristoylated PKC substrate (MARCKS) is involved in cell spreading, tyrosine phosphorylation of paxillin, and focal contact formation. FEBS Lett. 1997;419(1):95–98. doi: 10.1016/S0014-5793(97)01438-5
- Tang T, Rosenkranz A, Assmann KJM, et al. A Role for Mac-1 (CDIIb/CD18) in Immune Complex–stimulated Neutrophil Function in Vivo: Mac-1 Deficiency Abrogates Sustained Fcγ Receptor–dependent Neutrophil Adhesion and Complement-dependent Proteinuria in Acute Glomerulonephritis. J Exp Med. 1997;186(11):1853–1863. doi: 10.1084/jem.186.11.1853
- Zuchtriegel G, Uhl B, Pick R, et al. Vitronectin stabilizes intravascular adhesion of neutrophils by coordinating β2 integrin clustering. Haematologica. 2021;106(10):2641–2653. doi: 10.3324/haematol.2019.226241
- Maheshwari G, Brown G, Lauffenburger DA, et al. Cell adhesion and motility depend on nanoscale RGD clustering. J Cell Sci. 2000;113(10):1677–1686. doi: 10.1242/jcs.113.10.1677
- Zhou M-J, Brown EJ. CR3 (Mac-1, alpha M beta 2, CD11b/CD18) and Fc gamma RIII cooperate in generation of a neutrophil respiratory burst: requirement for Fc gamma RIII and tyrosine phosphorylation. J Cell Bio. 1994;125(6):1407. doi: 10.1083/jcb.125.6.1407
- Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity. 1996;5(6):653–666. doi: 10.1016/S1074-7613(00)80278-2
- Jones SL, Sharief Y, Chilcoat CD. Signaling mechanism for equine neutrophil activation by immune complexes. Vet Immunol Immunopathol. 2001;82(1–2):87–100. doi: 10.1016/S0165-2427(01)00350-6
- Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for β2 integrin–mediated leukocyte adhesion to endothelial cells. Nat Med. 2009;15(3):300–305. doi: 10.1038/nm.1921
- Graham IL, Lefkowith JB, Anderson DC, et al. Immune complex-stimulated neutrophil LTB4 production is dependent on beta 2 integrins. J Cell Biol. 1993;120(6):1509–1517. doi: 10.1083/jcb.120.6.1509
- Herlaar E, Brown Z. P38 MAPK signalling cascades in inflammatory disease. Mol Med Today. 1999;5(10):439–447. doi: 10.1016/S1357-4310(99)01544-0
- Lomakina EB, Waugh RE. Signaling and dynamics of activation of LFA-1 and Mac-1 by immobilized IL-8. Cell Mol Bioeng. 2010;3(2):106–116. doi: 10.1007/s12195-009-0099-x
- Lee SM, Suk K, Lee WH. Myristoylated alanine-rich C kinase substrate (MARCKS) regulates the expression of proinflammatory cytokines in macrophages through activation of p38/JNK MAPK and NF-κB. Cell Immunol. 2015;296(2):115–121. doi: 10.1016/j.cellimm.2015.04.004
- Jakus Z, Berton G, Ligeti E, et al. Responses of Neutrophils to Anti-Integrin Antibodies Depends on Costimulation through Low Affinity FcγRs: Full Activation Requires Both Integrin and Nonintegrin Signals. J Immunol. 2004;173(3):2068–77. doi: 10.4049/jimmunol.173.3.2068
- Tandon R, Sha’afi RI, Thrall RS. Neutrophil β2-integrin upregulation is blocked by a p38 MAP kinase inhibitor. Biochem Biophys Res Commun. 2000;270(3):858–862. doi: 10.1006/bbrc.2000.2540
- Bouaouina M, Blouin E, Halbwachs-Mecarelli L, et al. TNF-Induced β 2 Integrin activation Involves Src Kinases and a redox-regulated activation of p38 MAPK. J Immunol. 2004;173(2):1313–1320. doi: 10.4049/jimmunol.173.2.1313
- Guidetti GF, Canobbio I, Torti M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv Biol Regul [Internet]. 2015;59:36–52. Available from. doi: 10.1016/j.jbior.2015.06.001
- Laux T, Fukami K, Thelen M, et al. GAP43, MARCKS, and CAP23 Modulate PI(4,5)P 2 at Plasmalemmal Rafts, and Regulate Cell Cortex Actin Dynamics through a Common Mechanism. J Cell Biol. 2000;149(7):1455–1472. doi: 10.1083/jcb.149.7.1455
- Botelho RJ, Teruel M, Dierckman R, et al. Localized Biphasic Changes in Phosphatidylinositol-4,5-Bisphosphate at Sites of Phagocytosis. J Cell Bio. 2000;151(7):1353–1368. doi: 10.1083/jcb.151.7.1353
- Martel V, Racaud-Sultan C, Dupe S, et al. Conformation, localization, and integrin binding of talin depend on its interaction with Phosphoinositides. J Biol Chem. 2001;276:21217–21227. doi: 10.1074/jbc.M102373200
- Dietrich U, Krüger P, Gutberlet T, et al. Interaction of the MARCKS peptide with PIP2 in phospholipid monolayers. Biochim Biophys Acta. 2009;1788(7):1474–1481. doi: 10.1016/j.bbamem.2009.04.001
- Honda A, Nogami M, Yokozeki T, et al. Phosphatidylinositol 4-Phosphate 5-Kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 1999;99(5):521–532. doi: 10.1016/S0092-8674(00)81540-8
- Ziemba BP, Falke JJ, Obukhov AG. A PKC-MARCKS-PI3K regulatory module links Ca 2+ and PIP 3 signals at the leading edge of polarized macrophages. PLoS One. 2018;13(5):e0196678. doi: 10.1371/journal.pone.0196678
- Myat M, Anderson S, Allen L-A, et al. MARCKS regulates membrane ruffling and cell spreading. Curr Biol. 1997;7(8):611–614. doi: 10.1016/S0960-9822(06)00262-4
- Sheats MK, Sung EJ, Adler KB, et al. In Vitro Neutrophil Migration Requires Protein Kinase C-Delta (δ-PKC)-Mediated Myristoylated Alanine-Rich C-Kinase Substrate (MARCKS) Phosphorylation. Inflammation. 2015;38(3):1126–1141. doi: 10.1007/s10753-014-0078-9