
ABSTRACT
The aim of this study was to investigate how the concentration of interleukin-13 (IL-13) affects the regulation of endothelial cell migration after injury. The incubation of recombinant human interleukin-13 (rhIL-13) strongly increased the content of reactive oxygen species (ROS) in HUVECs via the JAK-1/STAT-3/NOX-4 signaling pathway. Antagonizing the high intracellular ROS that was induced by rhIL-13 promoted the migration of HUVECs. Furthermore, IL-13 neutralization not only inhibited intimal hyperplasia, but also promoted the migration of endothelial cells (ECs) after injury. The results suggest that IL-13 inhibition is a potential means of stimulating endothelial cells recovery after injury. Therefore, the attenuation of IL-13 activation may have therapeutic value for vascular disease.
GRAPHICAL ABSTRACT
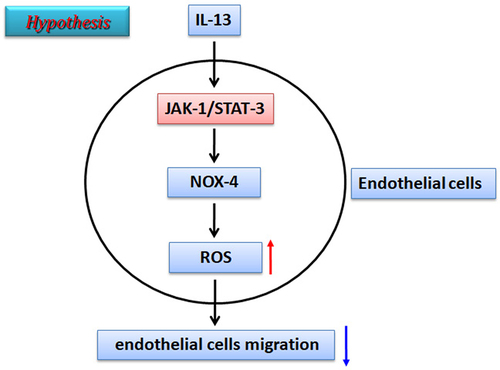
Introduction
Intimal hyperplasia (IH) is most commonly observed in vascular diseases [Citation1]. Studies indicate that IH induction is most commonly associated with mechanical trauma and high shear stress after injury [Citation2]. Research also indicates that many different cell types are linked to the pathogenesis of intimal hyperplasia [Citation3]. However, the underlying mechanism is still unknown.
Endothelial cells (ECs) compose the first solid layer between the blood and tissue, and perform diverse functions, such as modulating inflammation, controlling oxygen/nutrient supply and regulating coagulation [Citation4]. Studies have demonstrated that ECs remain quiescent under normal conditions, but acquire the ability to proliferation under pathological conditions [Citation5]. Evidences shows that this transition initially involves the damage of endothelial cells layer upon vascular injury, and this loss of ECs often subsequently leads to clot formation and immunocyte adhesion [Citation6,Citation7]. Hence, the re-endothelialization process that takes place after vascular injury may play a pivotal role in the recovery of vascular function [Citation8].
Interleukin-13 (IL-13) is a well-known anti-inflammatory cytokine produced by Th2 cells, mast cells and macrophages [Citation9]. Studies indicate that two kinds of IL-13 receptors exist: namely, the IL-13 receptor (IL-13 R) α1 subunit and the IL-13 receptor (IL-13 R) α2 subunit. Interestingly, IL-13 Rα2 is regarded as a decoy receptor for IL-13 [Citation10]. Based on existing knowledge, IL-13 is excreted as a result of Th2 cells activation. IL-13 signaling pathway activation is important in the process of tissue remodeling [Citation11]. Studies indicate that the JAK/STAT signaling pathway is the main downstream regulator of IL-13 [Citation12]. Further evidence strongly indicates that the persistent activation of JAK/STAT signaling induced by IL-13 is closely related to numerous diseases [Citation13].
A growing body of evidence indicates that immunity and inflammation play a major role in vascular disease, yet the mechanism remains unclear. Like IL-4, IL-13 is implicated allergic response [Citation14]. Additionally, IL-13 has the ability to promote smooth muscle cell contraction [Citation15], and plays a major role in fibro-proliferative disorders [Citation16]. Nevertheless, the role of IL-13 in intimal hyperplasia is unclear. The aim of this study was to therefore determine the role of IL-13 in the regulation of ECs migration after balloon injury. To provide a novel target for the treatment of intimal hyperplasia after stenting.
Materials and methods
Carotid balloon injury model
Male Sprague-Dawley rats weighing 280 to 300 g (total 50 rats were used and obtained from the Animal center of the second Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang Province, China). Rats were maintained in groups of five animals per cage on a 12 h light and 12 h dark circadian rhythm (23 ± 1°C and 55 ± 5% humidity) with water and food. The experimental protocol was designed in accordance with Institutional Laboratory Animal Care and Use Committee (ILACUC) standards. And all experimental procedures performed in studies involving animals participants were in accordance with the Institutional Animal Care and Use Committee of Harbin Medical University Cancer Hospital (Harbin, China; approval no. KY2016–16).
Animals were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg, i.p.). The left common carotid artery was exposed, and 2F Fogarty balloon embolectomy catheter (Edwards Lifesciences, Irvine, CA) was inserted via an external carotid arteriotomy incision. The catheter was advanced to the aortic arch, then inflated with 0.2 mL of air, and drawn back to the arteriotomy three times (). After the catheter was withdrawn, the proximal of the external carotid artery was ligated, and blood flow was restored. The surgical incision was closed, and the rats were allowed to recover from anesthesia. The right common artery was used as control tissue.
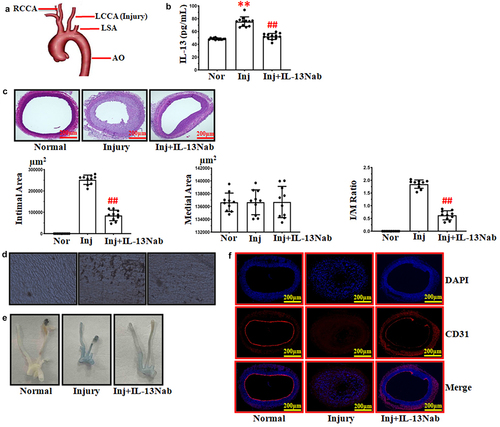
Figure 1. Neutralizing IL-13 stimulated the recovery of ECs after balloon injury. (a) schematic summary of the balloon injury model. (b) ELISA analysis of IL-13 serum level. **p < .01 vs. normal; ##p < .01 vs. injury. (c) treatment with IL-13Nab (30 ng) inhibited intimal hyperplasia following balloon injury after 14 days. ##p < .01 vs. injury. Scale bars: 200 μm. (d-e) silver andEvans blue staining to detect the recovery of endothelial cells after balloon injury. (f) immunofluorescence staining for CD31 (red) and DAPI (blue), scale bars: 200 μm. The results were derived from 3 independent experiments each performed in duplicate. Normal, nor. Injury, Inj. Neutralizing IL-13, IL-13Nab.
Rat neutralizing interleukin-13 antibody (IL-13Nab, 30 ng once a daily, R&D system, Cat#AF1945) or saline (injury group) treatment was begun 1 day after surgery by intraperitoneal injection, then continued for 14 days after surgery.
Histological evaluation
14 days after operation, rats were euthanized by chloralhydrate overdose. Carotid arteries were removed and harvest. Tissues were then embedded in paraffin, and four to five sections (5 μm) were cut at multiple levels. These sections were then stained with hematoxylin-eosin (H&E). Sections were examined microscopically, and the cross-sectional areas of the lumen, neointima and media were determined using Image J software. The intima to media ratio was then calculated from the determined mean. The data represent the mean ± SD.
Silver staining
After flushing the artery with PBS (pH 7.4), the vessel was perfused with silver nitrate (50 mL, 0.25%) for 30 seconds, then followed by a second PBS flush. The arteries were then perfusion fixed using 2% glutaraldehyde in phosphate buffer (pH 7.4) for 25 minutes. The left common carotid artery was excised and cut into four to five pieces. The artery was opened longitudinally and pinned, endothelium side up. Arteries were dehydrated in an alcohol series, incubated in xylene twice for 10 minutes, and mounted on glass slides under glass coverslips. Sections were examined microscopically
Evans blue staining
Briefly, mice was injected with 150 μl solution (5% Evans blue diluted in saline) through the tail vein 10 min before euthanasia and the perfusion was performed to fix inner wall of the carotid artery as following: the chest was opened immediately after death, and left ventricle of heart was punctured with a 25-gauge cannula for the infusion of saline for 10 min and then 4% phosphate-buffered formalin (pH 7.0) for 5 min under physiological pressure. Blood, saline, and fixative were removed through an incision in the right atrium. The left common carotid artery was dissected from the aortic arch to carotid bifurcation and the images of stained artery were got by using scanner.
Immunofluorescence
The sections were incubated with the primary antibody in 4°C by Rat anti-CD31 (CD31, 1:200; Cat#GB11063–2; Servicebio, China). The sections were washed three times with PBS. Then, sections were incubated with the FITC-labeled secondary antibody for 1 h at 37°C. The sections were then washed three times with PBS, followed by a 2-min incubation with DAPI (1 µg/µl in 1% BSA in PBS) at room temperature. Covers-lips were mounted with the washed sections and anti-fluorescence quenching sealant was applied. Images were acquired using an Olympus I×81upright fluorescence microscope and analyzed via Olympus CellSens Standard Software (Olympus Corporation).
F-actin cytoskeleton staining analysis
F-actin staining was performed according to the manufacturer’s instructions. Fixed cells were incubated with 100 nM working stock of Acti-stain 488 phalloidin (Cytoskeleton). Cells were counterstained with DAPI (Sigma) and imaged with a confocal laser-scanning microscope (Olympus FV1000, Japan).
ELISA
Blood samples were collected in anticoagulant-free tubes in model and neutralizing interleukin-13 groups. Plasma was separated by centrifugation at 3000 rpm for 10 minutes and was stored at −20°C for a maximum period of one month as per the manufacturer’s stability and storage instructions for the IL-13 ELISA kits (Cat#SP13766, Spbio, China). Briefly, the wells were covered with an adhesive strip and incubated for 2 h at room temperature on a horizontal orbital microplate shaker. Each well was washed three times with wash buffer. A total of 200 μL IL-13 conjugate was added to each well, covered with a fresh adhesive strip, and incubated for 2 h at room temperature on the shaker. The washing steps were repeated, then 200 μL substrate solution was added to each well and incubated for 30 min at room temperature in the dark. Stop solution (50 μL) was added to each well and the optical density of each well was determined within 30 min using an Infinite 200PRO microplate spectrophotometer (Tecan, Salzburg, Austria) set at 450 nm. All samples were run in duplicate.
Cultured cell
Human umbilical vein endothelial cells (HUVECs, obtained from American Type Culture Collection, ATCC) were maintained in RPMI-1640 medium (HyClone, UT, USA) supplemented with 10% fetal bovine serum (Gibco, CA, USA), 100 U/ml penicillin, and 100 mg/ml streptomycin (Gibco, CA, USA). HUVECs were cultured at 37°C in 5% CO2 environment. Cultured medium was renewed after 48 h and cells were further cultured for 24 h. When the cells reached 90% confluence, they were incubated with recombinant human interleukin-13 (rhIL-13, Cat#213-ILB, R&D Systems, China), recombinant human IL-13Ra1 Fc (Cat#146-IR, R&D Systems, China), Stattic (Cat#S7024, Selleck, China), Ruxolitinib (Cat#S5243, Selleck, China).
Measurement of cell viability
HUVECs were seeded into 96-well plates (5 × 10 [Citation5] cells/well) and incubated at 37°C for 24 h. On reaching 90% confluence, the cells were pre-treated with different concentrations of rhIL-13 for 24 h. Subsequently, 20 μL of MTT (pH 4.7) was added to each well, and the cells were incubated for another 4 h. Then, 100 μL of 10% sodium dodecyl sulfate (SDS) 0.01 M HCL was added, and the cells were incubated at 37°C overnight to dissolve the formazan crystals. Absorbance was measured at 570 nm.
EdU staining
Briefly, 2 × 10 [Citation3] cells/well was seeded into 96-well plates. Then, 10 nM EdU was added for 12 h. The wells were washed with PBS, and the cells were fixed in 4% paraformaldehyde and stained according to the manufacturer’s instructions. Cell proliferation was observed under a fluorescence microscope (Leica DMI6000B) and detected by flow cytometry (FACSCalibur, Becton Dickinson).
For flow cytometry, the dot plots in quadrants quantified the percentage of cells. The analysis was performed on a FACS Caliburflocytometer (BD Biosciences). All data were analyzed using FlowJo 10.8.1 (Tree Star Inc. Ashland, Oregon). All procedures were performed on ice until analysis.
Cell transfection
For siRNA transfection, HUVECs were plated at 3 × 10 [Citation5] cells/mL in OPTI-MEM serum-free medium and transfected of siRNA duplex using Lipofectamine RNAiMAX Reagent Agent (Life Technologies) according to the manufacturer’s instructions. The siRNA sequences are as described as follow: human NOX-4: GAAUUACAGUGAAGACUUU.
Reactive oxygen species (ROS) assay
HUVECs (5 × 10 [Citation3] cells/well in 96 well plates) were cultured in RPMI-1640 medium (10% FBS, 1% antibiotics). Cells were then treated with rhIL-13 and control group was set as blank. Intracellular ROS level was measured by 2’, 7’-dichlorofluorescein diacetate (DCFH, Cat#CA1410, Solarbio LIFE SCIENCE, China), which can be oxidized into fluorescent dichlorodihydrofluorescein (DCF) according to the manufacture. After fixing, the cells were washed in 1×PBS and then incubated in the dark for 30 min with 10 μM DCFH-DA. The DCF fluorescence was detected by fluorescence microscopy and flow cytometry (with an excitation of 485 nm and an emission of 520 nm).
Western blot
Briefly, frozen tissue was immerged into in 600 μL lysis buffer which contained 1% protease inhibitor solution. Then centrifuged at 13,500 r/min for 30 mins. Collected supernatant and stored at −80°C. Cells were washed with ice-cold PBS and centrifuged at 3000 r/min for 10 min. And then extracted in 70 µL of lysis buffer which contained 1% protease inhibitor solution, after pipetted 30 min on ice then centrifuged at 13,500 r/min for 30 mins, Collected supernatant and stored at −80°C. The protein content was determined with BCA Protein Assay Kit (Bio-Rad, Mississauga, ON, Canada). The samples were subjected to electrophoresed in 10% SDS-PAGE and separated proteins were transferred to nitrocellulose membrane. The membrane was blocked in 5% nonfat milk overnight at 4°C. Then incubated with the primary antibodies against p-JAK-1 (Cat#GB114584, Servicebio, China), p-STAT-3 (Cat#381552, ZenBio, China), NOX-4 (Cat#380874, ZenBio, China) and GAPDH (Cat#GB15002, Servicebio, China). After washing PBS-0.1% Tween-20 (PBST), membranes were incubated with fluorescence-conjugated goat anti-rabbit IgG secondary antibody (dilution 1:10000) which purchased from LI-COR Biosciences (Cat#926–32211) at room temperature. Then washing with PBS-0.1% Tween-20 (PBST). Western blot bands were quantified using Odyssey v3.0 software (LI-COR Bioscience) by measuring the densitometry for each group.
Cell migration assay
For the transwell assay, cell migration assays were performed using 6.5 mm diameter and 8.0 µm pore size transwells (Costar) coated with 0.5% gelatin. The lower chamber contained 15% serum as a chemoattractant. HUVECs were prepared in serum-free medium, and 5 × 10 [Citation4] cells were added to the upper chamber in migration buffer (M199 containing 0.1% BSA). After 4 h of incubation at 37°C, cells were removed from the upper surface of the membranes with a cotton swab, and cells that migrated to the lower surface were fixed with 4% paraformaldehyde for 30 mins and then stained with 0.1% crystal violet for 10 mins. Migrated cells were then counted under a microscope.
Scratch wound assay
For the scratch wound assay, 3 × 10 [Citation5] cells/well were plated into a 12-well plate and grown to confluence. The monolayer was scratched using a pipette tip and washed with serum-free medium to remove detached cells. HUVECs were photographed at 0 h, 12 h and 24 h after wounding.
Statistics
Obtained data were expressed as mean ± standard deviation. Data were assessed with SPSS 22.0 evaluation version (SPSS, Chicago, IL, USA) statistical programme with one-way analysis of variance, post hoc; and analyzed with Bonferroni test for multiple comparisons. P < .05 was considered significant.
Results
Neutralizing serum IL-13 promoted the migration of endothelial cells after balloon injury
In our experiments, we found that the serum content of IL-13 was increased after injury (). Treatment with neutralizing IL-13 antibody attenuated the high level of serum IL-13 that accompanied the suppression of intimal hyperplasia. In histological sections obtained from rats administered neutralizing IL-13 antibody, both intimal area and I/M ratio were reduced after 14 days when compared to the injury group. Treatment with IL-13Nab did not affect the medium area in either group ().
The loss of ECs after balloon injury occurs in the early phase of the intimal hyperplasia process.
Hence, introducing an endothelial cell layer is considered an optimal solution for the prevention of intimal hyperplasia. In our experiments, by employing silver staining, we observed that ECs of the left common carotid artery in normal groups were long and narrow, with their longitudinal axis parallel to the direction of blood flow (). After balloon injury, most ECs at the surface of the left common carotid artery had disappeared and the luminal surface was covered with platelets (). However, treatment with IL-13Nab strongly promoted ECs recovery on the surface of left common carotid artery (). According to our in vivo results, we speculated that neutralizing the high level of IL-13 after balloon injury might confer the benefit of reducing intimal hyperplasia via stimulating the recovery of ECs.
Recombinant human IL-13 (rhIL-13) inhibited HUVECs migration
Exposure to recombinant human IL-13 (rhIL-13) in concentration ranging from 25 to 100 ng/mL for 24 h was not cytotoxic with or without serum (). Therefore, all subsequent in vitro experiments were performed under serum-free conditions, which allowed for the use of rhIL-13 (100 ng/mL) [Citation17]. We detected that treatment with rhIL-13 did not promote the proliferation ().
Figure 2. rhIL-13 inhibited HUVECs migration. (a) rhIL-13 (from 12.5 to 100 ng/mL) did not affect the viability of cultured HUVECs in either the presence or absence of serum after 24 h, as measured by MTT. ***p < .001 vs. normal. (b-c) rhIL-13 (100 ng/mL) did not stimulate the proliferation of HUVECs. Scale bars: 200 μm. (d) the co-application of IL-13 Rα1 Fc (4 μg/mL) and rhIL-13 inhibited the rhIL-13 induced inhibition of cell migration in cultured HUVECs. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. Scale bars: 200 μm. The results were derived from 3 independent experiments each performed in duplicate. Data represent the mean±SD. Control, Ctrl.
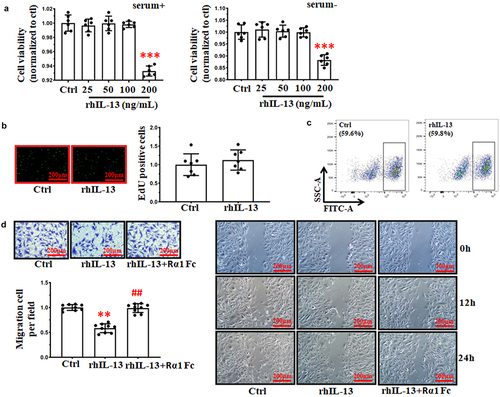
Next, we used transwell and scratch wound assay to detect whether rhIL-13 inhibited the migration of HUVECs in vitro. Compared to the control group, the average number of migrating HUVECs significantly decreased after rhIL-13 treatment. Co-application of rhIL-13 and IL-13 Rα1 Fc (4 μg/mL) reversed the attenuation of migration induced by rhIL-13 ().
rhIL-13 inhibited HUVECs migration through JAK-1/STAT-3 mediated ROS accumulation
It is well known that Janus kinase-1 (JAK-1)/signal transducer and activator of transcription 3 (STAT-3) is an important downstream factor of IL-13. Hence, we questioned whether rhIL-13 inhibited the migration of HUVECs through the JAK-1/STAT-3 signaling pathway.
In our experiments, incubation of rhIL-13 promoted the activation of the JAK-1/STAT-3 signaling pathway (). Antagonizing the activation of phospho-JAK-1 by the addition of ruxolitinib (5 μM) [Citation18] reversed the high protein level of phospho-STAT-3 induced by rhIL-13 (). However, incubation with rhIL-13 did not affect the expression levels of either total-STAT-3 and total- JAK-1 ().
Figure 3. rhIL-13 inhibited HUVECs migration via the JAK-1/STAT-3 signaling pathway. (a) rhIL-13 regulated the activation of the JAK-1/STAT-3 signaling pathway. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. (b) the co-application of stattic and rhIL-13 inhibited the rhIL-13 induced inhibition of cell migration in cultured HUVECs. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. Scale bars: 200 μm. (c) STAT-3 inhibition eliminated the accumulation of ROS induced by rhIL-13. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. Scale bars: 200 μm. The results were derived from 3 independent experiments each performed in duplicate. Data represent the mean±SD. Control, Ctrl. Negative control, NC. Ruxolitinib, Rux. Stattic, Stt.
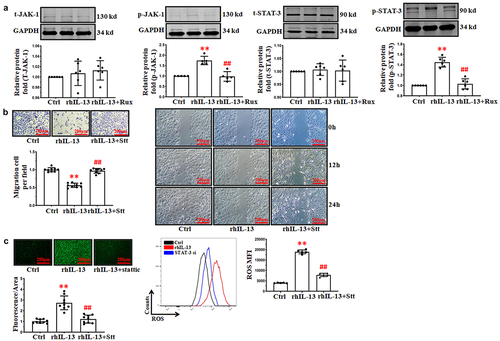
These results indicated that treatment with rhIL-13 promoted the activation of the JAK-1/STAT-3 signaling pathway. However, it remains unknown whether the activation of the JAK-1/STAT-3 signaling pathway plays an important role in the inhibition of HUVECs migration induced by rhIL-13. As shown in , we observed that co-application stattic (3 μM, the small molecule inhibitor of STAT-3) and rhIL-13 strongly antagonized the inhibition of HUVECs migration induced by rhIL-13 ().
Our previous study confirmed that the formation of reactive oxygen species (ROS) impeded HUVECs migration [Citation19]. According to our previous work, we hypothesized that oxidative stress may be a result of rhIL-13 inhibiting the migration of HUVECs via JAK-1/STAT-3 signaling pathway. As shown in , incubation with rhIL-13 induced ROS accumulation in HUVECs by DCFH-DA staining. By contrast, co-application of rhIL-13 and stattic attenuated rhIL-13 from increasing level of intracellular ROS in HUVECs.
rhIL-13 stimulated the accumulation of ROS via NOX-4 in HUVECs
NADPH oxidase-4 (NOX-4) has the ability to regulate oxidant species and maintain the redox balance of cells. Some research groups have reported that the expression of NOX4 is high in endothelial cells and may prevent endothelial dysfunction [Citation20]. Hence, we speculated that IL-13 stimulates the accumulation of ROS via NOX-4. Our results indicated that rhIL-13 increased the level of NOX-4, while the co-application of stattic and rhIL-13 attenuated the high protein level of NOX-4 induced by rhIL-13 used in isolation ().
Figure 4. rhIL-13 stimulated ROS accumulation via the NOX-4. (a) rhIL-13 stimulated the expression of NOX-4. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. (b) transfection efficiency of NOX-4 siRNA in cultured HUVECs. **p < .01 vs. Ctrl. (c) NOX-4 inhibition eliminated the accumulation of ROS induced by rhIL-13. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. Scale bars: 200 μm. (d) the co-application of NOX-4 siRNA and rhIL-13 inhibited the rhIL-13 induced inhibition of cell migration in cultured HUVECs. **p < .01 vs. Ctrl; ##p < .01 vs. rhIL-13. Scale bars: 200 μm. (e) F-actin staining after treatment with rhIL-13. Scale bars: 200 μm. The results were derived from 3 independent experiments each performed in duplicate. Data represent the mean±SD. Control, Ctrl. Negative control, NC.
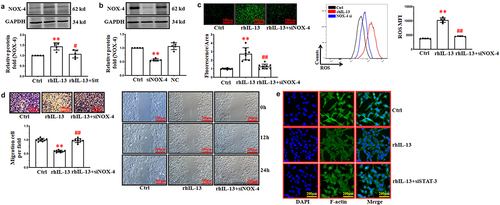
In our experiments, co-application of NOX-4 siRNA and rhIL-13 eliminated the high content of ROS induced by rhIL-13 (). The transfection of NOX-4 siRNA antagonized the inhibition of HUVECs migration induced by rhIL-13 ().
Alteration of cytoskeleton are associated with cell migration. Our previous work indicated that a high intracellular ROS content affects the formation of F-actin (the biomarker of cytoskeleton). We observed that treatment with rhIL-13 strongly decreased the formation of F-actin in HUVECs. Interestingly, co-application of NOX-4 siRNA and rhIL-13 reversed the low intracellular level of F-actin induced when rhIL-13 was applied in isolation ().
Discussion
A growing body of evidence supports the role of the IL-13 in numerous diseases [Citation21]. Based on current study, we provide strong evidence that antagonizing IL-13 significantly attenuates the recovery of endothelial cells after injury. These findings support the notion that the detrimental effect on endothelial cells recovery after injury that is caused by elevated IL-13 in the serum could be attributed to ROS formation mediated by STAT-3/NOX-4.
IL-13 is one of the key effectors in the process of disease induced by Th2 activation [Citation22]. Studies indicate that IL-13 also directly acts on other kinds of cells to regulate the cell cycle and other functions [Citation23]. In our experiments, we observed that a high level of IL-13 in the culture medium could inhibit ECs migration. The neutralization of this high serum level of IL-13 promoted the ECs migration after injury in our balloon injury model. According to these results, we have strongly demonstrated that a high level of IL-13 May be a major factor involved in the process of ECs recovery during wound healing. In our experiments, we only antagonized the function of the IL-13 receptor a1-subunit (IL-13Ra1), but not that of IL-13Ra2. It is worth noting that the IL-13Ra2 chain is closely related to IL-13 Rα1. However, IL-13Ra2 lacks a cytoplasmic domain. And hence, IL-13Ra2 does not function as a signal mediator [Citation24]. Previous research also provide strong evidence that IL-13Ra2 May function as a decoy receptor of IL-13, but not of IL-4 [Citation25]. This is why in our studies we have only investigated the IL-13/IL-13Ra1 pathway. In our experiments, we found that a low level of IL-13 (100 ng/mL) suppressed the HUVECs migration, but stimulated their apoptosis at high level. This result hints that IL-13 (especially in our in vivo experiments) may yield opposite effects in a concentration-dependent manner. Hence, further studies should be conducted to detect the underlying mechanisms of IL-13 at different concentration.
Oxidative stress is a significant contributor to various pathophysiological processes. Intracellular oxidative stress that is caused by increased ROS formation is associated with abnormal cellular function [Citation26,Citation27]. An interesting observation from our work herein was that antagonizing the IL-13/IL-13Ra1 pathway suppressed the accumulation of ROS. Based on the present study, level of ROS in ECs induced by rhIL-13 switched off signaling pathway transduction, which contributes to the cells migration. Studies have confirmed the major role of ROS in mediating different cellular processes, which expands the effect of ROS beyond their excess simply being harmful to normal cellular function. Due to this, the intracellular production of ROS is controlled by numerous enzymes [Citation28,Citation29]. In our study, the excess accumulation of ROS induced by rhIL-13 in HUVECs was associated with the expression of NOX-4 [Citation30,Citation31]. We observed that incubation with rhIL-13 promoted the production of ROS in HUVECs. According to our previously work, when the level of ROS results in the abnormal formation of the cytoskeleton, this has the ability to impede the migration of cells. Our in vitro results strongly support that IL-13 mediates the production of ROS to play a pivotal role in e cytoskeleton formation. Broadly speaking, besides activating the immune response, we speculate that IL-13 originating from immunocytes may also have the ability to regulate cytoskeleton formation. Our results also suggest that targeting cytoskeleton may be a useful strategy for developing novel therapeutics to support ECs recovery after vascular injury [Citation32].
The JAK/STAT pathway is stimulated by cytokines. To our knowledge, Janus kinase 1 (JAK-1)/signal transducer and activator of transcription (STAT) is the main down-regulating pathway of IL-13 [Citation33]. The activation of the JAK/STAT pathway induces an extracellular signal into a transcriptional response [Citation34]. In our experiments, we observed that the activation of STAT-3 had the effect of inducing the expression of NOX-4 after incubation rhIL-13, which then lead to the increased production of ROS in HUVECs.
In conclusion, the data presented herein strongly suggest that a high level of IL-13 suppresses endothelial cells migration by inducing ROS formation via the activation of STAT-3/NOX-4 signaling pathway. Furthermore, our experiments provide a new therapeutic strategy to stimulate the recovery of endothelial cells after injury. However, further questions are raised for future investigations to address: 1) IL-4 (also originating from Th2 cells) is closely related to IL-13. Further studies will need to determine whether IL-4 stimulates the production of ROS via the same signaling pathway. 2) It remains unknown whether IL-13 regulates the production of ROS in ECs only via NOX-4. As we know, NOX-1 and NOX-2 also play important roles in oxidative stress signaling. Addressing these questions will improve upon our current understanding and will help lay a more solid foundation on which to develop future therapeutic strategies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request
Additional information
Funding
References
- Jeong Y, Yao Y, Yim EKF. Current understanding of intimal hyperplasia and effect of compliance in synthetic small diameter vascular grafts. Biomater Sci. 2020;8(16):4383–4395. doi: 10.1039/d0bm0026g
- Liu B, Yan W, Luo L, et al. Macrophage membrane camouflaged reactive oxygen species responsive nanomedicine for efficiently inhibiting the vascular intimal hyperplasia. J Nanobiotechnology. 2021;19(1):374. doi: 10.1186/s12951-021-01119-5
- Melnik T, Jordan O, Corpataux JM, et al. Pharmacological prevention of intimal hyperplasia: a state-of-the-art review. Pharmacol Ther. 2022;235:108157. doi: 10.1016/j.pharmthera.2022.108157
- Krüger-Genge A, Blocki A, Franke RP, et al. Vascular endothelial cell biology: an update. Int J Mol Sci. 2019;20(18):4411. doi: 10.3390/ijms20184411
- Lee HW, Xu Y, He L, et al. Role of venous endothelial cells in developmental and pathologic angiogenesis. Circulation. 2021;144(16):1308–1322. doi: 10.1161/CIRCULATIONAHA.121.054071
- Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. 2018;16(2):231–241. doi: 10.1111/jth.13911
- Hao X, Gai W, Ji F, et al. Bovine serum albumin-based biomimetic gene complexes with specificity facilitate rapid re-endothelialization for anti-restenosis. Acta Biomater. 2022;142:221–241. doi: 10.1016/j.actbio.2022.02.005
- Lee CH, Hsieh M-J, Chang S-H, et al. Nanofibrous vildagliptin-eluting stents enhance re-endothelialization and reduce neointimal formation in diabetes: in vitro and in vivo. Int J Nanomedicine. 14 (2019) 7503–7513. 10.2147/IJN.S211898.
- Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. 2018;9:888. doi: 10.3389/fimmu.2018.00888
- Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–690. doi: 10.1067/mai.2003.1333
- Sicklinger F, Meyer IS, Li X, et al. Basophils balance healing after myocardial infarction via IL-4/IL-13. J Clin Invest. 2021;131(13):e136778. doi: 10.1172/JCI136778
- Shankar A, McAlees JW, Lewkowich IP. Modulation of IL-4/IL-13 cytokine signaling in the context of allergic disease. J Allergy Clin Immunol. 2022;150(2):266–276. doi: 10.1016/j.jaci.2022.06.012
- Roger I, Milara J, Montero P, et al. The role of JAK/STAT molecular pathway in vascular remodeling associated with pulmonary hypertension. Int J Mol Sci. 2021;22(9):4980. doi: 10.3390/ijms22094980
- Athari SS. Targeting cell signaling in allergic asthma. Signal Transduct Target Ther. 2019;4:45. doi: 10.1038/s41392-019-0079-0
- Wu Y, Huang Y, Zhang W, et al. The proprotein convertase furin inhibits IL-13-induced inflammation in airway smooth muscle by regulating integrin-associated signaling complexes. Am J Physiol Lung Cell Mol Physiol. 2021;321(1):L102–L115. doi: 10.1152/ajplung.00618.2020
- Nguyen JK, Austin E, Huang A, et al. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res. 2020;312(2):81–92. doi: 10.1007/s00403-019-01972-3
- Manson ML, Säfholm J, James A, et al. IL-13 and IL-4, but not IL-5 nor IL-17A, induce hyperresponsiveness in isolated human small airways. J Allergy Clin Immunol. 2020;145(3):808–817.e2. doi: 10.1016/j.jaci.2019.10.037
- Febvre-James A, Lecureur V, Augagneur Y, et al. Repression of interferon β-regulated cytokines by the JAK1/2 inhibitor ruxolitinib in inflammatory human macrophages. Int Immunopharmacol. 2018;54:354–365. doi: 10.1016/j.intimp.2017.11.032
- Li Q, Zhang M, Xuan L, et al. Anagliptin inhibits neointimal hyperplasia after balloon injury via endothelial cell-specific modulation of SOD-1/RhoA/JNK signaling in the arterial wall. Free Radic Biol Med. 2018;121:105–116. doi: 10.1016/j.freeradbiomed.2018.04.580
- Langbein H, Brunssen C, Hofmann A, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J. 2016;37(22):1753–1761. doi: 10.1093/eurheartj/ehv564
- Gandhi NA, Bennett BL, Graham NMH, et al. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. 2016;15(1):35–50. doi: 10.1038/nrd4624
- Ranasinghe C, Trivedi S, Wijesundara DK, et al. IL-4 and IL-13 receptors: roles in immunity and powerful vaccine adjuvants. Cytokine Growth Factor Rev. 2014;25(4):437–442. doi: 10.1016/j.cytogfr.2014.07.010
- Kasaian MT, Miller DK. IL-13 as a therapeutic target for respiratory disease. Biochem Pharmacol. 2008;76:147–155. doi: 10.1016/j.bcp.2008.04.002
- Fichtner-Feigl S, Strober W, Kawakami K, et al. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 pro-duction and fibrosis. Nat Med. 2006;12(1):99–106. doi: 10.1038/nm1332
- Chung KF. Targeting the interleukin pathway in the treatment of asthma. Lancet. 2015;386:1086–1096. doi: 10.1016/S0140-6736(15)00157-9
- Mittler R. ROS are good. Trends Plant Sci. 2017;22(1):11–19. doi: 10.1016/j.tplants.2016.08.002
- Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–27. doi: 10.1016/S0167-0115(00)00136-1
- Galasso M, Gambino S, Romanelli MG, et al. Browsing the oldest antioxidant enzyme: catalase and its multiple regulation in cancer. Free Radic Biol Med. 2021;172:264–272. doi: 10.1016/j.freeradbiomed.2021.06.010
- Belambri SA, Rolas L, Raad H, et al. NADPH oxidase activation in neutrophils: role of the phosphorylation of its subunits. Eur J Clin Invest. 2018;48(2):e12951. doi: 10.1111/eci.12951
- Peshavariya H, Jiang F, Taylor CJ, et al. Translation-linked mRNA destabilization accompanying serum-induced Nox4 expression in human endothelial cells. Antioxid Redox Signal. 2009;11:2399–2408. doi: 10.1089/ars.2009.2579
- Schieffer B, Luchtefeld M, Braun S, et al. Role of NAD(P)H oxidase in angiotensin II–induced JAK/STAT signaling and cytokine induction. Circ Res. 2000;87:1195–1201. doi: 10.1161/01.res.87.12.1195
- Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 2010;19:46–54. doi: 10.1183/09059180.00007609
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200
- Takeda Y, Matoba K, Kawanami D, et al. ROCK2 regulates monocyte migration and cell to cell adhesion in vascular endothelial cells. Int J Mol Sci. 2019;20(6):1331. doi: 10.3390/ijms20061331