
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Body pigmentation in insects and other organisms is typically variable within and between species and is often associated with fitness. Regulatory variants with large effects at bab1, t and e affect variation in abdominal pigmentation in several populations of Drosophila melanogaster. Recently, we performed a genome wide association (GWA) analysis of variation in abdominal pigmentation using the inbred, sequenced lines of the Drosophila Genetic Reference Panel (DGRP). We confirmed the large effects of regulatory variants in bab1, t and e; identified 81 additional candidate genes; and validated 17 candidate genes (out of 28 tested) using RNAi knockdown of gene expression and mutant alleles. However, these analyses are imperfect proxies for the effects of segregating variants. Here, we describe the results of an extreme quantitative trait locus (xQTL) GWA analysis of female body pigmentation in an outbred population derived from light and dark DGRP lines. We replicated the effects on pigmentation of 28 genes implicated by the DGRP GWA study, including bab1, t and e and 7 genes previously validated by RNAi and/or mutant analyses. We also identified many additional loci. The genetic architecture of Drosophila pigmentation is complex, with a few major genes and many other loci with smaller effects.
Abbreviations
| AIP | = | advanced intercross population |
| DGRP | = | Drosophila Genetic Reference Panel |
| GWA | = | genome wide association |
| LD | = | linkage disequilibrium |
| RNAi | = | RNA interference |
| xQTL | = | extreme quantitative trait locus |
Introduction
Body pigmentation contributes to the spectacular biodiversity present in nature and mediates mate choice, mimicry, and physiological functions such as thermoregulation and UV resistance. Thus, pigmentation is a significant contributor to fitness.Citation1 In order to understand how complex traits such as pigmentation evolve, we must first identify the genetic variants underlying phenotypic variation.
The amount of pigmentation on the abdomen of Drosophila melanogaster is a relatively simple morphological trait which serves as a model for mapping the genetic basis of variation in complex traits. Previous studies identified a handful of genes with alleles of large effect that contribute to variation in female D. melanogaster abdominal pigmentation. Kopp et al.Citation2 found that a quantitative trait locus (QTL) corresponding to the bric-a-brac locus (bab1 and bab2) contributes to ˜60% of the color variation recorded among a panel of recombinant inbred lines. In addition to bab1, studies using flies collected from natural populations have identified natural variants in tan (t)Citation3 and ebony (e).Citation3,4 Additional studies have confirmed the role of bab1 and bab2,Citation5,6 defined the functionCitation7 and cis-regulatory elements of eCitation8,9 and t,Citation10 and characterized involvement of the regulatory genes double sex (dsx) and Abdominal-B (Abd-B) in patterning the D. melanogaster abdomen.Citation11,12
After tergite development and spatial patterning,Citation13,14 some of the enzymes and color-giving dopamine derivatives must be transported from the underlying epidermal cells to and from the cuticle. The epidermis modulates the production of black melanin by the exo- and endocytosis of Megalin-dependent vesicles containing Yellow, a protein necessary for dark melanization.Citation7,15,16 Cell adhesion proteins and chitin synthesis and binding proteins such as krotzkopf verkehrt (kkv), knickkopf (knk) and retroactive (rtv), are necessary for proper epidermis and cuticle development.Citation17
In conjunction with classical mutant and biochemical analyses,Citation13,18-23 these studies have pieced together our current understanding of melanin (brown and black coloration) and sclerotin (yellow/ tan coloration) biosynthesis in D. melanogaster ().Citation1,24,25,17 However, this is only a small fraction of the 40+ genomic regions/ genes known to affect pigmentation from the catalog of mutant phenotypes previously reported.Citation12,17,18 Furthermore, genes outside of the pigment biosynthesis pathway may also contribute to natural variation in body coloration.
Figure 1. Construction of AIP for xQTL mapping. (A) Light and dark DGRP lines used for the diallel crosses. (B) Experimental protocol for maintaining the AIP.
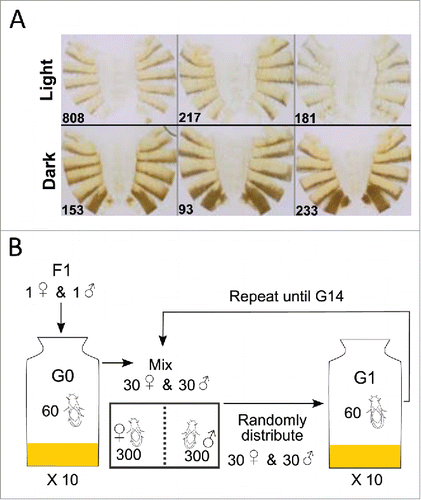
Dembeck et al.Citation26 assessed natural variation in female abdominal pigmentation in 175 sequenced inbred lines of the D. melanogaster Genetic Reference Panel (DGRP), derived from the Raleigh, NC population. The two most posterior abdominal segments, tergites 5 and 6 (T5, T6), were scored for the approximate percentage of the tergite colored with black or dark brown melanin. Consistent with other studies of D. melanogaster body pigmentation, there was significant genetic variation in the proportion of melanization and high broad-sense heritabilities for each tergite.Citation2
The genome-wide association (GWA) studies identified 155 DNA variants in or near (± 1 kb) 84 genes associated with the proportion of melanization on T5 (84), T6 (34), and the difference between T5 and T6 (35).Citation26 Several of the top variants associated with variation in pigmentation were in tan (t), ebony (e), and bric-a-brac1 (bab1). Mutational analyses and targeted RNAi-knockdown showed that 17 out of 28 (61%) novel candidate genes implicated by the genome-wide association study affected abdominal pigmentation. Many of the novel genes affect other well-studied pathways and phenotypes, such as wing and bristle development, providing evidence for widespread pleiotropy. We named 4 of the novel, computationally predicted genes based on their mutant and/or RNAi phenotypes: pinstripe (pns, CG7852), triforce (tfc, CG9134), plush (ph, CG1887), and farmer (frm, CG10625). Variation in the novel candidate genes identified by GWA analysis may serve as targets for adaptive evolution and sexual selection in D. melanogaster.
The GWA findings suggested that naturally occurring genetic variation in pigmentation may affect multiple steps in different pathways involved in tergite development and melanization. However, we reported all GWA results based on a nominal P < 10−5 since quantile-quantile plots indicated a systematic departure from random expectation below this value. Very few variants had P-values less than a Bonferroni correction for multiple tests. Thus, although the top variants are enriched for true positives, we need to perform validation experiments to test all candidate genes to discern which genes are true positives. Two methods are often used to provide additional support for the GWA results, functional testing such as RNAi or mutant analysis or replication in other populations. Dembeck et al.Citation26 assessed changes in pigmentation using RNAi knockdown and mutant alleles of selected candidate genes. This approach is limited by the availability of these resources. Further, failure to confirm a candidate gene using RNAi and mutations does not necessarily mean that the candidate allele segregating in the DGRP does not affect the trait. Here, we present the results of a complementary study, an extreme quantitative trait locus (xQTL) GWA analysisCitation27 in an outbred advanced intercross population (AIP) derived from dark and light DGRP lines. These data provide additional evidence supporting the results of the DGRP GWA analysis and previous functional validation experiments.Citation26
Methodology
Drosophila stocks and phenotyping
From the 175 DGRP lines scored by Dembeck et al.Citation26 we selected 3 lightly and 3 darkly melanized lines that were scored on a scale from 0 (no dark melanin) to 4 (100% pigmented with dark melanin) for the proportion of dark melanin on T5 and T6. The light lines were DGRP_217 (T5, T6 line means: 0.8, 0.45); DGRP_808 (0.7, 0.45); and DGRP_181 (1.15, s1). The dark lines were DGRP_93 (2.5, 3.85); DGRP_153 (1.45, 3.85); and DGRP_233 (1.8, 4). Each DGRP line was reared at a controlled adult density (CAD) of 10 males and 10 females on cornmeal-molasses-agar medium at 25°C, 75% relative humidity, and a 12-h light-dark cycle. The flies were allowed to lay eggs for 3 d Virgin males and females from each line were then crossed using a full-diallel design (excluding homozygous parental lines) and reared as described above. Ten virgin female F1 flies were aged to day 4 and phenotyped using the 0 – 4 proportion of melanization scale.
F1 virgin males and females were then used to establish Generation (G) 0 of an outbred AIP. Thirty males and 30 females (one male and female of each F1 genotype) were placed in each of 10 bottles (600 flies total). For G1, 30 male and 30 female virgins were collected from each G0 bottle, mixed according to sex, and then randomly distributed among 10 new rearing bottles. This cycle was then repeated until G14. At G10, G11, G13 and G14, ∼500 virgin females were collected and aged for 4 d The flies were phenotyped, and darkest ˜50 females and lightest ∼50 females were individually flash-frozen in a 0.2 mL microcentrifuge tube, and stored at −80°C until DNA extraction.
Extreme QTL mapping
DNA was extracted from pools of the darkest ˜50 females and lightest ˜50 females at G10, G11, G13 and G14. Sequence libraries were prepared from each DNA sample, barcoded, multiplexed, and sequenced on the Illumina HiSeq 2500 platform using 100bp paired-end sequencing. Sequence reads were mapped to the Drosophila melanogaster reference genome (BDGP5) using BWA-MEM 0.7.10,Citation28 locally realigned, masked for PCR duplicates, and recalibrated for base quality using GATK 2.4.9.Citation29 Allele counts were obtained at polymorphic SNP sites where the 6 parental lines segregated according their DGRP genotype calls. Finally, the difference in allele frequencies in the dark and light pools was called on each site using a test statistic Z = d/s with a 2-sided P-value obtained from the standard normal distribution, where d is the allele frequency difference p1 – p2, with p0 = (p1+p2)/2; n1, n2 the sequ-ence coverages, and h1, h2 the numbers of haploid genomes in the pooled samples respectively. To combine results across multiple generations, a combi-ned Z score was obtained
and the P-value drawn from standard normal, where the c1, …, c4 are the average coverages in the 4 generations. A total of 595,729 single nucleotide polymorphisms (SNPs) that remained polymorphic across all 4 generations were considered.
Results and Discussion
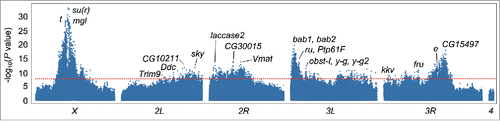
We created an outbred AIP from 3 darkly pigmented and 3 lightly pigmented DGRP lines. We initially crossed the lines in a full diallel design (excluding homozygous parental genotypes) to ensure equal representation of all lines, and maintained the populations at a census population size of 600 flies per generation, for a total of 14 generations (, , Fig. S1). At generations 10, 11, 13 and 14 we scored 500 females and selected the 10% darkest and lightest flies to use for xQTL mapping (Table S1). We sequenced the 8 DNA pools, and mapped QTLs affecting pigmentation by assessing the differences in allele frequency between the dark and light pools in each generation (Table S2). A qualitative visual assessment of the Manhattan plots showed that the QTL peaks are generally consistent across all analyses (Fig. S2). Therefore we combined data from all 4 generations and obtained a single P-value for each SNP (, Table S2). At a Bonferroni-corrected P-value of 8.39 × 10−8, we identified 7,069 variants in or near (± 1 kb) 1,884 genes from the xQTL analysis combined across generations. We mapped many genes and specific SNPs that overlap with the GWA results of Dembeck et al.,Citation26 providing independent validation for the results of this study. In addition, we also identified variants located within or near a new set of candidate genes that are biologically plausible with respect to variation in D. melanogaster pigmentation.
Figure 2. Manhattan plot for the xQTL mapping GWA analysis, combined across all generations. Gene symbols for loci implicated by the mapping are given. The Bonferroni corrected significance threshold (P = 8.39 × 10−8) is indicated by the horizontal dashed line.
Gene overlap between DGRP and xQTL GWA analyses
We compared the top candidate genes identified in the DGRP GWA analysis of Dembeck et al.Citation26 and the xQTL mapping GWA analysis for all generations combined, and identified 28 overlapping genes. These included genes previously known to affect Drosophila pigmentation (bab1, bab2, e, osa, t, and the 2 genes in the cis-regulatory region of t, Gustatory receptor 8a (Gr8a) and CG15370); and 11 of the 28 novel candidate genes identified and tested in Dembeck et al.Citation26: roughoid (ru), klarsicht (klar), multiple wing hairs (mwh), pns, ph, tfc, Vesicular monoamine transporter (Vmat), Glucose transporter 1 (Glut1), krotzkopf verkehrt (kkv), CG15803 and CG42340. All but 4 (Vmat, CG42340, CG15803, mwh) of the latter genes caused changes to T5 or T6 pigmentation when analyzed with RNAi and/or mutants. The additional genes that overlapped but were not previously tested were CG5888, CG30485, CG9194, CG42663, empty spiracles (ems), CG13330, Protein tyrosine phosphatase 61F (Ptp61F), CG2493, mir-288, RNA-binding protein 6 (Rbp6) and E5.
Although we had not previously tested Ptp61F, we note that it overlaps with ru, which led to large reductions in T5 and T6 pigmentation when knocked down. The variants in Ptp61F associated with pigmentation could well correspond to a cis-regulatory element for ru. We replicated effects on pigmentation in the xQTL mapping GWA analysis for which mutations and RNAi knockdown did not yield significant effects on pigmentation. We speculate that the naturally segregating variant(s) in these genes affect pigmentation and that the effects are not recapitulated by the available mutations or by reducing expression ubiquitously or with a pannier-GAL4 driver.
SNP overlap between DGRP and xQTL GWA analyses and other studies
Three SNPs located in the cis-regulatory region of t were mapped in the xQTL and DGRP GWA analyses and the xQTL study of variation in tergite 7 pigmentation of wild caught D. melanogaster females by Bastide et al.Citation3 One of these SNPS, X_9121177_SNP, as highly significant in the combined generations analysis (P = 1.34 × 10−29). Two other SNPs were significant following Bonferroni correction in G10 and G11 (X_9121129_SNP) or in G13 and G14 (X_9121094_SNP). These SNPs are promising causative variants given their replication across the 3 studies.
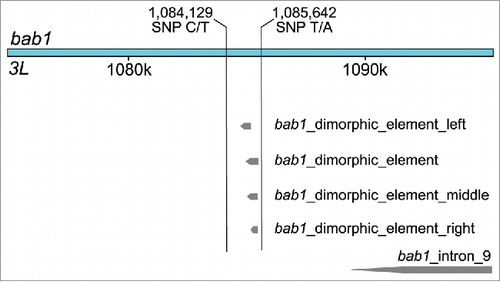
Four additional SNPs were shared by the DGRP GWAS and the combined generations xQTL analysis. Two SNPs, 3L_1085642_SNP (P = 1.56 × 10−14) and 3L_1084129_SNP (P = 1.08 × 10−11), are located within the intron of bab1. These SNPs fall on each side of the dimorphic element described by Williams et al.Citation11, which contains 3 other bab1 variants, 3L_1084980_SNP, 3L_1085137_SNP, and 3L_1085230_SNP mapped by Dembeck et al. and Rogers et al.Citation30 (). 3L_1084129_SNP is also within a dl transcription factor binding site; again highlighting the association of variants within dl binding sites as was seen in the DGRP GWAS. This overlap and the proximity of these associated SNPs to the cis-regulatory dimorphic element suggest that these SNPs may also functionally affect the regulatory region. 3L_1381936_SNP (P = 1.94 × 10−14) is located within the introns of the overlapping genes ru and Ptp61F. 2R_9404826_SNP is located downstream of Vmat (P = 9.42 × 10−9). These results provide additional support for variants with small effect sizes contributing to natural variation in female D. melanogaster pigmentation.
Figure 3. Schematic showing the location of the 2 SNPs in bric-a-brac1 that were associated with variation in body pigmentation in independent studies.
Even more candidate genes
One feature of the Manhattan plot from the combined xQTL analysis is the bifurcation of the large QTL peak on the X chromosome into 2 very close peaks. Upon inspection of the genes in these regions, it is evident that the first peak results from the association of genetic variants in t or its cis-regulatory regions. The second peak, somewhat to our surprise, is centered upon variants within or near megalin (mgl), a gene known to be involved in wing cuticle pigmentation in D. melanogaster. mgl mediates the endocytic removal of the Yellow protein, which is required to produce black melanin, from the developing cuticle. When mgl expression is reduced, Yellow accumulates, leading to the ectopic formation of abnormally large melanin granules.Citation15 To our knowledge, this is the first study to associate natural genetic variation in mgl with variation in abdominal pigmentation in D. melanogaster.
Surprisingly, the most significantly associated SNP on the X chromosome in the combined xQTL analysis was not in the cis-regulatory region of t or in mgl. Instead, the SNP (P = 6.4 × 10−34) was located in an intergenic region upstream of suppressor of rudimentary (su(r)). su(r) is located downstream from t and it is possible that the SNP may be located in another t cis-regulatory region. However, this intergenic region is a strong candidate as a cis-regulatory element for su(r). Insects use β-alanine for the synthesis of N-β-alanyl dopamine, which contributes to the yellowish-brown color of the cuticle. There are 2 primary pathways used to synthesize β-alanine. It can be formed from aspartate by aspartate decarboxylase (encoded by black) or from uracil by dihydropyrimidine dehydrogenase (Pyd1, encoded by su(r)), dihydropyrimidase (Pyd2), and β-ureidopropionase (Pyd3).Citation31 This supports our previous findings that genetic variation at a number of steps in biosynthetic, regulatory, and developmental pathways may contribute to natural variation in abdominal pigmentation.
The xQTL mapping analyses reveals 3 QTL peaks on chromosome 2R that are consistent in G10, G11 and G14 (Fig. S2). The first of these 2R QTL peaks contains a SNP within an intron of laccase2. laccase2 is a multicopper oxidase that is essential for cuticular sclerotinization.Citation20,32,33 As with mgl, to the best of our knowledge, this is the first study to associate natural genetic variation in laccase2 with variation in female abdominal pigmentation in D. melanogaster. Similarly, on chromosome 2L, there was a SNP (P = 6.30 × 10−10) 67-bp downstream of Dopa decarboxylase (Ddc), which could affect regulation of Ddc expression.
We identified several other notable and plausible candidate genes based on the xQTL mapping GWA analysis. One of these variants was in Tripartite motif containing 9 (Trim9) (P = 5.8 × 10−8). Trim proteins are found in all metazoans and have many intracellular functions. Trim9 binds to microtubules and regulates synaptic vesicle exocytosis.Citation34 The most strongly associated variant on chromosome 2L was in the intron of skywalker (sky) (P = 1.7 × 10−12). sky has a role in the negative regulation of synaptic vesicle recycling.Citation35 Trim9 and sky may be involved in the vesicle dynamics along with mgl and Vmat that transport pigment biosynthesis enzymes or dopamine-derivatives to and from the cuticle. Another gene implicated by a significant SNP (P = 1.2 × 10−11), CG10211, is located between Trim9 and sky. It is predicted to have peroxidase activity and reported to be lethal when gene expression is reduced using RNAi.Citation36,37 Peroxidases have been suggested to facilitate sclerotinization of the cuticle by catalyzing the oxidation of NADA and NBAD to ortho-quinones.Citation33 There are also several variants on 2L in and around CG9336. RNAi knockdown of this gene results in body color differences on the central notum and the surrounding area.Citation36
There were a handful of other candidate genes each containing several associated SNPs from the combined xQTL mapping analyses implicated by smaller QTL peaks on chromosomes 2R, 3L, and 3R. These include CG30015, fruitless (fru), and obstructor-I (obst-I), yellow-g and yellow-g2. While no functional annotation has been reported for CG30015, RNAi resulted in cuticle color differences on the notum.Citation36 fru has a vital role in sex determination and development and its variants may ultimately affect the expression of sex determination genes known to affect female pigmentation such as doublesex (dsx) and Abdominal-B (Abd-B). Abd-B was also implicated to affect pigmentation in the xQTL analysis. The three closely linked genes obst-I, yellow-g and yellow-g2 are all reported to be associated with cuticle development. obst-I is a chitin binding protein expressed primarily in the larval digestive system.Citation37 yellow-g and yellow-g2 are expressed in the female follicular cells during egg development and are likely to contribute to hardening the eggshell.Citation37 Given that these genes are not known be functional in adult cuticle pigmentation, we hypothesize that the SNPs in the region may indicate the presence of a cis-regulatory element for ecdysoneless, which is nearby and known to affect cuticle development.Citation37
Juvenile hormone (JH) has profound effects during insect development. It regulates embryogenesis, development and eclosion, and stimulates sexual maturation in adults.Citation38 Two SNPs in the haemolymph JH binding protein CG15497 were strongly associated (P = 1.1 × 10−18 and P = 1.3 × 10−18) with pigmentation in the combined xQTL mapping analysis. These proteins are responsible for transporting JH from the corpora allata through the haemolymph to specific target tissues.Citation39 As these variants are located over 400,000 bp away from the known pigmentation gene e, we feel confident that their association with pigmentation is not due to LD with e variants.
Genetic architecture of D. melanogaster abdominal pigmentation
The GWA analyses in the DGRP and the AIP derived from extreme DGRP lines have complementary properties that can be leveraged to replicate true positive candidate genes and variants in the case of additive gene action. The DGRP has the favorable properties of minimal relatedness and low local LD, but with ∼200 lines, only variants with large effects can be detected with high statistical confidence. xQTL mapping in an AIP can be done with very large numbers of individuals, such that top variants can achieve significance following a strict Bonferroni correction for multiple tests; but unless many generations of recombination are allowed, local LD is much higher than in the DGRP, decreasing the mapping precision. In addition, association mapping in the DGRP can only be performed using common variants (minor allele frequencies > 0.05),Citation40 whereas the effects of private variants in any of the 6 parental lines used to construct the AIP can be assessed. However, this replication approach will only work if gene action of the causal variants is additive; in the presence of epistatic gene action, differences in allele frequency between the DGRP and the AIP will lead to failure of replication.Citation6-8
Since we were able to replicate many of the GWA results of Dembeck et al.Citation26 at the levels of both genes and SNPs, we infer the genetic architecture of abdominal pigmentation is largely additive. Prior to the study of Dembeck et al.Citation26 and this report, the genetic basis of natural variation in D. melanogaster abdominal pigmentation was thought to be oligogenic, controlled by variants with large effects at a few loci. Our studies indicated that reveal a more complex genetic basis than this, with many additional loci with smaller effects. Replicated associations of genetic variants in the genes that were tested previouslyCitation26 using RNAi knockdown or mutant analyses but that did not affect pigmentation suggest that these genes may indeed contribute to variation in pigmentation, and that naturally occurring variants can cause subtle phenotypic effects that cannot be reproduced by mutagenesis or RNAi knockdown. Since most of the associated SNPs are in intronic and intergenic regions, we hypothesize that they affect tissue- or stage-specific gene regulatory motifs. These results highlight the known and potential contributions of cis-regulatory evolution, which likely limits negative pleiotropic effects, to phenotypic variation within the DGRP population.
Previously,Citation26 we hypothesized that Efa6, Klp61F, klar, and pns facilitate vesicle transport of enzymes and/or dopamine-derivatives to and from the cuticle. The replication of the association of variants in these genes in addition to those in Vmat, mgl, sky, and Trim9 provide more support for this hypothesis. Together these genes may represent components of the transport mechanism for cuticle sclerotization and melanization in D. melanogaster.
In summary, we confirm our previous results and provide additional evidence that genetic variation at a number of steps in biosynthetic, regulatory, developmental, and transport may pathways contribute to natural variation in abdominal pigmentation. Our results hold open the door for new hypotheses to be tested on the epithelial cell vesicle dynamics, the locations of cis-regulatory elements, and how metabolic and hormonal regulation can lead to variation in pigmentation. Future studies will provide much needed mechanistic inferences about these novel candidate genes and SNPs and their contributions to the evolution of pigmentation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1102807_Supplemental_Material.zip
Download Zip (3.5 MB)Funding
This work was funded by National Institutes of Health grant R01 GM45146 to TFCM.
Related Research Data
References
- True JR. Insect melanism: the molecules matter. Trends Ecol Evol 2003; 18:640-7; http://dx.doi.org/10.1016/j.tree.2003.09.006
- Kopp A, Graze RM, Xu S, Carroll SB, Nuzhdin SV. Quantitative trait loci responsible for variation in sexually dimorphic traits in Drosophila melanogaster. Genetics 2003; 163:771-87; PMID:12618413
- Bastide H, Betancourt A, Nolte V, Tobler R, Stöbe P, Futschik A, Schlötterer C. A genome-wide, fine-scale map of natural pigmentation variation in Drosophila melanogaster. PLoS Genet 2013; 9:e1003534; PMID:23754958; http://dx.doi.org/10.1371/journal.pgen.1003534
- Pool JE, Aquadro CF. The genetic basis of adaptive pigmentation variation in Drosophila melanogaster. Mol Ecol 2007; 16:2844-51; PMID:17614900; http://dx.doi.org/10.1111/j.1365-294X.2007.03324.x
- Kopp A, Duncan I, Godt D, Carroll SB. Genetic control and evolution of sexually dimorphic characters in Drosophila. Nature 2000; 408:553-9; PMID:11117736; http://dx.doi.org/10.1038/35046017
- Bickel RD, Kopp A, Nuzhdin SV. Composite effects of polymorphisms near multiple regulatory elements create a major-effect QTL. PLoS Genet 2011; 7:e1001275; PMID:21249179; http://dx.doi.org/10.1371/journal.pgen.1001275
- Wittkopp PJ, True JR, Carroll SB. Reciprocal functions of the Drosophila Yellow and Ebony proteins in the development and evolution of pigment patterns. Development 2002; 129:1849-58; PMID:11934851
- Williams TM, Carroll SB. Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat Rev Genet 2009; 10:797-804; PMID:19834484; http://dx.doi.org/10.1038/nrg2687
- Richardt A, Kemme T, Schwarzer D, Mohamed A, Hovemann BT, Wagner S, Marahiel MA. Mechanisms of signal transduction: Ebony, a novel nonribosomal peptide synthetase for β -alanine conjugation with biogenic amines in Drosophila. J Biol Chem 2003; 278:41160-6; PMID:12900414; http://dx.doi.org/10.1074/jbc.M304303200
- Jeong S, Rebeiz M, Andolfatto P, Werner T, True J, Carroll SB. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell 2008; 132:783-93; PMID:18329365; http://dx.doi.org/10.1016/j.cell.2008.01.014
- Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, Carroll SB. The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 2008; 134:610-23; PMID:18724934; http://dx.doi.org/10.1016/j.cell.2008.06.052
- Wittkopp PJ, Carroll SB, Kopp A. Evolution in black and white: genetic control of pigment patterns in Drosophila. Trends Genet 2003; 19:495-504; PMID:12957543; http://dx.doi.org/10.1016/S0168-9525(03)00194-X
- True JR, Edwards KA, Yamamoto D, Carroll SB. Drosophila wing melanin patterns form by vein-dependent elaboration of enzymatic prepatterns. Curr Biol 1999; 9:1382-91; PMID:10607562; http://dx.doi.org/10.1016/S0960-9822(00)80083-4
- Kopp A, Blackman RK, Duncan I. Wingless, decapentaplegic and EGF receptor signaling pathways interact to specify dorso-ventral pattern in the adult abdomen of Drosophila. Development 1999; 126:3495-507; PMID:10409497
- Riedel F, Vorkel D, Eaton S. Megalin-dependent Yellow endocytosis restricts melanization in the Drosophila cuticle. Development 2011; 138:149-58; PMID:21138977; http://dx.doi.org/10.1242/dev.056309
- Walter MF, Black BC, Afshar G, Kermabon AY, Wright TR, Biessmann H. Temporal and spatial expression of the yellow gene in correlation with cuticle formation and dopa decarboxylase activity in Drosophila development. Dev Biol 1991; 147:32-45; PMID:1879614; http://dx.doi.org/10.1016/S0012-1606(05)80005-3
- Moussian B. Recent advances in understanding mechanisms of insect cuticle differentiation. Insect Biochem Mol Biol 2010; 40:363-75; PMID:20347980; http://dx.doi.org/10.1016/j.ibmb.2010.03.003
- Wright TR. The genetics of biogenic amine metabolism, sclerotization, and melanization in Drosophila melanogaster. Adv Genet 1987; 24:127-222; PMID:3124532; http://dx.doi.org/10.1016/S0065-2660(08)60008-5
- Hodgetts RB, Konopka RJ. Tyrosine and catecholamine metabolism in wild-type Drosophila melanogaster and a mutant, ebony. J Insect Physiol 1973; 19:1211-20; PMID:4196594; http://dx.doi.org/10.1016/0022-1910(73)90205-9
- Arakane Y, Muthukrishnan S, Beeman RW, Kanost MR, Kramer KJ. Laccase 2 is the phenoloxidase gene required for beetle cuticle tanning. Proc Natl Acad Sci U S A 2005; 102:11337-42; PMID:16076951; http://dx.doi.org/10.1073/pnas.0504982102
- Kramer KJ, Hopkins TL. Tyrosine metabolism for insect cuticle tanning. Arch Insect Biochem Physiol 1987; 6:279-301; http://dx.doi.org/10.1002/arch.940060406
- True JR, Yeh S-D, Hovemann BT, Kemme T, Meinertzhagen IA, Edwards TN, Liou S-R, Han Q, Li J. Drosophila tan encodes a novel hydrolase required in pigmentation and vision. PLoS Genet 2005; 1:e63; PMID:16299587; http://dx.doi.org/10.1371/journal.pgen.0010063
- Greenspan RJ. Fly Pushing: The theory and practice of Drosophila genetics, Part 7. Cold Spring Harbor Laboratory Press; 1997.
- Hopkins TL, Kramer KJ. Insect cuticle sclerotinization. Annu Rev Entomol 1992; 37:273-302; http://dx.doi.org/10.1146/annurev.en.37.010192.001421
- Payre F. Genetic control of epidermis differentiation in Drosophila. Int J Dev Biol 2004; 48:207-15; PMID:15272387; http://dx.doi.org/10.1387/ijdb.15272387
- Dembeck LM, Huang W, Magwire MM, Lawrence F, Lyman RF, Mackay TFC. Genetic architecture of abdominal pigmentation in Drosophila melanogaster. PLOS Genet 2015; 11:e1005163; PMID:25933381; http://dx.doi.org/10.1371/journal.pgen.1005163
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature 2010; 464:1039-42; PMID:20393561; http://dx.doi.org/10.1038/nature08923
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013. Available from: http://arxiv.org/abs/1303.3997.
- DePristo MA, Banks E, Poplin R, Garimella K V, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43:491-8; PMID:21478889; http://dx.doi.org/10.1038/ng.806
- Rogers WA, Salomone JR, Tacy DJ, Camino EM, Davis KA, Rebeiz M, Williams TM. Recurrent modification of a conserved cis-regulatory element underlies fruit fly pigmentation diversity. PLoS Genet 2013; 9:e1003740; PMID:24009528; http://dx.doi.org/10.1371/journal.pgen.1003740
- Borycz J, Borycz JA, Edwards TN, Boulianne GL, Meinertzhagen IA. The metabolism of histamine in the Drosophila optic lobe involves an ommatidial pathway: β-alanine recycles through the retina. J Exp Biol 2012; 215:1399-411; PMID:22442379; http://dx.doi.org/10.1242/jeb.060699
- Niu B-L, Shen W-F, Liu Y, Weng H-B, He L-H, Mu J-J, Wu Z-L, Jiang P, Tao Y-Z, Meng Z-Q. Cloning and RNAi-mediated functional characterization of MaLac2 of the pine sawyer, Monochamus alternatus. Insect Mol Biol 2008; 17:303-12; PMID:18477244; http://dx.doi.org/10.1111/j.1365-2583.2008.00803.x
- Andersen SO. Insect cuticular sclerotization: a review. Insect Biochem Mol Biol 2010; 40:166-78; PMID:19932179; http://dx.doi.org/10.1016/j.ibmb.2009.10.007
- Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem 2006; 281:8970-80; PMID:16434393; http://dx.doi.org/10.1074/jbc.M512755200
- Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell 2011; 145:117-32; PMID:21458671; http://dx.doi.org/10.1016/j.cell.2011.02.039
- Mummery-Widmer JL, Yamazaki M, Stoeger T, Novatchkova M, Bhalerao S, Chen D, Dietzl G, Dickson BJ, Knoblich JA. Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature 2009; 458:987-92; PMID:19363474; http://dx.doi.org/10.1038/nature07936
- St Pierre SE, Ponting L, Stefancsik R, McQuilton P. FlyBase 102–advanced approaches to interrogating FlyBase. Nucleic Acids Res 2014; 42:D780-8; PMID:24234449; http://dx.doi.org/10.1093/nar/gkt1092
- Kolodziejczyk R, Bujacz G, Jakób M, Ozyhar A, Jaskolski M, Kochman M. Insect juvenile hormone binding protein shows ancestral fold present in human lipid-binding proteins. J Mol Biol 2008; 377:870-81; PMID:18291417; http://dx.doi.org/10.1016/j.jmb.2008.01.026
- Kołodziejczyk R, Kochman M, Bujacz G, Dobryszycki P, Ożyhar A, Jaskolski M. Crystallization and preliminary crystallographic studies of juvenile hormone-binding protein from Galleria mellonella haemolymph. Acta Crystallogr Sect D Biol Crystallogr 2003; 59:519-21; http://dx.doi.org/10.1107/S0907444902022904
- Huang W, Massouras A, Inoue Y, Peiffer J, Rámia M, Tarone A, Turlapati L, Zichner T, Zhu D, Lyman R, et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res 2014; 24:1193-208; PMID:24714809; http://dx.doi.org/10.1101/gr.171546.113