
ABSTRACT
The growth of epithelial tumors is often governed by cell interactions with the surrounding stroma. Drosophila has been instrumental in identifying the relevant molecular elements mediating these interactions. Of note is the role of the TNF ligand Eiger, released from recruited blood cells, in activating the JNK tumor-promoting pathway in epithelial tumors. JNK drives the transcriptional induction of mitogenic molecules, matrix metalloproteases and systemic signals that lead to tumor growth, tissue invasiveness and malignancy. Here we review our findings on a tumor-intrinsic, Eiger- and stroma-independent mechanism that contributes to the unlimited growth potential of tumors caused either by chromosomal instability or impaired cell polarity. This newly identified mechanism, which was revealed in an experimental condition in which contacts between tumor cells and wild-type epithelial cells were minimized, relies on interactions between functionally distinct tumor cell populations that activate JNK in a cell-autonomous manner. We discuss the impact of cell interaction-based feedback amplification loops on the unlimited growth potential of epithelial tumors. These findings are expected to contribute to the identification of the relevant cell populations and molecular mechanisms to be targeted in drug therapy.
Drosophila in cancer research
The earliest evidence that flies develop tumors came from studies in the 1910s conducted by Mary Stark, a student of Thomas Morgan, who was analyzing a mutant isolated by Calvin Bridges, the l(1)7. She observed the development of melanotic tumors in the larvae of this mutant and showed that these tumors were inherited [Citation40]. This finding strengthened the notion that cancer is a genetic disease at a time in which this hypothesis was just emerging. It was later, in the 1970s, when experiments performed by Elisabeth Gateff put Drosophila on the map in cancer research. Gateff was studying another mutation isolated by Bridges, the lethal giant larvae (lgl). In pioneer allograft experiments, she showed that fragments of tumors transplanted into the abdomens of adult flies proliferate indefinitely, invade organs and eventually kill the host [Citation18]. A second generation of studies were conducted decades later with the development of new technologies and genetic tools [Citation4,Citation15,Citation33]. Work from these and other labs demonstrated that Drosophila tumors reproduce the key hallmarks of cancer, namely self-sufficiency in growth signals, insensitivity to growth inhibitory signals, evasion of programmed cell death, limitless replicative potential, tissue invasion and metastasis, and malignancy [Citation20]. Drosophila was thus established as a useful model system in which to characterize the mechanisms of tumor initiation and progression (reviewed in [Citation5,Citation19,Citation34]).
Imaginal discs to characterize the biology of epithelial tumors
Imaginal discs, mono-layered epithelial sacs that grow a 1000 fold in tissue size and cell number in a matter of days within the feeding larvae, are probably one of the most useful tissues in which to identify and molecularly characterize the relevant elements driving tumorigenesis in proliferating epithelia. These discs give rise to ectodermal adult structures such as the eyes, legs and wings [Citation8] and have been classically used to identify and dissect the major signaling pathways mediating cell interactions in development and disease, and conserved from flies to humans. Many of the pathways with a relevant role in human cancer were also genetically identified and their elements dissected in these primordia, as mutations that caused the loss of their activity induce easy-to-score alterations in the size, pattern or shape of the resulting adult structures. Over the last 15 years, this accumulated knowledge, together with a variety of genetic tools available in Drosophila, have contributed to the dramatic increase in the use of Drosophila imaginal discs as model systems through which to unravel the relevance of cell interactions in driving tumorigenesis and the pivotal activity of the JNK signaling pathway in promoting tumor growth, invasive behavior and malignancy [reviewed in [Citation5,Citation34]]. JNK exerts these actions by activating a transcriptional program that includes the expression of mitogenic molecules, matrix metalloproteases and systemic signals [Citation4,Citation16,Citation23,Citation44]. Interestingly, JNK plays only a minor role in the development of fly appendages [Citation46], but its activation in damaged tissues or upon neoplastic transformation is an absolute requirement for both tissue regeneration and tumorigenesis [Citation2,Citation4,Citation23,Citation44]. Wing and eye primordia were used as models for tumorigenesis for the first time in genetic screenings designed to identify mutations that drive neoplastic transformation when combined with oncogene expression. Loss of cell polarity determinants scribbled or Disc large was shown to drive neoplastic transformation, tumor growth, tissue invasiveness and organismal lethality when combined with the expression of the Ras-V12 oncogene [Citation4,Citation33]. Since those studies, a plethora of genes have been identified as key drivers of oncogene cooperation, and the relevant cell populations and signaling pathways have been characterized. While recruited hemocytes expressing Eiger activate JNK in transformed cells [Citation9], cancer cells engage neighboring epithelial cells as indispensable contributors of their own growth through the non-autonomous induction of autophagy, thus providing nutrients for the tumor [Citation24]. Also recently, wild-type resident myoblasts have been reported to contribute to the oncogene-induced transformation of the wing epithelium [Citation22]. All together, these results have revealed the relevance of cell interactions and the unmodified microenvironment in the initiation and progression of epithelial tumors.
Genomic instability-induced epithelial tumors
Imaginal discs have also contributed to shed new light on the role of genomic instability in tumorigenesis. Genomic instability was originally proposed to cause cancer over 100 years ago [Citation3,Citation14,Citation45], and aneuploidy—an abnormal number of whole chromosomes or parts of them—has been observed in most sporadic carcinomas [Citation13], the most common type of cancer in humans and derived from putative epithelial cells. However, aneuploid genotypes compromise cell proliferation and growth in yeast, flies and human cells [Citation37,Citation41,Citation43]. Research in imaginal discs has contributed to solve this controversy and has proposed that aneuploidy exerts stress-induced tumor-promoting activity [Citation11,Citation28]. Indeed, forty years ago, John L. Haynie and Peter J. Bryant unraveled the deleterious effects of ionizing radiation (IR) on the proliferation dynamics of Drosophila wing cells and proposed that these effects were “possibly due to radiation-induced aneuploidy” [Citation21]. When IR-treated cells were maintained in the tissue by additional blockade of the apoptotic pathway, a JNK-dependent tumor-like overgrowth was observed [Citation10,Citation35]. A recent analysis of the effects of Chromosomal Instability (CIN)—the continuous change in chromosome number—on the larval wing disc confirmed the deleterious effects of aneuploidy at the cellular and tissue levels. The depletion of Spindle Assembly Checkpoint genes induces CIN in epithelial cells and highly aneuploid cells—the latter resulting from chromosome missegregation events—delaminate from the epithelium, most probably as a result of DE-cadherin delocalization, and die by apoptosis [Citation11,Citation12,Citation29]. In contrast to mammalian cells, CIN-induced apoptosis in fly epithelia is independent of the activity of the tumor suppressor gene Dp53 and dependent on the activation of the JNK pathway [Citation11]. A recent analysis of gene expression data of aneuploid cells in several organisms, including yeast, plants, mice, and humans, has unraveled a consistent upregulation of genes involved in the stress response in all these species [Citation39]. The stress caused by protein stoichiometry imbalance has been confirmed as being responsible for some aneuploidy-related phenotypes [Citation32]. It has been proposed that the formation of protein aggregates in aneuploid cells is caused by the limitation of protein quality-control systems, including the proteasome and the chaperone systems, and an increase in proteasomal degradation by deletion of the gene encoding the de-ubiquitinating enzyme Ubp6 improves the proliferative capacity of these cells [Citation42]. Whether aneuploidy-induced protein stoichiometry imbalances contribute to JNK activation and tumor-like overgrowth in Drosophila epithelial tissues remains to be elucidated. However, chromosome-wide gene dosage imbalances are known to drive JNK activation and apoptosis in highly aneuploid cells in the fly [Citation7]. Another common feature of CIN-tissues in flies and mammals is the generation of Radical Oxygen Species (ROS [Citation17]). In fly tissues, these compounds contribute to JNK activation upon CIN [Citation7]. The acquisition of aneuploidy-tolerating mutations improves the fitness of multiple aneuploidies [Citation42] and contributes to CIN-induced tumorigenesis in mammalian in vivo models. Indeed, mutations in p53, which drives CIN-induced apoptosis in mammalian cells, accelerate tumor development in two independent CIN models [Citation27]. Similarly, the maintenance of aneuploid cells in the fly epithelium by means of blocking the apoptotic pathway at different levels elicits CIN-induced tumorigenic behavior [Citation11,Citation12,Citation29]. Aneuploid cells delaminating from the epithelium activate a JNK transcriptional program that leads to their neoplasic transformation.
Analyzing the growth potential of epithelial tumors in the absence of a tumor microenvironment
The similarities between CIN-tumors and those produced by the oncogenic cooperation between Ras-V12 and loss of cell polarity determinants (polarity impaired tumors) are striking. In both cases, neoplastic transformation is determined by the activity of JNK, and the transcriptional program that leads to tumor growth, invasiveness and malignancy is almost identical [Citation6,Citation7,Citation25]. The production of the mitogenic molecule Wingless and the Unpaired cytokines in the two cases contributes to tumor growth. The roles of Eiger and the tumor microenvironment to the progression of polarity-impaired tumors have been extensively reported in a situation in which tumor cells are confronted to nearby wild type cells by the use of clonal analysis [Citation9]. Indeed, a feedback amplification loop between blood cells, tumor cells and wild-type cells has recently been proposed to contribute to tumor growth. This feedback loop involves Caspase activation in tumor cells downstream of JNK, the production of ROS by the cells thus helping recruiting hemocytes, and hemocytes signaling back to tumor cells through Eiger [Citation36]. In a recent report [Citation30], we combined the GAL4/UAS and LexA/LexO transactivation systems with allograft transplantations in adult hosts to genetically ablate the components of the tumor microenvironment in both polarity-impaired and CIN-tumors and then analyzed the impact on JNK activation and tumor initiation and progression. To prevent interactions between tumor cells and nearby wild-type cells, we used the GAL4/UAS system to drive the expression of tumor-promoting transgenes in large territories of the wing primordium, instead of the regularly used FRT-mediated clonal analysis. After implantation in the abdomens of young females, the tumors were induced by means of the thermo-sensitive version of the Gal80 protein (Gal80ts). We then examined the impact of genetic ablation of adult hemocytes on the growth of these tumors and on the levels of JNK activation. Interestingly, none of these features was visibly affected by the absence of hemocytes. Using LexA/LexO, we also demonstrated that tumor growth and JNK activation were not affected by the genetic ablation of the myoblast population or by the removal of hemocytes and myoblasts in allograft transplantations. These results strongly supported the notion that JNK is not induced by an exogenous source of Eiger in these tumors under these circumstances. Consistently, tumor growth and JNK levels were not altered in individuals mutant for eiger or grindelwald, the latter encoding for the Eiger receptor in larval tissues [Citation1]. These results indicate that the tumor-promoting relevance of Eiger and Grindewlald is determined by the number of interactions between tumor cells and the nearby wild-type epithelium () and that other mechanisms are responsible for JNK activation and tumor growth when a low number of these interactions occur.
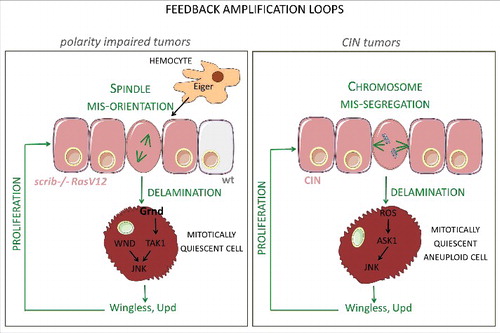
Figure 1. Feedback amplification loops in epithelial tumors. Schematic diagram depicting the molecular elements and cellular events that underlie the feedback amplification loops that drive unlimited growth in tumors of epithelial origin induced either by the oncogenic cooperation between Ras-V12 and loss of polarity determinants (left) or by the induction of chromosomal instability (CIN) through depletion of Spindle Assembly Checkpoint genes (right).
Unraveling a tumor-intrinsic mechanism in CIN- and polarity-impaired epithelial tumors
So what are the molecular inputs to JNK activation in CIN-tumors and polarity-impaired tumors? Are interactions between tumor cell populations responsible for the unlimited growth potential of these tumors, as shown by their ability to grow 1000 fold in size in allograft transplantations in a matter of days [Citation11,Citation30]?. While we identified two distinct kinases of Drosophila JNKK (Hemipterous)—Ask1 in CIN tumors and Wallenda in polarity-impaired tumors—as those responsible for driving JNK activation in a tumor-dependent manner, the same type of interactions were found to play a relevant role in promoting tumor growth (). The two types of tumor were shown to have a mitotically quiescent delaminating cell population characterized by high levels of JNK activity and acting as a potent source of mitogenic Wingless, and knock down of the wg gene was observed to rescue tumor growth [Citation7, Citation11, Citation30]. While cell delamination in CIN-tumors is a result of aneuploidy [Citation7], in the case of polarity-impaired tumors it is caused, among others, by errors in the planar orientation of the mitotic spindle [Citation31]. We thus reasoned that the mitogenic activity of Wingless and Upd emanating from delaminating cells would increase proliferation in the epithelium and that this proliferation might augment the number of cells with chromosome missegregation (in CIN tumors) or mitotic spindle orientation errors (in polarity-impaired tumors). Consequently, the number of cells expressing mitogenic molecules would be expected to increase. This proposal was experimentally tested and successfully validated by genetic manipulation of the G2/M transition through expression of Cdc25/String or its negative regulator Tribbles, in the tumors. These data unveil a feedback amplification loop between two functionally distinct cell populations that increase each other's size through proliferation and delamination. Furthermore, this feedback can explain the unlimited growth potential of epithelial tumors caused by CIN or loss of apical-basal polarity. Remarkably, mammalian cells with complex karyotypes caused by chromosome segregation defects are cell-cycle arrested, exhibit features of senescence, and produce pro-inflammatory signals [Citation38], as occurs in epithelial models of CIN in Drosophila [Citation11,Citation30]. Most interestingly, CIN drives tumorigenesis in mammals (e.g [Citation26].). These results demonstrate the potential translational impact of the use of Drosophila as a model in cancer research.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Lada Murcia for comments. M.M. is an ICREA Research Professor. This work was funded by the SIGNAGROWTH-BFU2013-44485 and INTERGROWTH-BFU2016-77587-P grants from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO), and ERDF “Una manera de hacer Europa”. We gratefully acknowledge institutional funding from MINECO through the Centres of Excellence Severo Ochoa Award, and from the CERCA Programme of the Catalan Government. We apologize to those colleagues whose work could not be cited because of space restrictions.
Additional information
Funding
References
- Andersen DS, Colombani J, Palmerini V, et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature. 2015;522(7557):482–486. doi:10.1038/nature14298
- Bosch M, Serras F, Martín-Blanco E, et al. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol. 2005;280(1):73–86. doi:10.1016/j.ydbio.2005.01.002
- Boveri T. Zur Frage der Entstehung Maligner Tumoren [Origin of malignant tumors]. Gustav Fisher. 1914; Jena:1–64.
- Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. Embo J. 2003;22(21):5769–5779. doi:10.1093/emboj/cdg548
- Brumby AM, Richardson HE. Using Drosophila melanogaster to map human cancer pathways. Nat Rev Cancer. 2005;5(8):626–639. doi:10.1038/nrc1671
- Bunker BD, Nellimoottil TT, Boileau RM, et al. The transcriptional response to tumorigenic polarity loss in Drosophila. Elife. 2015;4. doi:10.7554/eLife.03189.
- Clemente-Ruiz M, Murillo-Maldonado JM, Benhra N, et al. Gene Dosage Imbalance Contributes to Chromosomal Instability-Induced Tumorigenesis. Dev Cell. 2016;36(3):290–302. doi:10.1016/j.devcel.2016.01.008
- Cohen SM. Imaginal disc development. Drosophila Development. A. Martinez-Arias and M. Bate. Cold Spring Harbor, Cold Spring Harbor Press. 1993;2:747–841.
- Cordero JB, Macagno JP, Stefanatos RK, et al. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell. 2010;18(6):999–1011. doi:10.1016/j.devcel.2010.05.014
- Dekanty A, Barrio L, Milán M. Contributions of DNA repair, cell cycle checkpoints and cell death to suppressing the DNA damage-induced tumorigenic behavior of Drosophila epithelial cells. Oncogene. 2015;34(8):978–985. doi:10.1038/onc.2014.42
- Dekanty A, Barrio L, Muzzopappa M, et al. Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc Natl Acad Sci U S A. 2012;109(50):20549–20554. doi:10.1073/pnas.1206675109
- Dekanty A, Milan M. Aneuploidy, cell delamination and tumorigenesis in Drosophila epithelia. Cell Cycle. 2013;12(5):728–731. doi:10.4161/cc.23949
- Duijf PH, Benezra R. The cancer biology of whole-chromosome instability. Oncogene. 2013;32(40):4727–4736. doi:10.1038/onc.2012.616
- Duijf PH, Schultz N, Benezra R. Cancer cells preferentially lose small chromosomes. Int J Cancer. 2013;132(10):2316–2326. doi:10.1002/ijc.27924
- Ferres-Marco D, Gutierrez-Garcia I, Vallejo DM, et al. Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature. 2006;439(7075):430–436. doi:10.1038/nature04376
- Figueroa-Clarevega A,, Bilder D. Malignant Drosophila Tumors Interrupt Insulin Signaling to Induce Cachexia-like Wasting. Dev Cell. 2015;33(1):47–55. doi:10.1016/j.devcel.2015.03.001
- Foijer F, Xie SZ, Simon JE, et al. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc Natl Acad Sci U S A. 2014;111(37):13427–13432. doi:10.1073/pnas.1400892111
- Gateff E. Malignant neoplasms of genetic origin in Drosophila melanogaster. Science. 1978;200(4349):1448–1459. doi:10.1126/science.96525
- Gonzalez C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer. 2013;13(3):172–183. doi:10.1038/nrc3461
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
- Haynie JL, Bryant PJ. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing discs of Drosophila melanogaster. Wilhelm Roux's Arch. 1977;183:85–100. doi:10.1007/BF00848779
- Herranz H, Weng R, Cohen SM. Crosstalk between epithelial and mesenchymal tissues in tumorigenesis and imaginal disc development. Curr Biol. 2014;24(13):1476–1484. doi:10.1016/j.cub.2014.05.043
- Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol. 2006;16(11):1139–1146. doi:10.1016/j.cub.2006.04.042
- Katheder NS, Khezri R, O'Farrell F, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541(7637):417–420. doi:10.1038/nature20815
- Kulshammer E, Mundorf J, Kilinc M, et al. Interplay among Drosophila transcription factors Ets21c, Fos and Ftz-F1 drives JNK-mediated tumor malignancy. Dis Model Mech. 2015;8(10):1279–1293. doi:10.1242/dmm.020719
- Levine MS, Bakker B, Boeckx B, et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev Cell. 2017;40(3):313–322e315. doi:10.1016/j.devcel.2016.12.022
- Li M, Fang X, Baker DJ, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(32):14188–14193. doi:10.1073/pnas.1005960107
- Milan M, Clemente-Ruiz M, Dekanty A, et al. Aneuploidy and tumorigenesis in Drosophila. Semin Cell Dev Biol. 2014;28C:110–115. doi:10.1016/j.semcdb.2014.03.014
- Morais da Silva, S., Moutinho-Santos T, Sunkel CE. A tumor suppressor role of the Bub3 spindle checkpoint protein after apoptosis inhibition. J Cell Biol. 2013;201(3):385–393. doi:10.1083/jcb.201210018
- Muzzopappa M, Murcia L, Milán M, et al. Feedback amplification loop drives malignant growth in epithelial tissues. Proc Natl Acad Sci U S A. 2017;114(35):E7291–E7300. doi:10.1073/pnas.1701791114
- Nakajima Y, Meyer EJ, Kroesen A, et al. Epithelial junctions maintain tissue architecture by directing planar spindle orientation. Nature. 2013;500(7462):359–362. doi:10.1038/nature12335
- Oromendia, A. B., Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012;26(24):2696–2708. doi:10.1101/gad.207407.112
- Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302(5648):1227–1231. doi:10.1126/science.1088474
- Pastor-Pareja JC, Xu T.. Dissecting social cell biology and tumors using Drosophila genetics. Annu Rev Genet. 2013;47:51–74. doi:10.1146/annurev-genet-110711-155414
- Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131(22):5591–5598. doi:10.1242/dev.01432
- Perez E, Lindblad JL, Bergmann A. Tumor-promoting function of apoptotic caspases by an amplification loop involving ROS, macrophages and JNK in Drosophila. Elife. 2017;6. doi:10.7554/eLife.26747.
- Ripoll P. Effect of terminal aneuploidy on epidermal cell viability in Drosophila melanogaster. Genetics. 1980;94:135–152.
- Santaguida, S., Richardson A, Iyer DR, et al. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell. 2017;41(6):638–651e635. doi:10.1016/j.devcel.2017.05.022
- Sheltzer JM, Torres EM, Dunham MJ, et al. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci U S A. 2012;109(31):12644–12649. doi:10.1073/pnas.1209227109
- Stark MB. An hereditary tumor in the fruit fly, Drosophila. J Cancer Res. 1918;3:279–301.
- Stingele S, Stoehr G, Peplowska K, et al. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:608. doi:10.1038/msb.2012.40
- Torres EM, Dephoure N, Panneerselvam A, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143(1):71–83. doi:10.1016/j.cell.2010.08.038
- Torres EM, Sokolsky T, Tucker CM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317(5840):916–924. doi:10.1126/science.1142210
- Uhlirova M, Bohmann D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. Embo J. 2006;25(22):5294–5304. doi:10.1038/sj.emboj.7601401
- von Hansemann D. Ueber asymmetrische Zelltheilung in epithel Krebsen und deren biologische Bedeutung [On the asymmetrical cell division in epithelial cancers and its biological significance]. Virchow's Arch Path Anat. 1890;119:299. doi:10.1007/BF01882039
- Willsey, HR, Zheng X, Carlos Pastor-Pareja J, et al. Localized JNK signaling regulates organ size during development. Elife. 2016;5. doi:10.7554/eLife.11491.