
ABSTRACT
Drosophila melanogaster has proven to be a powerful genetic model to study human disease. Approximately 75% of human disease-associated genes have homologs in the fruit fly and regulatory pathways are highly conserved in Drosophila compared to humans. Drosophila is an established model organism for the study of genetics and developmental biology related to human disease and has also made a great contribution to epigenetic research. Many key factors that regulate chromatin condensation through effects on histone post-translational modifications were first discovered in genetic screens in Drosophila. Recently, the importance of chromatin regulators in cancer progression has been uncovered, leading to a rapid expansion in the knowledge on how perturbations of chromatin can result in the pathogenesis of human cancer. In this review, we provide examples of how Drosophila melanogaster has contributed to better understanding the detrimental effects of mutant forms of histones, called ‘oncohistones’, that are found in different human tumours.
Drosophila as an animal model for cancer
As discussed by Villegas[Citation1] and Gonzalez[Citation2], Mary Stark’s work[Citation3] over a century ago on the lethal(l)7 strain, where larvae develop intense black spots in their bodies before they die prior to eclosion, pioneered the use of Drosophila as a cancer model. Stark produced comprehensive work on these dark bodies and even (unsuccessfully) tried to transfer them to healthy animals, which was the first attempt at tumour transplantation in Drosophila. Many years later, Elizabeth Gateff and Howard Schneiderman discovered the first Drosophila tumour suppressor gene, lethal giant larvae (lgl). Homozygous lgl mutant larvae develop malignant tumours while heterozygotes do not, therefore, lgl behaves as a tumour suppressor gene. Gateff developed fly models for cancer research for use in both genetic and epigenetic studies and what she discussed more than 40 years ago, on the need to better understand the implication of both genetics and epigenetics in the pathogenesis of cancer[Citation4], has now been confirmed by the discovery of many inactivating mutations in genes that control the epigenome in human cancers[Citation5].
Later, the development of sophisticated genetic tools in flies, such as the UAS/Gal4 system[Citation6] and the FLP-FRT system[Citation7], enabled study of tissues formed by wild-type and oncogenic mutant clones, leading to important discoveries in signalling pathways that very often came before the discovery of the link to cancer of the human orthologs, and therefore contributed to understanding the biology of tumorigenesis. It is also important to note that around 75% of genes causing human diseases are thought to have a functional homolog in flies[Citation8], underscoring the utility of Drosophila as a disease model.
In this review, we will discuss oncohistones, focusing on the two best characterized mutations H3K27M and H3K36M, and illustrating the contribution of Drosophila to better understanding the mechanistic effects of these mutations. We propose how the Drosophila model can be very helpful in studying many new histone mutations reported since. We will start by giving a short background on chromatin and histone proteins, in particular histone variant H3.3 which has been most frequently linked to cancer and which undergoes important regulatory post-translational modifications. We will then focus on two residues affected by oncogenic mutations, H3K27 and H3K36, and on hypotheses as to how those mutations can lead to genetic dysregulation and cancer.
Chromatin, the main substrate for epigenetics
Chromatin is a dynamic structure that compacts the genome to fit into the nucleus and also regulates the accessibility of DNA for transcription, replication and DNA repair. The different chromatin components are histone proteins that form the nucleosome, the basic repeating unit of chromatin, enzymes that covalently modify histones and DNA, and ATP-dependent remodellers of nucleosome structure[Citation9]. Chromatin organization in the nucleus provides additional information to that encoded genetically and plays a major role in determining cellular state by influencing cell identity and plasticity. In contrast to the genetic code, epigenetic features of chromatin are dynamic and can be reversed, allowing genetic reprogramming. In the past decade, the importance of chromatin regulators in cancer progression has been uncovered, leading to a rapid expansion in the knowledge on how perturbations of chromatin can result in the pathogenesis of human cancer[Citation10].
The histone variant H3.3
Histone proteins can be classified into two groups: replication-dependent ‘canonical histones’ or replication-independent ‘histone variants’. Replication-dependent histones are transcribed at high levels during S phase from clusters of intron-less genes, whereas histone variants are usually synthesized throughout the cell cycle and are incorporated into the genome in a replication-independent manner. Canonical histone genes in Drosophila are located in a single cluster on the left arm of chromosome 2 called HisC which comprises ~100 copies of the histone gene unit His-GU that contains genes encoding the four canonical core histones, H2A, H2B, H3, and H4, and the linker histone H1[Citation11].
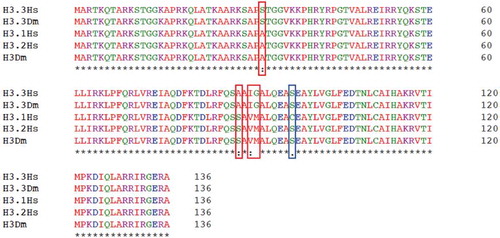
The histone variant H3.3 is highly conserved in evolution. In Drosophila it is encoded by two genes, and it differs from the canonical histone H3 by substitutions in four amino acids. There are two canonical H3 proteins in humans called H3.1 and H3.2, they differ by only one amino acid and H3.2 is identical to the single canonical H3 in Drosophila. H3.3 is also identical in humans and in flies (). The H3.3-specific amino acids contribute to the localization of H3.3 on chromatin, as mutating these residues prevents the incorporation of the replication-independent H3.3 into nucleosomes[Citation12]. Histone chaperones that bind to H3.3 specifically target these amino acids and direct its localization to specific parts of the genome. H3.3 is assembled into chromatin by the histone regulator A (HIRA) complex[Citation13] independently of DNA synthesis. In both human cells and Drosophila, H3.3 is deposited by this complex into DNA-regulatory elements, promoters and transcribed genes[Citation14], reflecting a close association between H3.3 incorporation and transcriptional activity. Other histone chaperone complexes participate in the deposition of H3.3 in other regions of the genome and the DAXX/ATRX complex[Citation15] is responsible for its incorporation at telomere and pericentric heterochromatin.
Figure 1. Histone H3 and H3.3 are conserved in humans and flies
Histone post translational modifications
Histone post-translational modifications define the ‘histone code’[Citation16] and are marks that enable recruitment of downstream reader or effector proteins. Histone H3 and its variant H3.3 are the histones that undergo the largest number of post-translational modifications.
Histone H3K27 methylation
Histone H3K27 can be mono, di or tri-methylated or acetylated. H3K27 di- and tri-methylation are associated with transcriptional repression and methylation of this residue is regulated by Polycomb group (PcG) proteins. PcG proteins are essential epigenetic regulators that were first discovered in Drosophila for their role in establishing the stable repression of homoeotic (Hox) genes throughout development[Citation17] thus controlling segment identity. PcG proteins have key roles in development, pluripotency, senescence, and cancer[Citation18]. They function within multiprotein complexes that maintain gene silencing of important development regulators. PcG proteins form evolutionarily conserved multimeric repressive complexes called Polycomb repressive complex 1 and 2 (PRC1 and PRC2), that can function together to silence PcG target genes[Citation19]. Conversely the Trithorax group (TrxG) of proteins have opposing effects on Hox genes and many members of this group activate transcription. TrxG proteins are also important regulators of development and differentiation ().
Table 1. A comparison of selected chromatin remodellers in humans and flies
PRC2 comprises as core components Esc, Su(z)12, Caf1-55, Jing, and the lysine methyltransferase E(z). It sets up stable repression of target genes by trimethylation of histone H3 at lysine 27 (H3K27me3). PRC1, which is composed of Pc, Ph, Scm, Ring1, and either of the two closely related proteins Su(z)2 or Psc, maintains the silenced state of chromatin through the recognition and binding of H3K27me3 by the chromodomain protein Pc, which enables the mono-ubiquitination of Lys118 on H2A (H2AK118ub) by other PRC1 subunits. H2AK118ub is associated with chromatin compaction, and repression of the underlying genes, thereby establishing strong transcriptional repression [Citation20–22]. In Drosophila, the recruitment of PcG complexes is regulated by DNA regulatory elements called PREs[Citation23] (Polycomb response elements). Hierarchical models have been proposed for recruitment of PRC1 and PRC2 to these elements with different sequences of events [Citation24,Citation25]. However, in cultured cells from Drosophila embryos which are deficient for Su(z)12 and therefore lack PRC2, PRC1 is nevertheless recruited to PREs[Citation26]. Similarly, PRC2 is recruited to approximately one-third of PREs in cultured embryonic cells lacking the PRC1 components Psc and Su(z)2. These results argue for multiple mechanisms for PcG recruitment to PREs.
Histone H3K36 methylation
Nucleosomes hinder the recruitment or movement of RNA polymerase II (RNAPII) and the associated transcription machinery. ATP-dependent chromatin remodellers and post-translational modifications of histone proteins help to overcome these challenges. Although transcription initiation plays an essential role in the regulation of gene expression, transcription elongation is also an important step in this regulation, and the methylation of H3K36 by SETD2 is a key modification in the process of transcription elongation[Citation27]. Methylation of H3K36 is a hallmark of active transcription but has also other roles in DNA repair and RNA splicing.
SETD2 catalyzes the trimethylation of H3K36. This modification is enriched in the gene bodies of actively transcribed genes, but H3K36 trimethylation by SETD2 is thought to be repressive to transcription[Citation28]. Studies in budding yeast have established a model for the recruitment of the SETD2 ortholog which interacts with the phosphorylated C-terminal domain of elongating RNAPII leading to the deposition of H3K36 methylation co-transcriptionally throughout gene coding regions. This mark is recognized by Eaf3, a component of the Rpd3C(S) histone deacetylase (HDAC) complex, which binds to methylated H3K36[Citation29]. Histone deacetylation is associated with inhibition of transcription by promoting chromatin compaction[Citation30], therefore preventing cryptic transcription.
In metazoans, multiple H3K36 methyltransferases exist with distinct functions (). SETD2 is the sole enzyme responsible for producing H3K36me3, whereas MES-4 in C. elegans and NSD proteins in Drosophila and mammals catalyse mono- and/or dimethylation of H3K36. Ash1, another H3K36 methyltransferase and a TrxG protein, is involved in the dimethylation of H3K36 at specific regions of the genome[Citation31]. Ash1 works in a complex with Mrg15 and Caf1 to prevent H3K27me3 deposition by the PRC2 complex and antagonizes repression at Hox genes through H3K36 dimethylation [Citation32,Citation33]. The role of Ash1 in counteracting Polycomb repression has been established but whether it is through H3K36 methylation remains unclear. Recent work[Citation34] showed that Ash1 is the only H3K36 specific methyltransferase required to counteract Polycomb repression of homoeotic genes and that NSD and Set2 did not have any role in this repression. Unexpectedly, Ash1-mediated opposition to Polycomb repression was independent of H3K36 methylation yet still required the SET domain of Ash1.
Oncohistones
Mutations in chromatin-modifying enzymes and remodellers have been found many times in different cancer types[Citation10], but more recently, mutations within histone proteins themselves have been reported in different tumours including gliomas, sarcomas and head and neck squamous carcinomas [Citation35–39]. These mutations occur in the N-terminal tail of histone H3 (more often the histone H3.3 variant) and are found at or near key regulatory post-translational modification sites, therefore likely altering the writing, reading and erasing of these marks, consequently changing the conformation of chromatin and disrupting regulation of gene expression[Citation40]. Mutated histones with amino acid substitutions that are expressed in tumours are called ‘oncohistones’ and are considered to be drivers of tumorigenesis.
H3K27M and H3K36M oncohistones and molecular mechanisms
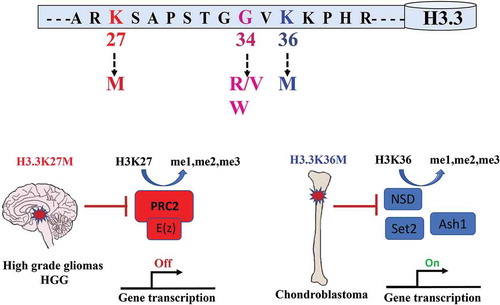
Substitutions of methionine to lysine on H3.3K27 or H3.3K36 are the most common alterations in cancer, occurring in diffuse intrinsic pontine gliomas and chondroblastomas respectively, and these oncohistones affect the function of PRC1 and PRC2 directly or indirectly [Citation37,Citation41].()
Figure 2. Oncogenic mutations in Histone H3.3 affect its post-translational modification
H3K27M molecular mechanisms
K27M mutations occurs mainly on one of the two genes encoding H3.3, and more rarely on the canonical histones H3.1 and H3.2. This mutation is present in 80% of DIPGs[Citation41] (diffuse intrinsic pontine gliomas), particularly aggressive paediatric brain tumours that are localized to a specific region of the brain. H3.3K27M is associated with mutations in TP53 and PDGFRA[Citation35] (platelet-derived growth factor receptor A), while H3.1K27M is more often associated with mutations in ACVR1[Citation42] (Activin A Receptor Type 1).
H3K27M causes a global decrease in H3K27 methylation and has a dominant negative role on the catalytic subunit of PRC2 EZH241. A proposed model to explain how the H3K27M mutation plays its dominant effect is by stabilizing the binding of PRC2 to the mutant histone. EZH2 affinity for H3K27M peptide is 20-fold higher than for wild-type peptide[Citation43], thus sequestering the EZH2 methyltransferase and blocking the genome-wide deposition of H3K27me2 and H3K27me3. Additionally, H3K27M affects PRC2 automethylation[Citation44], thus decreasing its histone methyltransferase activity. Automethylation of PRC2 occurs on EZH2 and SUZ12, the two core subunits. Two residues in EZH2, EZH2-K510 and EZH2-K514, have been identified as major sites for automethylation. This mechanism increases the histone methyltransferase activity required to reach proper H3K27me3 levels. EZH2 automethylation is significantly reduced in the presence of H3K27M, potentially disrupting the intrinsic activity of PRC2 and leading to global decrease in H3K27me3 levels. Interestingly, Chan et al[Citation45]. showed that this global decrease in H3K27 methylation levels was also associated with an important enrichment of H3K27me3 and EZH2 at specific loci which sets limits to the proposed model. ChIP-seq analysis of mouse neural stem cells expressing H3.3K27M and PDGFB[Citation46], where the distribution of H3K27me3 was mapped genome-wide, showed that H3K27me3 is retained and even accumulates at several loci. Moreover, epigenome profiling of H3K27M-mutant DIPG cells found that H3K27M associates with increased H3K27 acetylation (H3K27ac)[Citation47], and that heterotypic H3K27M-K27ac nucleosomes colocalized at actively transcribed genes from where PRC2 was excluded suggesting that K27M does not sequester PRC2. Work by Tatavosian et al[Citation48] brought some insight on how H3K27M reduces global H3K27 methylation, but retains H3K27me3 at a subset of genes in vivo. Using live-cell single molecule tracking, they determined the binding and search kinetics of PRC2 for its targets and how H3K27M impacts these parameters. H3K27M alters the binding and target search mechanism by increasing the residency time of EZH2 on chromatin and the search time for a specific target by 1.5-fold compared to wild-type. This lengthened search time has a differential effect on PRC2 targets, with lesser effects on strong Polycomb targets (more binding sites), but greater effects on weaker Polycomb targets (fewer binding sites).
H3K36M molecular mechanisms
Similarly to H3K27M which inhibits its specific methyltransferase (EZH2), H3K36M mutations inhibit H3K36 methyltransferases, more specifically SETD2 and NSD2 which are responsible for producing H3K36me3 and H3K36me2. H3K36M has no effect on two other methyltransferases, ASH1L and NSD1. This leads to global H3K36 hypomethylation and a subsequent increase in H3K27 methylation. ChIP-sequencing analysis in H3K36M-mutant mesenchymal precursor cells identified an intergenic gain of H3K27 trimethylation (H3K27me3) where H3K36me2 was significantly reduced. The increase in intergenic H3K27me3 leads to the redistribution of Polycomb-repressive complex 1 (PRC1) from its target genes and aberrant gene activation[Citation37]. Similar results were observed by Fang et al[Citation49], showing that H3.3K36M mutant proteins inhibit NSD2 and SETD2 to reduce H3K36 methylation and also affect various H3K36 methyltransferases differently, with no inhibitory effect on ASH1L and NSD1 activities.
Other histone mutations
Mutations in H3.3 were also reported in cortical high grade glioma (HGG) in which G34 is substituted by arginine or valine (H3.3G34R/V)[Citation35] and in a benign tumour, giant cell of the bone, where glycine 34 is replaced by a tryptophan (H3.3G34W)[Citation39]. As glycine is not post-translationally modified, these mutations are thought to affect post-translational modifications of the neighbouring residues K27 and K36 [Citation41,Citation50].
A recent large-scale analysis of sequences of 183 specific tumour types from 3074 unique patients revealed 4205 missense mutations in all core histone proteins[Citation40]. This greatly expands the potential set of oncohistones and it will be very important to evaluate the potential roles of these mutations in the development of tumours and whether they are drivers of the tumorigenic process or merely passengers. These histone mutations are located both in the N-terminal tails and in the globular histone fold domains. Among these newly identified mutants are H3K4M and H3K4I. H3K4 methylation plays an important role in transcriptional activation and H3K4M/I could act by blocking methylation of this residue and repressing gene expression. Another interesting observation is the presence of mutations within a nucleosome anchoring point called the ‘acidic patch’ where chromatin remodellers bind to move nucleosomes around and change chromatin conformation. These mutations could potentially affect many essential chromatin mediated processes and by doing so promote oncogenesis. Putative neomorphic mutations were also reported, where the mutation produces an amino acid that can undergo PTMs, leading to the aberrant recruitment of writers or reader complexes. Examples are H3E73K and H3K73Q, where glutamic acid, which cannot be modified, is replaced with lysine, which can be acetylated, or with glutamine, which is an acetyl mimic.
Oncohistones in Drosophila melanogaster
As Drosophila has played such an important role in investigating chromatin biology, it is unsurprising that several groups have used it to study the effects of oncohistones on histone post-translational modifications and gene expression. Importantly, the amino acid sequence of H3.3 is identical in Drosophila and human, so the effects of oncogenic histone mutations can be directly studied in flies. In addition, Drosophila provides opportunities to examine the effects of histone mutations that have not been, at least thus far, identified in humans, but that may nonetheless be informative for studying histone function.
One such example is H3K27R, which is not post-translationally modified. Flies were constructed that express only H3K27R, and these fail to repress genes that are normally repressed by PRC2, resulting in homoeotic transformations[Citation51]. Shortly after the report of these previously unknown mutations in histone proteins, Herz et al[Citation52] established a fly model expressing the human H3.3K27M oncohistone. Overexpression of H3K27M resembled the PRC2 loss of function phenotype, causing derepression of PRC2 target genes and developmental perturbation. They also studied H3K9M mutants (to date not reported in any tumour) to better understand the role of H3K9 methylation. Indeed, substitution of H3K9 by a methionine (H3K9M) depleted H3K9 methylation levels, disrupting HP1 interaction which is enriched at telomeres and centromeres[Citation53] and therefore suppressing PEV (position effect variegation) in different Drosophila tissues. This work confirmed Drosophila as a robust in vivo model to study K to M histone H3.3 mutants, to gain molecular insights into chromatin signalling pathways.
Sarthy et al[Citation54] used Drosophila to show that inhibition of H3K27 trimethylation occurs only when H3K27M oncohistones are deposited into chromatin. Drosophila S2 cells were transfected with either H3.3K27M or H3.2K27M mutants or only the corresponding N-terminal tail (residues 1–44). A decrease in H3K27me3 levels was only observed with the full-length proteins even though the truncated proteins lacking the histone fold domain (HFD) also localized to nuclei, arguing that incorporation of H3K27M into nucleosomes is necessary to influence H3K27 methylation. Taking advantage of the well-characterized larval eye disc system, unique to Drosophila, and using the GMR-Gal4 driver to express H3K27M for only one more division before differentiation into photoreceptors, they further demonstrated that H3.3K27M inhibition of H3K27 trimethylation is only effective in proliferating cells. Expression of H3K27M under the control of the GMR-Gal4 driver did not produce any morphological defects on its own. However, when the cyclin E inhibitor p21, which arrests cell division at late S phase, is concomitantly expressed with H3K27M, H3K27 trimethylation is no longer inhibited. This further supports the conclusion that H3K27M only inhibits H3K27 trimethylation in proliferating cells.
However, recently published work by Jain et al[Citation55] challenges the conclusion that incorporation of H3K27M into chromatin is essential to affect H3K27 methylation levels. They expressed mutant forms of H3.3 in which residues L126 and I130, which are required for H3-H3 dimerization are mutated to alanine. H3.3L126A/I130A inhibited H3-H4 tetramer formation in vitro, and this mutant histone was, as expected, absent from chromatin. Surprisingly, expression of triple-mutant transgene H3.3K27M/L126A/I130A in mouse embryonic fibroblasts, reduced global H3K27me2/me3 levels. H3.3K27M/L126A/I130A also immunoprecipitated PRC2 subunits from cell lysates, linking this reduction to a direct interaction with PRC2 in vivo even though it is not deposited into chromatin. Additionally, they used Drosophila to study EZHIP (EZH inhibitory protein) a newly discovered protein associated with posterior fossa A (PFA) ependymomas[Citation56]. EZHIP has been shown to act as an H3K27M mimic [Citation53,Citation54] and expression of this human placental protein in wing discs repressed H3K27me3. This demonstrated that EZHIP, which is not found outside of mammals, nevertheless inhibits Drosophila PRC2 through a conserved mechanism.
In our recent work[Citation57], we also aimed to identify pathways involved in the biology of H3.3 mutations by using a transgenic Drosophila model of oncohistones H3.3G34R and H3.3K27M. Ubiquitous expression of these transgenes is lethal at the pupal stage respectively, and the H3.3K27M mutant is also lethal when expressed in glia cells. We decided to focus on the H3.3K27M oncohistone and conducted a genetic modifier screen to look for genes that would rescue or positively shift the phenotype to later stages of development by knocking down candidate genes involved in glial cell biology and/or affecting H3.3. To do this we used the UAS-Gal4 system and publicly-accessible libraries of fly strains expressed interfering RNA targeting essentially all Drosophila genes[Citation58]. Such a forward-genetic screen is prohibitively expensive and laborious in a mammalian animal model. We found that downregulation of PRC2 components E(z), Su(z)12 and Caf1-55 shifted the lethal phenotype to earlier stages of development, underlying a synergistic effect with H3.3K27M. Interestingly, knockdown of some PRC1 components (Psc, ph-d and Su(z)2) suppressed the H3.3K27M phenotype, shifting the lethality to later stages and demonstrating a functional involvement for PRC1 in H3.3K27M detrimental effects. We then looked to the expression levels of human orthologs of the identified genes in the screen in H3.3K27M glioma samples and confirmed that these genes were significantly upregulated as a set when compared to wild-type gliomas or normal human brain samples. Some of the suppressors we identified in Drosophila are cell cycle regulators such as CycD which was also significantly upregulated in H3.3K27M HGG compared to normal brain samples. Therefore, Drosophila provided a unique and unbiased approach to identifying genes that are functionally involved in the H3K27M phenotype. Furthermore, some of these candidates have already been targeted for other purposes by FDA-approved drugs, leading to potential new therapeutic approaches for HGGs.
Conclusion
In this review, we have reported and discussed various roles of histone H3 proteins and their PTMs in the regulation of gene expression, and how the fruit fly contributed to identify and understand the role of writers, erasers and readers of these PTMs, underlying the tremendous contribution of Drosophila in epigenetics research. With the development of new techniques and strategies, such as CRISPR-Cas9 technology[Citation59] for precise gene editing and new chromatin immuno-precipitation techniques such as CUT&RUN[Citation60] which provides high quality data from as few as 100 cells, Drosophila melanogaster will continue to play a major role in epigenetic research.
For instance, Zhang et al[Citation61]. used CRISPR-Cas9 and the attP/attB double integration system in Drosophila to alter histone gene number and sequences, generating 40 histone mutants, where each known modified residue of H3 and H4 was substituted with an alanine. Many of these mutations compromised viability and fertility, uncovering novel essential functions for particular amino acids in histones. For example, H4K16 was required for expression of male X-linked genes and male viability, and replacing all H3K4 by alanine produced embryonic lethality. Additionally, Zhang et al. developed FLP/FRT transgenic flies to perform mosaic analysis of these mutants, a valuable technique in Drosophila that allows comparison of mutant and wild-type cells in the same tissues, and also enables analysis of mutations that are lethal when present throughout the organism.
Similar experiments using Drosophila would be a very useful system to rapidly assess, in a controlled genetic background, the effects of the large number of histone mutants recently uncovered in human tumours[Citation40], perhaps focusing first on H3K4M/I and H3E73K/Q where hypotheses for a functional mechanism exist.
Acknowledgments
P.L. acknowledges support from the Canadian Rare Disease Models and Mechanisms (RDMM) Network and from a Visiting Professorship from the Radboud University Excellence Program.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Villegas SN. One hundred years of Drosophila cancer research: no longer in solitude. DMM Dis Model Mech. 2019;12:dmm039032.
- Gonzalez C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer. 2013;13:172–183.
- Stark MB. An hereditary tumor in the fruit fly drosophila. J Cancer Res. 1918;3:279–301.
- Gateff E. The genetics and epigenetics of neoplasms in Drosophila. Biol Rev Camb Philos Soc. 1978;53:123–168.
- You JS, Jones PA. Cancer Genetics and Epigenetics: two Sides of the Same Coin? Cancer Cell. 2012;22(1):9–20.
- Brand A, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415.
- Golic KG, Lindquist S. The FLP recombinase of yeast catalyzes site-specific recombination in the drosophila genome. Cell. 1989;59(3):499–509.
- Ugur B, Chen K, Bellen HJ. Drosophila tools and assays for the study of human diseases. DMM Dis Model Mech. 2016;9(3):235–244.
- Yadav T, Quivy J-P, Almouzni G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science. 2018;361(6409):1332–1336.
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55.
- McKay DJ, Klusza S, Penke TR, et al. Interrogating the function of metazoan histones using engineered gene clusters. Dev Cell. 2015;32(3):373–386.
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9(6):1191–1200.
- Tagami H, Ray-Gallet D, Almouzni G, et al. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116(1):51–61.
- Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9:15–26.
- Drané P, Ouararhni K, Depaux A, et al. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24(12):1253–1265.
- Jenuwein T. Translating the histone code. Science. 2001;293(5532):1074–1080.
- Schuettengruber B, Bourbon H-M, Di Croce L, et al. Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell. 2017;171(1):34–57.
- Loubiere V, Martinez A-M, Cavalli G. Cell fate and developmental regulation dynamics by Polycomb proteins and 3D genome architecture. BioEssays. 2019;41(3):e1800222.
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708.
- Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043.
- Wang H, Wang L, Erdjument-Bromage H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431(7010):873–878.
- Gil J, O’Loghlen A. PRC1 complex diversity: where is it taking us? Trends Cell Biol. 2014;24(11):632–641.
- Müller J, Kassis JA. Polycomb response elements and targeting of Polycomb group proteins in Drosophila. Curr Opin Genet Dev. 2006;16:476–484.
- Wang L, Brown JL, Cao R, et al. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell. 2004;14:637–646.
- Blackledge NP, Farcas A, Kondo T, et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014;157(6):1445–1459.
- Dorafshan E, Kahn TG, Schwartz YB. Hierarchical recruitment of Polycomb complexes revisited. Nucleus. 2017;8(5):496–505.
- Venkatesh S, Workman JL. Recognizing methylated histone variant H3.3 to prevent tumors. Cell Res. 2014;24(6):649–650.
- Strahl BD, Grant PA, Briggs SD, et al. Set2 Is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 2002;22(5):1298–1306.
- Keogh M-C, Kurdistani SK, Morris SA, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123(4):593–605.
- Butler JS, Dent SYR. Chromatin ‘resetting’ during transcription elongation: a central role for methylated H3K36. Nat Struct Mol Biol. 2012;19:863–864.
- Tanaka Y, Katagiri ZI, Kawahashi K, et al. Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene. 2007;397:161–168.
- Huang C, Yang F, Zhang Z, et al. Mrg15 stimulates Ash1 H3K36 methyltransferase activity and facilitates Ash1 Trithorax group protein function in Drosophila. Nat. Commun. 2017;8:1649.
- Schmähling S, Meiler A, Lee Y, et al. Regulation and function of H3K36 di-methylation by the trithorax-group protein complex AMC. Dev. 2018;145(7):dev163808.
- Dorafshan E, Kahn TG, Glotov A, et al. Ash1 counteracts Polycomb repression independent of histone H3 lysine 36 methylation. EMBO Rep. 2019;20:e46762.
- Schwartzentruber J, Korshunov A, Liu X-Y, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231.
- Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253.
- Lu C, Jain SU, Hoelper D, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352(6287):844–849.
- Papillon-Cavanagh S, Lu C, Gayden T, et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. . Nature Genetics. 2017;49(2):180–185.
- Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013;45(12):1479–1482.
- Nacev BA, Feng L, Bagert JD, et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature. 2019;567(7749):473–478.
- Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861.
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014;46(5):462–466.
- Justin N, Zhang Y, Tarricone C, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016;7(1):11316.
- Lee C-H, Yu J-R, Granat J, et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev. 2019;33(19–20):1428–1440.
- Chan K-M, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27(9):985–990.
- Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017;23(4):483–492. .
- Piunti A, Hashizume R, Morgan MA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017;23(4):493–500.
- Tatavosian R, Duc HN, Huynh TN et al. Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat Commun. 2018;9:2080.
- Fang D, Gan H, Lee JH, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 2016;352:1344–1348.
- Shi L, Shi J, Shi X, et al. Histone H3.3 G34 mutations alter histone H3K36 and H3K27 methylation in cis. J Mol Biol. 2018;430(11):1562–1565.
- Pengelly AR, Copur Ö, Jäckle H, et al. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor polycomb. Science. 2013;339:698–699.
- Herz H-M, Morgan M, Gao X, et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 2014;345(6200):1065–1070.
- Danzer JR, Wallrath LL. Mechanisms of HP1-mediated gene silencing in Drosophila. Development. 2004;131:3571–3580.
- Sarthy JF, Meers MP, Janssens DH, et al. Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones. Elife. 2020;9:e61090.
- Jain SU, Rashoff AQ, Krabbenhoft SD, et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol Cell. 2020;80(4):1–10.
- Hübner JM, Müller T, Papageorgiou DN et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 2019;21:878–889.
- Berlandi J, Chaouch A, De Jay N, et al. Identification of genes functionally involved in the detrimental effects of mutant histone H3.3-K27M in Drosophila melanogaster. Neuro Oncol. 2019;21(5):628–639.
- Dietzl G, Chen D, Schnorrer F, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448(7150):151–156.
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821.
- Skene PJ, Henikoff JG, Henikoff S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018;13(5):1006–1019.
- Zhang W, Zhang X, Xue Z, et al. Probing the function of metazoan histones with a systematic library of H3 and H4 mutants. Dev Cell. 2019;48(3):406–419.e5.