
ABSTRACT
Drosophila pseudoobscura is a classic model system for the study of evolutionary genetics and genomics. Given this long-standing interest, many genome sequences have accumulated for D. pseudoobscura and closely related species D. persimilis, D. miranda, and D. lowei. To facilitate the exploration of genetic variation within species and comparative genomics across species, we present PseudoBase, a database that couples extensive publicly available genomic data with simple visualization and query tools via an intuitive graphical interface, amenable for use in both research and educational settings. All genetic variation (SNPs and indels) within the database is derived from the same workflow, so variants are easily comparable across data sets. Features include an embedded JBrowse interface, ability to pull out alignments of individual genes/regions, and batch access for gene lists. Here, we introduce PseudoBase, and we demonstrate how this resource facilitates use of extensive genomic data from flies of the Drosophila pseudoobscura subgroup.
Introduction
Flies of the Drosophila pseudoobscura subgroup have a long and rich history as model systems in evolutionary genetics and genomics. The pseudoobscura subgroup is particularly recognized for its roles in the study of the population genetics of chromosomal inversions and the evolutionary genetics of species barriers. In the early 1900s, the development of cytogenetic methods that visualized karyotypes of polytene chromosomes enabled analysis of variation in Drosophila chromosome structure. These cytogenetic approaches enabled a wide range of genetic analyses, including early genetics-based species phylogenies based on inversion differences between closely related species of the pseudoobscura subgroup [Citation1]. Surveys of natural populations of the pseudoobscura subgroup quantified extensive variation within and between populations as well as fixed structural differences between species [Citation2–4]. In early analyses, inversion differences were often thought to be neutrally evolving, based on the logic that inversions can change gene order without disrupting gene content. With the rapid expansion of DNA sequencing technology in the early 2000s, the pseudoobscura subgroup played a key role in the resurgence of empirical and theoretical research on inversion polymorphisms [Citation5]. Today, inversions are recognized for the diverse roles across various taxa in local adaptation, divergence, and speciation [Citation6,Citation7,Citation8,Citation9,Citation10].
Flies of the pseudoobscura group remain prominent model systems for understanding the evolutionary genetics of differences between species. Thus, many genome sequences have accumulated both for D. pseudoobscura and for its close relative species. D. pseudoobscura and its naturally-hybridizing sister species, D. persimilis, provide a model for understanding how inversions shape speciation. Nearly all reproductive barriers between these species map to the fixed or nearly fixed inversions that differ between them [Citation10,Citation11]. A subspecies of D. pseudoobscura, D. pseudoobscura bogotana, also provides important insights in speciation research. There are two named subspecies of D. pseudoobscura – D. pseudoobscura pseudoobscura and D. pseudoobscura bogotana. Throughout this article, we use D. pseudoobscura to refer to both subspecies, and we specify D. pseudoobscura pseudoobscura or D. pseudoobscura bogotana when we are specifically referring to only one of the two subspecies. D. pseudoobscura bogotana is found near Bogota, Colombia, and does not exchange genes with allopatric D. pseudoobscura pseudoobscura or D. persimilis. Thus, D. pseudoobscura bogotana is an important point of genomic comparison for D. pseudoobscura pseudoobscura and D. persimilis, and has served as a model for the early stages of speciation [Citation12–15]. Additional important points of comparison for D. pseudoobscura evolutionary genetics include D. lowei and D. miranda. D. lowei and D. pseudoobscura likely diverged 5–11 million years ago [Citation16], and D. lowei often serves as an outgroup species for comparative genetics and genomics within the pseudoobscura subgroup [Citation17–19]. D. miranda is also often leveraged as a point of comparison for D. pseudoobscura [Citation19,Citation20] and has become particularly known as a model for sex chromosome evolution [Citation21,Citation22].
As is often the case with classic study systems, multiple laboratories have sequenced a variety of strains of D. pseudoobscura and closely related species. To facilitate the exploration of genetic variation within and among these species, we present PseudoBase as a resource that presents publicly available genomic data via an intuitive graphical interface, accessible to students or researchers without any prior experience in working with genomic data. Our intention is to provide a resource that maximizes the utility of sequence data by lowering barriers to working with these data. PseudoBase originated in 2012 as a resource primarily for the Noor laboratory at Duke University, for a handful of other laboratories, and for use as a classroom tool. The ongoing accumulation of new genomic data for these species as well as a desire for broader functionality recently prompted a redesign of the original interface. We released ‘PseudoBase 2.0ʹ, http://pseudobase.biology.duke.edu/, to include a broader sampling of publicly available pseudoobscura subgroup genomes, update the underlying D. pseudoobscura reference genome, and provide a significantly improved user interface. The new user interface includes embedded JBrowse visualization tools, the ability to download FASTA formatted alignments of individual genes/regions, and batch access for gene lists. Here, we provide an overview of the underlying data within PseudoBase, we introduce its features and functionalities, and we illustrate how this public resource supports the use of flies of the pseudoobscura subgroup for biological discovery and education.
Materials and methods
Sequencing information and variant calling
PseudoBase aggregates whole genome paired-end Illumina experiments from multiple laboratory groups and experiments [Citation17,Citation19,Citation23–25]. Raw data and associated details are available on the NCBI Short Read Archive under the sample accessions indicated on PseudoBase and listed in . When more than one whole genome experiment was available for a given strain (e.g., D. pseudoobscura Flagstaff 14), we included the one sequenced using the most recent technology (which happened to also provide the highest coverage). We did not include genomes resulting from crosses of multiple laboratory strains.
Table 1. Genomes represented in the PseudoBase 2.0 release
The pipeline used for genome alignment and variant calling is available on GitHub (https://github.com/kkorunes/PseudobaseScripts). We first used BWA-0.7.17 [Citation26] to align all sequences to the D. pseudoobscura genome assembly (Dpse_3.04: GCA_000001765.2), obtained from FlyBase [Citation27]. We next used Picard command line tools to mark adapters and duplicates (http://broadinstitute.github.io/picard/). Variants were called and filtered using GATK v4.1.1 [Citation28,Citation29]. We filtered SNPs and indels separately, according to the hard filtering recommendations provide by GATK. Specifically, we excluded SNPs with QualByDepth (QD) < 2.0, FisherStrand Bias (FS) > 60, and StrandOddsRatio (SOR) > 3.0, MQ < 40, MQRankSum < −12.5, ReadPosRankSum < −8. Indels were filtered to exclude variants with QualByDepth (QD) < 2.0, FisherStrand (FS) > 200, and StrandOddsRatio (SOR) > 10.0, ReadPosRankSum < 20.
Database architecture
PseudoBase server-side code is written in Python using the Django Framework. Sequence alignments are stored on the server as a series of indexed files (one indexed file per chromosome/strain), and indexes are created within each file for every reference sequence base position. This allows for fast retrieval of both aligned and unaligned sequences for any specifically requested chromosome region or gene searches. PseudoBase utilizes a mySQL database to store a) strain metadata, b) pointers to indexed sequence alignment file locations on the server for each chromosome/strain, and c) pointers to gene locations within sequences, to assist with optimizing search queries. Importantly, new strains can be readily added to PseudoBase as they become available. This is facilitated via an import mechanism which utilizes standard Variant Call Format (VCF) files as input. Each imported VCF file contains called indels and SNPs for each chromosome/chromosome group for a strain.
PseudoBase is served as a browser-based web app compatible with all major browsers. An embedded JBrowse instance is fully integrated within the PseudoBase application, allowing browsing to specific genes/regions [Citation30]. All strains imported into PseudoBase are automatically made available for browsing within JBrowse utilizing ‘HTMLVariant’ JBrowse track types with store class of ‘VCFTabix’, while any other supplementary tracks/track types useful for analysis can also be uploaded directly to the JBrowse interface using standard JBrowse import mechanisms. The following JBrowse plugins are activated within PseudoBase: HierarchicalCheckBoxPugin, NeatHTMLFeatures and HideTrackLabels.
Results and discussion
Interface and key features
The PseudoBase site, http://pseudobase.biology.duke.edu/, is designed to be simple and intuitive. The landing page is divided into six tabs: ‘Home’, ‘Browse’, ‘Info’, ‘Links’, ‘Updates’, and ‘Contact Us’. The homepage () allows the user to select the species of interest and readily pull up genetic information by gene name or by genomic region. PseudoBase is currently configured to accept the following types of gene identifiers: D. pseudoobscura IDs prefixed with GA- (e.g., GA26895) and D. persimilis IDs prefixed with GL- (e.g., GL15062), D. melanogaster IDs prefixed with CG- or FBgn- (e.g., CG10064 or FBgn0035724), and gene name abbreviations when available (e.g., atl or Adh). D. melanogaster orthologs in other sequenced Drosophila genomes are reported by FlyBase as determined by OrthoDB, and PseudoBase uses this ortholog report to display the relevant orthologous D. pseudoobscura gene when a D. melanogaster gene identifier is entered [Citation31, Citation27, FlyBase file ‘dmel_orthologs_in_drosophila_species_fb_2020_04.tsv.gz’]. We also use this ortholog report to look up gene identifiers of D. persimilis, by first determining the D. melanogaster ortholog, then looking up the D. pseudoobscura ortholog. We note that these search functions will be an important area for future PseudoBase updates, as more orthology predictions become available and as the maintenance of D. pseudoobscura annotations shifts from FlyBase to GenBank, as discussed further below.
Figure 1. | Overview of the PseudoBase interface. (a) The PseudoBase homepage allows the user to query by gene (or genes if the user uploads a batch query) or by chromosomal region. In this example, the gene Adh (alcohol dehydrogenase) is entered. By selecting one or more species of interest, the user can either access a FASTA-formatted alignment or navigate to the JBrowse interface (snapshots in B-D) to explore the genomic region. (b) Track selection in the JBrowse interface enables the user to toggle tracks on or off to add or remove strains from the view. (c) An overview of the genomic region includes annotations from FlyBase. Clicking on any of these features brings up detailed information, including coordinates, the feature length, any aliases, the full nucleotide sequence, and the nucleotide sequence of each subfeature (e.g., introns). (d) JBrowse allows the user to visualize SNP and indels specific to each selected track. The zoomed view of a portion of the ‘San Antonio, NM, Pikes Peak 1137’ strain shows SNPs highlighted in green. Clicking on any of these SNPs brings up further details, such as the specific allele and its attributes (e.g., sequencing depth)
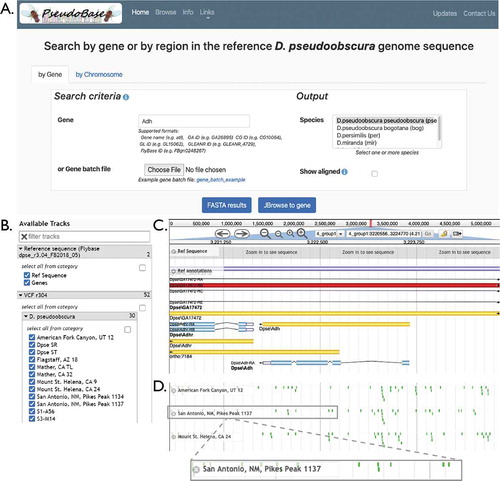
Once the species of interest and the gene/region are indicated, the user has the option of either generating an alignment or navigating to the relevant JBrowse view. The ‘FASTA results’ option generates a FASTA formatted output (aligned or unaligned), which can be downloaded for downstream analyses. Alternatively, the ‘JBrowse to gene’ option allows the user to navigate to the region of interest within the JBrowse interface (). This interface can also be accessed from the ‘Browse’ tab. The user can select and deselect genomes of interest (), enabling comparative genomics between genomes of D. pseudoobscura pseudoobscura, D. pseudoobscura bogotana, D. persimilis, D. miranda, and D. lowei. Navigating to a particular region in the JBrowse interface allows the user to view any FlyBase annotations contained within the region ( [Citation27];. This interface also allows the user to visualize the genomic context of SNPs and indels in the genomes of interest ().
The other tabs on the homepage (‘Info’, ‘Links’, ‘Updates’, and ‘Contact Us’) provide documentation and help. The ‘Info’ tab provides users with general information about PseudoBase including an overview of PseudoBase, tool documentation, variant calling details, and a list of available strains and their accessions. The ‘Links’ tab consists of a dropdown menu with related external resources. ‘Updates’ serves as a location for Release Notes, and will summarize future changes to the site. Contact information for comments or assistance is located in the ‘Contact Us’ tab as well as the footer on each page.
Data content and data types
PseudoBase 2.0 includes a total of 61 sequenced genomes from D. pseudoobscura, D. persimilis, D. miranda, and D. lowei (). All genome alignment and variant calling was performed through a standardized workflow and uses a common genomic coordinate system based on the D. pseudoobscura genome assembly (initial version published in [Citation32]). This structure contributes to the simplicity of the site and enables comparative genomics across species and strains (see Methods). Further, to our knowledge, this reference genome has received the most independent (not including reference-based assemblies to other species) assembly effort out of the pseudoobscura subgroup. The database was constructed to accommodate future additions of additional sequencing data and variant calls. The gene model annotations available through PseudoBase are pulled from FlyBase [Citation27]. Importantly, the genome assembly and annotations obtained from FlyBase are static within PseudoBase, rather than being dynamically updated when FlyBase is updated. Recent releases of FlyBase are moving the focus of FlyBase away from non-melanogaster species (see Release Notes at flybase.org). As a result, FlyBase no longer maintains the D. pseudoobscura assembly and annotations. We plan to import future updates from GenBank, where the current assembly is maintained (GCA_000001765.2). However, we note that the static nature of assembly and annotations within PseudoBase gives us the flexibility to import future assemblies and annotations from other sources if they become available.
Applications
One of the key features of PseudoBase that sets it apart from other interactive tools for interfacing with Drosophila genomic data is its simplicity. We provide a simple user interface and a relatively focused dataset representing only genomic variation within the pseudoobscura subgroup. By lowering the barriers to working with genomic data, we provide a widely accessible tool particularly useful for pilot analyses, data checks, and educational purposes. Anyone can take advantage of this database without the burden of obtaining and downloading raw data, assembling genomes, or calling variants. For example, the ‘FASTA results’ option available on the homepage can generate a FASTA formatted alignment specifically for any gene of interest without requiring the user to handle full genomes. Such alignments can be readily downloaded for downstream analyses. The JBrowse feature further allows selection of specific strains as well as visualization of all polymorphisms or just SNPs or indels, thereby simplifying the process for rapid marker development.
While this database is useful for Drosophila researchers, it also offers educational opportunities. Simple bioinformatic exercises can be designed where students or trainees extract variation for genes of interest. Indeed, PseudoBase has already been leveraged extensively for many years by undergraduates in our research team as well as for an introductory-level college course laboratory exercise [Citation33]. In sum, the accessibility of PseudoBase makes it useful to both the community of Drosophila researchers and to those who lack extensive computational or genomic analysis expertise but wish to do simple population genetic analyses or develop genetic markers.
Acknowledgments
The authors wish to thank past and present members of the Noor Lab whose contributions have facilitated the creation and improved the functionality of PseudoBase. Helpful feedback from two anonymous peer reviewers improved both the manuscript and the functionality of the site. The original version of PseudoBase was funded by NIH award GM086445 to MAFN. The current update was funded through NSF grants DEB-1754022 and DEB-1754439 to MAFN. KLK was supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE 1644868.
Disclosure statement
The authors have no conflicts of interest to declare.
Additional information
Funding
References
- Sturtevant AH, Dobzhansky T. Inversions in the third chromosome of wild races of Drosophila pseudoobscura, and their use in the study of the history of the species. Proc Nat Acad Sci. 1936;22:448–450.
- Dobzhansky T. Genetics of natural populations IX. Temporal changes in the composition of populations of Drosophila pseudoobscura. Genetics. 1943;28(2):162–186.
- Dobzhansky T, Sturtevant AH. Inversions in the chromosomes of Drosophila pseudoobscura. Genetics. 1938;23(1):28–64.
- Lewontin R, Hubby J. A molecular approach to the study of genic heterozygosity in natural populations II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudobscura. Genetics. 1966;54(2):565–609.
- Fuller ZL, Koury SA, Phadnis N, et al. How chromosomal rearrangements shape adaptation and speciation: case studies in Drosophila pseudoobscura and its sibling species Drosophila persimilis. Mol Ecol. 2018;28(6):1283–1301.
- Fishman L, Stathos A, Beardsley PM, et al. Chromosomal rearrangements and the genetics of reproductive barriers in Mimulus (monkey flowers). Evolution. 2013;67(9):2547–2560.
- Hoffmann AA, Rieseberg LH. Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu Rev Ecol Evol Syst. 2008;39:21–42.
- Kirkpatrick M, Barrett B. Chromosome inversions, adaptive cassettes and the evolution of species’ ranges. Mol Ecol. 2015;24(9):2046–2055.
- Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173(1):419–434.
- Noor MA, Grams KL, Bertucci LA, et al. Chromosomal inversions and the reproductive isolation of species. Proc Nat Acad Sci. 2001;98(21):12084–12088.
- Noor MAF, Grams KL, Bertucci LA, et al. The genetics of reproductive isolation and the potential for gene exchange between Drosophila pseudoobscura and D. persimilis via backcross hybrid males. Evolution. 2001;55:512–521.
- Brown KM, Burk LM, Henagan LM, et al. A test of the chromosomal rearrangement model of speciation in Drosophila pseudoobscura. Evolution. 2004;58(8):1856–1860.
- Chang AS, Noor MAF. The genetics of hybrid male sterility between the allopatric species pair Drosophila persimilis and D. pseudoobscura bogotana: dominant sterility alleles in collinear autosomal regions. Genetics. 2007;176(1):343–349.
- Kulathinal RJ, Stevison LS, Noor MAF. The genomics of speciation in Drosophila: diversity, divergence, and introgression estimated using low-coverage genome sequencing. PLoS Genet. 2009;5(7):e1000550.
- Phadnis N, Orr H. A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science. 2009;323(5912):376–379.
- Beckenbach AT, Wei YW, Liu H. Relationships in the Drosophila obscura species group, inferred from mitochondrial cytochrome oxidase II sequences. Mol Biol Evol. 1993;10(3):619–634.
- Korunes KL, Machado CA, Noor MA. Inversions shape the divergence of Drosophila pseudoobscura and D. persimilis on multiple timescales. BioRxiv. 2019;842047. DOI:https://doi.org/10.1101/842047
- Manzano-Winkler B, McGaugh SE, Noor MAF. How hot are Drosophila hotspots? Examining recombination rate variation and associations with nucleotide diversity, divergence, and maternal age in Drosophila pseudoobscura. PLoS ONE. 2013;8(8):e71582.
- McGaugh SE, Heil CSS, Manzano-Winkler B, et al. Recombination modulates how selection affects linked sites in Drosophila. PLoS Biol. 2012;10(11):e1001422.
- Smukowski Heil CS, Ellison C, Dubin M, et al. Recombining without hotspots: A comprehensive evolutionary portrait of recombination in two closely related species of Drosophila. Genome Biol Evol. 2015;7(10):2829–2842.
- Bachtrog D, Charlesworth B. Reduced adaptation of a non-recombining neo-Y chromosome. Nature. 2002;416(6878):323–326.
- Mahajan S, Wei KH, Nalley M, et al. De novo assembly of a young Drosophila Y chromosome using single-molecule sequencing and chromatin conformation capture. PLoS Biol. 2018;16(7):e2006348.
- Fuller ZL, Leonard CJ, Young RE, et al. Ancestral polymorphisms explain the role of chromosomal inversions in speciation. PLoS Genet. 2018;14(7):e1007526.
- McGaugh SE, Noor MAF. Genomic impacts of chromosomal inversions in parapatric Drosophila species. Philos Trans R Soc B. 2012;367(1587):422–429.
- Samuk K, Manzano-Winkler B, Ritz KR, et al. Natural selection shapes variation in genome-wide recombination rate in Drosophila pseudoobscura. Curr Biol. 2020;30(8):1517–1528.E6.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25(14):1754–1760.
- Thurmond J, Goodman JL, Strelets VB, et al. FlyBase 2.0: the next generation. Nucleic Acids Res. 2019;47(D1):D759–D765.
- McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303.
- Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1):11.10.1–11.10.33.
- Buels R, Yao E, Diesh CM, et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016;17(66). DOI:https://doi.org/10.1186/s13059-016-0924-1
- Kriventseva EV, Tegenfeldt F, Petty TJ, et al. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015;43(Database issue):D250–6. Epub 2014 Nov 26. PMID: 25428351; PMCID: PMC4383991
- Richards S, Liu Y, Bettencourt BR, et al. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 2005;15(1):1–18.
- Noor JKF, Noor MAF. Finding selection in all the right places: a college genetics laboratory inquiry-based learning exercise. Genet Soc Am Peer-Reviewed Educ Portal. 2013;2013:1.