
Abstract
Glutamate is the major excitatory neurotransmitter in the vertebrate nervous system and is used for communication between neurons at specialized points of contact named synapses. Neural activity (depolarization) at glutamatergic synapses leads to presynaptic release of glutamate, which binds to ionotropic glutamate receptors (iGluRs) on postsynaptic neurons. Binding of glutamate gates open an integral cation-conducting channel pore, allowing cations to enter the postsynaptic cell resulting in rapid depolarization. The strength of glutamatergic transmission depends on the number of postsynaptic AMPA receptors (AMPARs) – a subtype of iGluRs. Accumulating evidence suggests that experience-dependent changes in the number of synaptic AMPARs are key cellular correlates of learning and memory. For example, long-term potentiation or depression (LTP or LTD) refer to long-lived changes in synaptic strength (synaptic plasticity) that can be elicited by specific patterns of neuronal stimulation.
Plasticity is most intensely studied in the CA1 region of the mammalian hippocampus, where LTP and LTD reflect long-lasting, experience-dependent changes in the number of postsynaptic AMPARs. Such changes can occur at any of the thousands of synapses along the dendritic processes of a neuron. While certain molecules are clearly critical for these phenomena, in particular the NMDA subtype of iGluR and the Ca2+- and calmodulin-dependent kinase, CaMKII, we still lack a mechanistic understanding of how neuronal activity leads to synapse-specific changes in receptor number. Disruption of synaptic signaling is associated with many psychiatric disorders and is often the earliest change associated with neurodegenerative disorders.Citation1 Thus, a major challenge for neuroscience is to gain a mechanistic understanding of how AMPARs are delivered to and maintained at synapses.
Synaptic proteins, especially iGluRs, appeared early in evolution and are highly conserved.Citation2 Thus, genetic studies in simpler invertebrate model organisms, such as C. elegans, can provide important mechanistic insights into synaptic function. This transparent organism has a well-described nervous system containing 302 neurons and about 7000 identified synapses. In vivo confocal microscopy enables the direct study of the complement of iGluRs at individual synapses and standard electrophysiological techniques can be used to measure neuronal function. Using these approaches along with genetic analysis, we revealed an unanticipated role for microtubule-dependent kinesin-1 motors in the delivery, removal and redistribution of synaptic AMPARs.Citation3 What we found particularly striking was the rapid, bidirectional flux of AMPARs along the processes of a pair of interneurons (AVA) that contribute to the control of locomotion. Thus, any given synapse in AVA was only seconds removed from an AMPAR transport vesicle. This result suggested that motor-driven transport of AMPARs might also contribute to synaptic strength and plasticity.
To address whether neuronal activity modulated the transport of AMPARs in the adult nervous system, we ectopically expressed a histamine-gated chloride channelCitation4 in either the postsynaptic AVA neurons or the presynaptic glutamate releasing neurons. By treating worms with histamine we could selectively hyperpolarize (silence) either the presynaptic or postsynaptic neurons. We found that presynaptic or postsynaptic silencing dramatically reduced the kinesin-mediated flux of AMPARs. Thus, we concluded that the size of the mobile pool of AMPARs was regulated by postsynaptic activity.Citation5
By taking advantage of available C. elegans mutants, we uncovered the relevant signaling pathway that linked postsynaptic activity to AMPAR flux. Thus, flux was also decreased in CaMKII loss-of-function (lf) mutants and in mutants with defective voltage-gated calcium channels (VGCCs). Selectively expressing either C. elegans CaMKII or vertebrate αCaMKII in the AVA neurons of CaMKII(lf) mutants was sufficient to restore both AMPAR flux and glutamate-gated current. We found fascinating that the CaMKII signaling pathway regulating AMPAR flux is evolutionarily conserved and functions cell autonomously.
How might CaMKII signaling regulate the flux of AMPARs? We found that a gain-of-function (gf) mutation in CaMKII increased AMPAR transport and that the increase was dependent on kinesin-1 motors. These results suggested that kinesin-1 functions downstream of CaMKII and might be a target of CaMKII phosphorylation. Kinesin-1 motors are heteromeric complexes composed of a light chain and a heavy chain, and in C. elegans are encoded by klc-2 and unc-116, respectively. We found that mutating 2 consensus CaMKII phosphorylation sites on KLC-2 suppressed AMPAR transport in CaMKII(gf) mutants. Together, these results suggest that in the absence of CaMKII AMPAR cargo are not loaded onto kinesin-1 motors.
CaMKII is found in the cell body and throughout the AVA neuronal processes where it is enriched at synapses. To better evaluate the role(s) of CaMKII, we used Chromophore Assisted Light Inactivation (CALI)Citation6 to selectively inactivate CaMKII at specific subcellular domains. With this method we demonstrated that CaMKII was required in the cell body for AMPAR export, and in the processes for the removal of AMPARs from synapses. These findings suggested a comparable role for CaMKII in the cell body and at synapses: it is required for some step in the loading of AMPAR cargo onto motors. Additionally, we found that CaMKII was required for coordinating the delivery and insertion of AMPARs at synapses.
Our studies of AMPAR transport and synaptic function complement and extend ongoing efforts in vertebrate systems. We demonstrated that neuronal activity regulates AMPAR transport to and from synapses via a CaMKII-dependent signaling pathway (Fig. 1). In our model, overall neuronal activity regulates the size of the mobile pool of AMPARs, thus providing a rapid-response mechanism to deliver or remove synaptic AMPARs. To test whether synaptic plasticity also depends on CaMKII-regulated transport of AMPARs, we used an optogenetic strategy to activate specific synaptic inputs to AVA and demonstrated that repeated presynaptic activation led to an increase in postsynaptic AMPARs. As predicted by our model, this plasticity was not observed in CaMKII(lf) mutants.
We can now build on these studies by taking a systematic genetic approach to uncover the signaling molecules upstream and downstream of CaMKII that contribute to the regulated delivery and removal of AMPARs, and thus synaptic plasticity and learning. Our findings also provide a new conceptual framework for synaptic plasticity. In our model of LTP, an activated synapse has a greater probability of capturing AMPARs transported on motors that pass by every few seconds. This rapid delivery system can increase the number of receptors at the synapse, which in turn increases both current and CaMKII activity, thus enabling long-term changes in the balance of AMPAR delivery and removal. In this model, a “synaptic tag” might simply be an increase in AMPAR-mediated current at a given synapse, and the increased synaptic strength a consequence of a new balance between CaMKII-mediated delivery and removal of synaptic AMPARs. Disruption in the delivery or removal of AMPARs would strongly affect synaptic strength, plasticity and circuit function, and thus might contribute to neuropsychiatric or neurodegenerative disorders. Clearly, we are still at the early stages of identifying the complex signaling components regulating the delivery and removal of AMPARs.
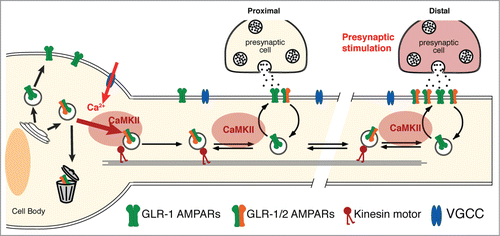
Figure 1. CaMKII and neuronal activity regulate transport, delivery and removal of synaptic AMPARs. Presynaptic glutamate release depolarizes the postsynaptic cell, opening VGCCs and activating CaMKII. This signaling promotes the loading of AMPARs onto kinesin-1 motors at both the cell body and at synapses. Thus, CaMKII is required for the export of AMPARs from the cell body, the delivery of receptors to synapses and the removal of synaptic AMPARs.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Maricq laboratory for comments on the manuscript and the Caenorhabditis Genetics Center.
Funding
The Caenorhabditis Genetics Center is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This research was made possible by support from NIH Grants NS35812 and DA035080 (A.V.M.), and by postdoctoral fellowships from the Swiss National Science Foundation (F.J.H.).
References
- Grant SG. Curr Opin Neurobiol 2012; 22:522–9; PMID:22409856; http://dx.doi.org/10.1016/j.conb.2012.02.002
- Emes RD, et al. Annu Rev Neurosci 2012; 35:111–31; PMID:22715880; http://dx.doi.org/10.1146/annurev-neuro-062111-150433
- Hoerndli FJ, et al. Neuron 2013; 80:1421–37; PMID:24360545; http://dx.doi.org/10.1016/j.neuron.2013.10.050
- Pokala N, et al. Proc Natl Acad Sci U S A 2014; 111:2770–5; PMID:24550306; http://dx.doi.org/10.1073/pnas.1400615111
- Hoerndli FJ, et al. Neuron 2015; 86(2):457–74;PMID:25843407; http://dx.doi.org/10.1016/j.neuron.2015.03.011
- Lin JY, et al. Neuron 2013; 79:241–53; PMID:23889931; http://dx.doi.org/10.1016/j.neuron.2013.05.022