
ABSTRACT
TRPV3 is a non-selective cationic channel and is important for several physiological functions. It can be activated by physiological temperature and selective endogenous and exogenous compounds. TRPV3 is one of the key ion channel involved in Ca2+-signaling in keratinocyte and thus involved in skin-related functions. Recently, naturally occurring mutations in TRPV3, namely G573A, G573S, G573C and W692G have been detected which are linked with the development of pathophysiological conditions such as Olmsted Syndrome (OS) and other skin disorders. Our qualitative and quantitative data suggests that these naturally occurring TRPV3 mutants are mainly restricted in the ER. Expression of OS-mutants cause impaired vesicular trafficking resulting reduced surface localization of these mutants and other membrane proteins too. OS-mutants also cause reduced cell adhesion, altered distribution and less number of lysosomes. Our data confirms that TRPV3 is a lysosomal protein suggesting that Olmsted Syndrome is a lysosomal disorder. These findings may have a broad implication in the context of keratinocyte functions, skin-degeneration and in skin-cancer.
Introduction
Olmsted Syndrome (OS) is a rare genetic disorder first reported by Olmsted in 1927.Citation1 So far nearly 75 patients worldwide have this syndrome. OS is a disorder of keratinocytes characterized by defective keratinization appearing at birth or in early infancy.Citation2 These patients develop clinical symptoms of bilateral mutilating palmoplantar keratoderma and periorifacial keratotic plaques.Citation3-9 Severe itching at the affected regions, impaired hair growth, hearing loss, and loss of terminal portion of the limbs, auto-amputation of digits, loss-of-bones and/or osteonecrosis are other specific features of OS.Citation9-11 There is no satisfactory treatment for this disease as different agents such as salicylic acid, urea, boric acid, shale oil, retinoic acids, corticosteroids, anti-microbial drugs, antihistamines, vitamin-E, vitamin-A and other emollients failed to improve the pathological conditions.Citation11 Surgical removals of affected skin and autografting have also been unsuccessful due to the recurrence problem.Citation12-16 The hallmarks of OS represent an exceptional case that excludes this disorder from other syndromes of keratoderma. OS shares etiological similarities with symptoms of carcinoma and indeed co-occurrence of squamous cell carcinoma and adenocarcinoma of the lung in certain cases of OS is also reported.Citation17 OS-patients have higher susceptibility to develop epidermal tumors.Citation16 All these factors render OS as a clinically important etiology for its uniqueness.
Though initially believed to be limited within male, so far few OS cases have been reported in females too.Citation4-5,8,11,17,18 The exact mode of inheritance of OS is not well established. Sporadic occurrence, autosomal-dominant, X-linked dominant and X-linked recessive modes of inheritance had been proposed. It is primarily believed to be a disorder of keratinization as expression of mature epidermal keratins (type 1 and 10) are defective.Citation12 OS has also been referred as hyper proliferative disorder of the epidermis.Citation3 Recently, abnormality of the immune systems, especially hyper IgE production, elevated eosinophil and follicular T-cells in the peripheral blood have been linked with the OS.Citation19
Transient Receptor Potential Vanilloid sub-type 3 (TRPV3) is a non-selective cation channel and member of TRP super family of ion channels.Citation20 Different point mutations in TRPV3, namely in G573S, G573C, G573A, and W692G have been linked with the development of OS.Citation19,21,22 Deleterious effect of these mutations are also in line with the expression of TRPV3 in keratinocytes and its involvement in skin barrier formation, cutaneous growth and survival, skin inflammation, cutaneous pain and Puriceptive itch.Citation23 Notably, electrophysiological experiments suggest that G573S, G573C and W692G conduct more currents and thus behave as “gain-of-function” mutants.Citation21 Though these reports strongly suggest the involvement of TRPV3 in this disorder, the exact molecular mechanism by which these TRPV3 mutations induce Olmsted Syndrome is not clear.
In this work we explored the role of OS-causing TRPV3 mutants in Keratinocytes and demonstrate that these mutants have impaired trafficking, reduced surface expression, impaired cell adhesion and abnormal lysosomal functions. We demonstrate that TRPV3 is a lysosomal protein, thereby suggesting that Olmsted Syndrome is associated with lysosomal disorder.
Results
Wild type TRPV3 localizes at cell-cell contact sites and OS-mutants are retained in ER
Localization of TRPV3 in keratinocytes were studied by expressing TRPV3-Wt in HaCaT cells as a GFP-tagged protein. TRPV3-Wt localizes primarily in the plasma membrane and is often enriched in the cell-cell contact sites (). Recently four point mutations in TRPV3 have been linked with the development of Olmsted Syndrome and these mutations are located at the intracellular loop region between 4th and 5th TM region as well as in the TRP domain (). To explore the localization pattern of these TRPV3 mutants, full-length TRPV3 carrying the different point mutations (G573S, G573C, G573A and W692G; collectively termed as “OS-mutants”) individually were expressed in HaCaT cells as GFP-tagged proteins. These OS-mutants are localized in the perinuclear regions and are mainly restricted to the ER and in the nuclear envelope ().
Figure 1. OS-mutants but not TRPV3-Wt have reduced surface expression. (A) Shown are the 3D-reconstituted confocal images of HaCaT cells transiently expressing TRPV3-Wt-GFP. The enlarged image demonstrates the presence of TRPV3 at cell-cell contact sites. (B) Positions of point mutations (G573A, G573S, G573C and W692G) are indicated. (C) The GFP fluorescence (green) alone or merged with DIC are shown. OS-mutants show no localization at the cell surface (cell boundary is indicated by dotted line) and accumulate at the ER. (D) An extracellular loop-specific antibody detects TRPV3 available at the cell surface (red) only in un-permeabilized cells expressing TRPV3-Wt but not in un-permeabilized cells expressing OS-mutants. Intensity of TRPV3staining is provided in the right side. (E-G) expressing OS-mutants have much reduced cell periphery (E), area (F) and are more round in shape (in each case n = 70 cells and p value < 0.001 is considered as significant).
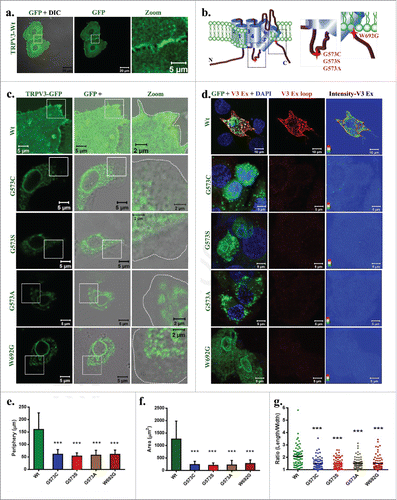
OS-mutants have altered surface expression and affect cell size
To explore if the surface expression of OS-mutants is indeed reduced, we used an antibody (Alomone Labs, recognizes AA 464–478) specific for the extracellular loop region of TRPV3 and thus this antibody is able to detect TRPV3 present in the plasma membrane without permeabilizing the cells. This antibody readily detected TRPV3 surface expression only in the non-permeabilized HaCaT cells transiently expressing TRPV3-Wt-GFP but not in the cells expressing the other OS-mutants (). This antibody does not detect the non-transfected HaCaT cells, most likely due to very low level endogenous expression of TRPV3. This result suggests that only TRPV3-Wt-GFP but not the OS-mutants are present in the surface.
To characterize the effect of expression of OS-mutants in a more precise manner, we have quantified morphological parameters (such as cell periphery and area) of HaCaT cells that transiently express TRPV3-Wt-GFP or OS-mutants (). Cells expressing OS-mutants have significantly smaller cell periphery and smaller cell size when compared with the cells expressing TRPV3-Wt-GFP. Cells expressing OS-mutant are also more round-shaped as observed in the length by width ratio ().
OS-mutants Co localize with ER but not with membrane protein
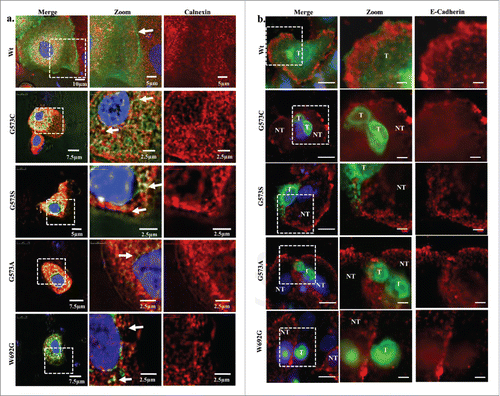
To further validate that we used an antibody specific for ER (calnexin) and stained the transfected cells which express TRPV3-Wt and OS mutants. TRPV3-Wt expressing cells show discrete staining for ER and TRPV3-Wt shows localization on plasma membrane but most of the OS mutants show co localization with calnexin suggesting that these mutants are primarily restricted in the ER and/or are unable to reach to the plasma membrane (). To further confirm this aspect, we explored the status of E cadherin by using specific antibody. Our results show that the TRPV3-Wt is present on membrane while the OS mutants are mostly undetectable in the membrane (). These data strongly suggests that cell expressing OS mutants have altered membrane trafficking.
Figure 2. OS-mutants co localize with calnexin and not with E cadherin. (A) Shown are the confocal images of HaCaT cells transiently expressing TRPV3-Wt-GFP or OS-mutants. Cells were fixed within 36 hours after transfection and the cells were stained for anti Calnexin antibody. TRPV3-Wt shows discrete ER labeling and TRPV3 localization on membrane, while OS mutants shows co localization with ER (Merge image) suggesting that OS mutants have reduced surface expression and are primarily retained in ER. (B) Immunolocalization of TRPV3-Wt-GFP and OS mutants with membrane marker E cadherin is shown. TRPV3-Wt-GFP shows proper E cadherin labeling and proper TRPV3 localization on membrane, while OS mutants are not localize on the membrane. In most cases, cells expressing OS-mutants have much reduced E cadherin staining (T and NT represent transfected and non-transfected cells respectively). Scale bar is 10µm and 5µm for merge and zoom images.
To further confirm that OS mutants have reduced surface expression, we have quantified more than 100 cells in each category and classified the localization of TRPV3 in 4 types. Results confirm that OS mutants are primarily restricted in ER and in nuclear envelope while TRPV3-Wt is primarily localized in plasma membrane and in sub-membranous region (). Analysis of relative presence of TRPV3-Wt or OS-mutants in the membrane regions were analyzed by quantifying the fluorescence intensity from plasma-membrane regions of GFP-tagged TRPV3 protein as well as antibody labeling of surface localized TRPV3 in unpermeabilized cells. This analysis also confirms that the presence of TRPV3-Wt at the plasma membrane is predominant while the presence of OS mutants are much less there ().
Figure 3. OS mutants reveal reduced surface expression. (A) Percentage scoring of cells expressing TRPV3-Wt-GFP or OS-mutants are shown. Cells were classified according to the localization of TRPV3, i.e.: exclusively on the membrane (A-type), in membrane and in the sub-membranous region (B-type), in cytoplasm but not on the membrane (C-type), and exclusively in the ER and in nuclear envelope (D-type). TRPV3-Wt-GFP is primarily localized on membrane and sub membranous region, while OS mutants are mainly restricted in the ER and in the nuclear envelop. (B) Quantification of the amount of TRPV3 (GFP fluorescence) present in plasma membrane per unit surface area are shown. Minimum 10 or more numbers of region-of-interests (ROI) in the membrane region from individual cells (n = 20 or more) in each group was considered and plotted. (C) Quantification of the total surface expression of TRPV3 is shown. Amount of TRPV3 present in the surface is quantified by calculating total intensity of fluorescence in un-permeabilized cells detected by extracellular loop-specific antibody raised against TRPV3. Microscopic images of similar experiments are represented in . (D) Percentage expression of TRPV3 at the surface is estimated by quantifying the total GFP fluorescence (considered as 100%) present in individual cells and the fluorescence of TRPV3 that can be labeled by extracellular-loop specific antibody. In each case, the p value <0.001 is considered as significant.
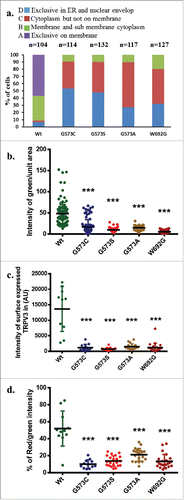
To further calculate the extent of surface localization, we have labeled the TRPV3 expressing cells with antibody specific for extracellular loop region of TRPV3. Cells without any detergent permeabilization were used for this analysis. Calculation of florescence intensity indicates that the total antibody labeling was performed. This analysis also suggests that OS mutants have much reduced surface expression (). In addition we analyzed the percentage of total TRPV3 that are surface expressed. For that TRPV3-Wt or OS-mutants-tagged with GFP are expressed (considered as 100%) and the cells were further stained with the extracellular loop-specific antibody detected as Red fluorescence. Ratio of the red florescence (from labeled antibody) with green fluorescence (from GFP-tagged protein) obtained from multiple cells were calculated. This analysis suggests that approximately 50% of the total TRPV3-Wt-GFP are present in the plasma membrane while this number is much less for OS-mutants (). Taken together all these analysis strongly suggests that OS-mutants have much reduced surface expression than that of TRPV3-Wt-GFP.
OS-mutants affect surface expression of other proteins and cell adhesion
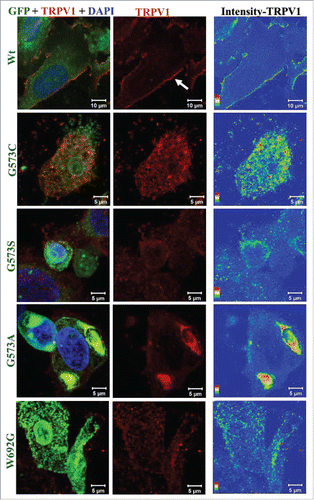
We explored if expression of OS-mutants affect the surface localization of these mutants only or affect other membrane proteins as well. Therefore, wild-type TRPV1 along with TRPV3 (wild-type or OS-mutants) were co-expressed in 5:1 ratio (plasmid DNA in order to express more TRPV1 and less TRPV3) since it is reported that both TRPV1 and TRPV3 form heteromers.Citation20,24 TRPV1 (a close homolog of TRPV3) localizes to the plasma membrane efficiently in cells expressing TRPV3-Wt-GFP but not in cells expressing OS-mutants (). These results suggest that OS-mutants affect the surface expression of other membrane proteins as well.
Figure 4. OS-mutants show impaired trafficking for membrane proteins. Confocal images of HaCaT cells transiently expressing TRPV1 along with either TRPV3-Wt-GFP or OS-mutants are shown. Cells were fixed 36 hours after transfection and were stained for TRPV1 using an antibody that is raised against extracellular loop of TRPV1. This antibody detects TRPV1 at the surface (red) only in cells expressing TRPV3-Wt-GFP but much less or not at all in cells expressing OS-mutants. Fluorescence of TRPV3-Wt-GFP or OS-mutants are represented in green and TRPV1 is represented in red. Intensity of surface-expressed TRPV1 is indicated in the rainbow scale (right panel).
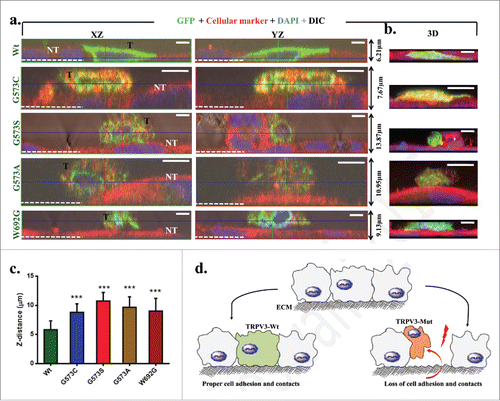
Gradual loss of keratinocytes from skin is a common feature of Olmsted Syndrome. Therefore we have characterized the adhesion of cells expressing TRPV3-Wt-GFP or OS-mutants by allowing the cells to grow on uncoated glass surface and measured the Z-distance of the individual cells. Cells expressing TRPV3-Wt-GFP are tightly attached to the glass surface and reveal very flat morphology. In contrast, cells expressing these OS-mutants are either not attached to the glass surface and/or loosely attached to other non-transfected cells as observed by the 3D-confocal microscopy. The cells expressing OS-mutants are more round-shaped in their morphology (). The average Z-distance of cells expressing OS-mutants are higher than the cells expressing TRPV3-Wt-GFP. These results suggest that OS-mutants affect cell adhesion and cell-to-cell contact properties as well.
Figure 5. Expression of OS-mutants induce impaired cell adhesion. (A) Shown are the XZ, YZ plane and 3D (right panel) confocal images of representative HaCaT cells that transiently express either TRPV3-Wt or OS-mutants. The GFP fluorescence (green) distinguishes the transfected cells (T) from non-transfected (NT) cells (red) and the white dotted line indicate the Z-position of the uncoated glass coverslip. While cells expressing TRPV3-Wt are flat and tightly adhere to the glass surface, the cells expressing OS-mutants are spherical, loosely attached to the glass surface and mostly grow on other non-transfected cells. Scale bar (5 μm). (C) OS-mutants reveal increment in Z-distance than the cells that express TRPV3-Wt (n = 20 cells in each case, P-value < 0.001). (D) A schematic diagram demonstrating the loss of cell adhesion and cell-to-cell contacts due to expression of OS-mutants.
Expression of OS-mutants alter distribution of ER and Golgi bodies
As expression of OS-mutants reveal reduced surface localization and are mainly restricted in the perinuclear regions, we tested if these mutants cause abnormalities in the ER distribution and functions. For that purpose, we expressed TRPV3-Wt-GFP as well as OS-mutants transiently in HaCaT cells and immunostained with anti-KDEL antibody (a marker for ER-targeted proteins). We noted that cells expressing OS-mutants reveal aggregated KDEL staining while cells expressing TRPV3-Wt-GFP reveal normal distribution of KDEL staining (Fig. S1a). Both KDEL immunostaining, and the localization of TRPV3 strongly suggest that OS-mutants induce mislocalization of ER-targeted proteins in general and development of aggregated ER. The abnormalities in the ER functions is also prominent by ER-tracker staining, as the cells expressing OS-mutants are generally weakly labeled by ER-tracker dye (data not shown).
As OS-mutants are largely accumulated in the ER regions, we explored the vesicular distribution in these cases. We labeled cells with anti-GM130 antibody (specific for Golgi complex). We noted that well distributed Golgi bodies in cells expressing TRPV3-Wt-GFP (Fig. S1b). In contrast, cells expressing OS-mutants show either much reduced levels or clustered Golgi bodies suggesting a general vesicular trafficking problem. Cells expressing OS-mutants also reveal aggregation of mitochondria (Fig. S2a, b).
OS-mutants have altered lysosomal distribution and functions
Distribution and function of lysosomes were measured by staining with anti-Lamp1 antibody (as lysosomal marker). Cells expressing TRPV3-Wt-GFP reveal well-distributed lysosomes, while cells expressing OS-mutants show aggregated lysosomes (Fig. S3). Lysotracker Red imaging confirmed well-distributed and well-labeled lysosomes in nontransfected cells, and in cells which express either GFP or TRPV3-Wt-GFP (Fig. S3). In contrast, cells expressing OS-mutants have less number of Lysotracker Red labeled lysosomes which mainly appear as big clusters.
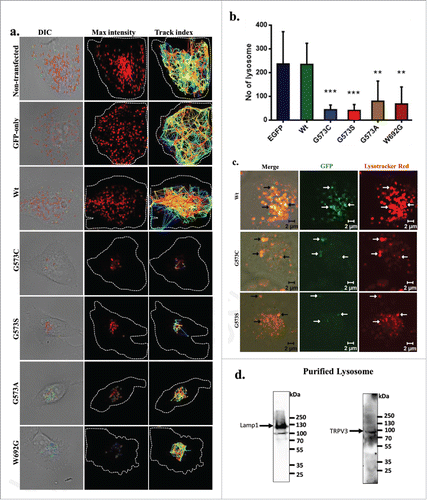
Next, we measured the distribution and number of functional lysosomes in live cells. For that purpose, we prepared cells that express only GFP or TRPV3-Wt-GFP or OS-mutants after a stable selection. We labeled these cells with Lysotracker Red and performed live cell imaging. We monitored the movement of functional lysosomes also. Cells that are either non-transfected, or are expressing only GFP or expressing TRPV3-Wt-GFP (after stable selection) have several lysosomes which are well distributed and move fast, and travel long distances (; Movies S1–5). In contrast, cells expressing OS-mutants have either very less number of lysosomes and/or aggregated lysosomes. Very less or no movement at all of lysosomes was detected for cells that are expressing OS-mutants (). No-significant difference in lysosomal number is observed between cells that express TRPV3-Wt-GFP and cells that are either non-transfected or are expressing GFP only (). These results suggest a strong correlation between expressions of OS-mutants with the malfunction of lysosomes (as Lysotracker Red labeling is largely dependent on the lysosomal functions).Citation25
Figure 6. TRPV3 is present in lysosomes and OS-mutants alter lysosomal numbers and movement. (A) Time-series confocal images were acquired for analyzing lysosomal movements in stable cells expressing TRPV3-Wt –GFP or OS-mutants. DIC images (white dotted lines show the periphery of the cells) superimposed with the florescence images (for Lysotraker-red labeled Lysosomes, red circles). The track-index of Lysosomal movements are indicated by lines (Right panel). For more details see movies (S1-S5). (B) HaCaT cells stably expressing OS-mutants have lower number of lysosomes (labeled with Lysotracker red) than the cells expressing TRPV3-Wt-GFP (P-values: *** < 0.001, ** < 0.01 TRPV3-G573C or TRPV3-G573S; n = 20 cells in each case). (C) Confocal images of stable HaCaT cells show that both TRPV3-Wt-GFP and OS-mutants:localize (white arrows) in lysosomes in live cells. Colocalization experiments with TRPV3-G573A and TRPV3-W692G failed mainly due to the undetectable level of tagged proteins and absence of lysosomes properly labeled with Lysotracker red. (D) Western blot analysis of lysosomal fraction isolated from goat brain shows the presence of endogenous TRPV3 there (indicated by arrow) and enrichment of lysosomal fraction shown with lysosomal marker.
TRPV3 is a lysosomal protein
As mutation in TRPV3 leads to reduced lysosomal numbers and functions, we explored if TRPV3 is present in the lysosomes. We could detect TRPV3-Wt-GFP in the lysosomes in stable cells (). In spite of low number of lysosomes, we could also detect the presence of few OS-mutants too in the lysosomes suggesting that these TRPV3-mutants are also localizes in the lysosomes (). To explore if endogenous TRPV3 is present in lysosomes, we have purified lysosomal fractions isolated from goat brain. By Western blot analysis, TRPV3-specific immunoreactivity is detected in lysosomal fractions ().
Taken together, our data unravel new aspects of TRPV3 in the context of cellular functions and subcellular characterization indicating that impaired lysosomal functions are linked with the development of Olmsted Syndrome.
Discussion
Olmsted Syndrome represents a unique case where keratinocytes in terminal organs are affected and the cellular - molecular mechanisms involved in this manifestation are largely unknown. At genetic level too, very little is known for this syndrome. The LOR gene was previously implicated in the pathogenesis of several hereditary diseases with mutilating Palmoplantar Keratoderma (PPK), a disorder which has certain similarity with Olmsted Syndrome.Citation26 Notably, LOR gene codes for loricrin, the main component of the cornified cell envelop present iN-terminally differentiated epidermis, and it is also associated with inherited skin diseases, namely with Vohwinkel's Syndrome and progressive symmetric erythrokeratoderma.Citation27 However, specific screening studies have failed to detect any mutations in LOR gene in OS patients.Citation17 No mutations have been detected in 3 other genes namely KRT1 (Keratin 1), GJB2 (Gap junction protein β 2) and SLURP1 (Secreted Ly-6/uPAR-related protein 1); which are otherwise involved in other skin-related problems.Citation4,17 A missense mutation in membrane-bound transcription factor protease site 2 (MBTPS2) is involved in OS, though the molecular mechanisms remain uncharacterized.Citation28 Though involvement of other candidate genes in OS cannot be ruled out at this stage; involvement of TRPV3 is highly significant.
Though expression and functions of TRPV3 have been mainly characterized in the context of peripheral neurons and in the context of nociception, expression of TRPV3 has been demonstrated in other non-neuronal tissues too, such as in testis, stomach, trachea, small intestine, placenta, fibroblasts and immune cells present in the dermis.Citation29-31 In fact, expression of TRPV3 is more in skin than in sensory neurons.Citation20,29 TRPV3 also regulates hair follicle, hair growth and keratinocyte functions.Citation23 For example, constitutively active TRPV3 leads to hair loss and skin dermatitis.Citation32,33 TRPV3 is important for EGFR-mediated regulation of hair morphogenesis and skin barrier formation.Citation34 TRPV3 is highly expressed in skin keratinocytes, and particularly in differentiated basal layer and undifferentiated suprabasal layer of mouse epidermis.Citation33 In this context, our results unravel subcellular and molecular phenomena by which TRPV3 regulates different functions in keratinocytes and possibly in other cell types as well.
Our qualitative and quantitative data suggest that OS-mutants are primarily restricted in the ER regions and have much reduced surface expression. Accordingly, HaCaT cells expressing OS-mutants have much reduced vesicular trafficking and abnormal lysosomal functions. Though all these quantifications are from immunofluorescence images and are semi-quantitative in nature, different quantifiable parameters correlate well and strongly suggest that the OS-mutants have much reduced surface expression than TRPV3-Wt-GFP. These OS-mutants also induce abnormal cellular conditions where surface expression of other membrane proteins are also affected. Such situations may explain well about the low cell adhesion and cell-cell contacts and such defects correlate well with the loss of keratinocytes as observed in OS patients, these defects could be the consequence of gain of function mutants which has seen in some of preliminary calcium imaging experiments.
These TRPV3 mutants are constitutively active in their electrophysiological properties and thus categories as “gain-of-function mutants”.Citation21 These “gain-of-function” mutants affect different subcellular organelles, at least ER, Golgi and Lysosomes. This work demonstrates that endogenous TRPV3, as well as TRPV3-Wt-GFP or OS-mutants are physically present in lysosome. Our data suggest that TRPV3 is a lysosomal protein and mutations in TRPV3 seem to cause lysosomal disorders. Our results are in accordance with recent reports suggesting other TRP channels are also present in lysosomes. For example, a report suggest that TRPV3 interacts with Nexin 11, a lysosomal protein.Citation35 TRPML members belong to TRP superfamily are also present in the lysosome and late endosomes; and defects in these TRPMLs also result in lysosomal storage disorders leading to impaired trafficking of lipids and proteins.Citation36 Similarly, using patch clamp method, presence of TRPV2 in the endosome has also been confirmed.Citation37 Highly Ca2+-selective channels (such as TRPV5 and TRPV6) are specifically present in the Rab11-positive recycling endosomes.Citation37 In spite of low numbers, still we could detect OS-mutants in the lysosomes confirming that OS-mutants are also targeted to the lysosomes. How mutations in TRPV3 actually leads to these lysosomal disorders is not known.
Collectively this work demonstrate that TRPV3 is a lysosomal protein and we conclude that TRPV3 mutation mediated Olmsted Syndrome is a lysosomal disorder.
Materials and methods
Reagents
DAPI, Lysotracker Red, and Lipofectamine were purchased from Invitrogen (Carlsbad, California, USA). Fluoromount-G was purchased from Southern Biotech (Birmingham, US).
Constructs, cells and transfection
TRPV3-Wt-GFP, TRPV3-G573S-GFP, TRPV3-G573C-GFP and constructs (cloned in pCMV6-AC-GFP vector) used in this study were kindly provided by Dr. Yong Yang.Citation21 The other constructs, namely TRPV3-G573A-GFP and TRPV3-W692G-GFP were prepared by using site directed mutagenesis kit (Stratagene) using different primer sets [for TRPV3-G573A: 5′CGGGGTTTCCAGTCCATGGCCATGTACAGC3′ and 5′GCTGTACATGGCCATGGACTGGAAACCCCG3′; for TRPV3-W692G:5′GAGAGCGAACGCATCGGGCGCCTGCAGAG3′ and 5′CTCTGCAGGCGCCCGATGCGTTCGCTCTC3′]. All these TRPV3-constructs were verified by restriction digestion and also by sequencing. Full-length TRPV1 (rat) inserted in pCDNA 3.1 vector was used as described previously.Citation38 In this study TRPV3-G573S-GFP, TRPV3-G573C-GFP, TRPV3-G573A-GFP and TRPV3-W692G-GFP are collectively termed as OS-mutants. Human keratinocyte cell line (HaCaT) was obtained from NCCS (Pune, Maharashtra, India) and maintained in Dulbecco's modified Eagle's medium (HiMedia, Bangalore, India) with 10% fetal bovine serum (HiMedia). All media were supplemented with L-glutamine (2mM), streptomycin (100 mg/ml) and penicillin (100 mg/ml) (HiMedia). Cells were maintained in a humidified atmosphere at 5% CO2 and 37°C. Cells stably expressing TRPV3-Wt-GFP or different OS-mutants were selected by antibiotics and subsequently maintained in selection media (Neomycin, 100µg/ml).
Immunocytochemistry
For immunocytochemical analysis, cells were fixed with 4% PFA for 5 minutes and were permeabilized with 0.1% Triton X-100 in PBS (5 min) and subsequently blocked with 5% BSA. For co-immunostaining with subcellular markers, specific antibodies were used [(for Golgi rabbit anti-GM130, Sigma-Aldrich, 1:500), (for mitochondria mouse anti-HSP60, Abcam, 1:500), (for ER mouse anti-KDEL antibody, Abcam, 1:500 and rabbit anti calnexin antibody, Abcam 1:500), (for lysosome rabbit anti-Lamp1 antibody, Sigma-Aldrich 1:500 and rat anti E cadherin antibody, cell signaling technology, 1:500)]. Anti-TRPV3 antibody [which recognizes the 1st extracellular loop (464–478aa) of hTRPV3] and anti-TRPV1 antibody [which recognizes the C-terminus (824–838aa) of rat TRPV1] were purchased from Alomone Labs (Jerusalem, Israel). Another antibody which recognize the C-terminus of TRPV3 was purchased from Sigma-Aldrich. Alexa-594-labeled anti rabbit and Alexa-647-labeled anti- mouse were used as the secondary antibodies. For comparative analysis, all imaging parameters were kept constant and images were acquired by confocal microscope (LSM780, Zeiss) or with Fluorescence microscope (IX83, Olympus).
Live cell imaging
For visualizing the localization and intracellular dynamics of Wt as well as different OS-mutants as GFP-tagged proteins, live cell imaging was performed after selection of stable cells in antibiotics for 20 d. During imaging, cells were maintained at a constant temperature (25°C) and in PBS (pH 7.5) if not mentioned otherwise. Images were acquired by a confocal microscope (LSM 780, Zeiss) using a 63X objective.
The dynamics of lysosomes in live cells were visualized by labeling cells with Lysotracker Red. Cells were grown on glass cover slips (25 mm) overnight and subsequently labeled with Lysotracker Red (100 nM) for 20 minutes.
Image analysis
Calculation of cellular size, shape and other images processing was done by using LSM image examiner software (Zeiss), Image J and by Adobe Photoshop. Quantitative analysis for mitochondrial potentiality, lysosomal movement, Trackmate plugin was used for analyzing Lysosomal dynamics.
Cell size and shape calculation
Cellular size and shape were calculated manually for each image using LSM image examiner software (Zeiss). Parameters such as periphery, area and shape were calculated by manual drawing on images (n = 70), and labeled the cells with different subcellular marker such as golgi (anti GM130), mitochondria (anti HSP60), ER (anti KDEL), and lysosome (anti Lamp1).
Fluorescence intensity calculation
To calculate the surface expression of TRPV3-Wt-GFP or OS mutants, total fluorescence intensity (of GFP-tagged TRPV3 or TRPV3 labeled by antibody specific for extracellular loop) of TRPV3 was measured. The fluorescence intensities were measured from at least 10 (or more) square-shaped region-of-interests (ROI) per cells (from 20 or more cells). In all cases, the ROI was placed on the plasma membrane regions. Fluorescence intensity was measured by using Fiji software tool ROI manager.
Statistical analysis
Graph pad prism software was used for statistical analysis. One way ANOVA (Dunnett's multiple comparisons test) was performed for each set of data. The P-values are as follows *** <0.001, ** <0.01, * <0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KCHL_S_1249076.zip
Download Zip (49 MB)Acknowledgments
We thank Dr. Yong Yang for sending us TRPV3-Wt-GFP, TRPV3-G573C-GFP and TRPV3-G573S-GFP constructs.Citation21 Imaging facility from NISER is appreciated. We thank all the lab members for their support.
Funding
Financial support from NISER and ICMR is appreciated.
Related Research Data
References
- Olmsted HC. Keratoderma palmaris et plantaris congenitalis: report of a case showing associated lesions of unusual location. Am J Dis Child 1927; 33:757-64; https://doi.org/10.1001/archpedi.1927.04130170055008
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis 2015; 10:33; PMID:25886873; https://doi.org/10.1186/s13023-015-0246-5
- Requena L, Manzarbeitia F, Moreno C, Izquierdo MJ, Pastor MA, Carrasco L, Farina MC, Martin L. Olmsted syndrome: report of a case with study of the cellular proliferation in keratoderma. Am J Dermatopathol 2001; 23:514-20; PMID:11801792; https://doi.org/10.1097/00000372-200112000-00003
- Mevorah B, Goldberg I, Sprecher E, Bergman R, Metzker A, Luria R, Gat A, Brenner S. Olmsted syndrome: mutilating palmoplantar keratoderma with periorificial keratotic plaques. J Am Acad Dermatol 2005; 53:S266-72; PMID:16227106; https://doi.org/10.1016/j.jaad.2005.03.036
- Tharini GK, Hema N, Jayakumar S, Parveen B. Olmsted syndrome: report of two cases. Indian J Dermatol 2011; 56:591-3; PMID:22121289; https://doi.org/10.4103/0019-5154.87166
- Vosynioti V, Kosmadaki M, Tagka A, Katsarou A. A case of Olmsted syndrome. Eur J Dermatol 2010; 20:837-8; PMID:20876042
- Yaghoobi R, Omidian M, Sina N, Abtahian SA, Panahi-Bazaz MR. Olmsted syndrome in an Iranian family: report of two new cases. Arch Iran Med 2007; 10:246-9; PMID:17367233
- Kumar P, Sharma PK, Kar HK. Olmsted syndrome. Indian J Dermatol 2008; 53:93-5; PMID:19881998; https://doi.org/10.4103/0019-5154.41657
- Batra P, Shah N. Olmsted syndrome–a rare syndrome with oral manifestations. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2004; 97:599-602; PMID:15153872; https://doi.org/10.1016/j.tripleo.2003.10.025
- Poulin Y, Perry HO, Muller SA. Olmsted syndrome–congenital palmoplantar and periorificial keratoderma. J Am Acad Dermatol 1984; 10:600-10; PMID:6232300; https://doi.org/10.1016/S0190-9622(84)80264-9
- Al-Mutairi N, Sharma AK, Nour-Eldin O, Al-Adawy E. Olmsted syndrome: report of a new case with unusual features. Clin Exp Dermatol 2005; 30:640-2; PMID:16197376; https://doi.org/10.1111/j.1365-2230.2005.01871.x
- Kress DW, Seraly MP, Falo L, Kim B, Jegasothy BV, Cohen B. Olmsted syndrome. Case report and identification of a keratin abnormality. Arch Dermatol 1996; 132:797-800; PMID:8678572; https://doi.org/10.1001/archderm.1996.03890310083012
- Atherton DJ, Sutton C, Jones BM. Mutilating palmoplantar keratoderma with periorificial keratotic plaques (Olmsted's syndrome). Br J Dermatol 1990; 122:245-52; PMID:2138494; https://doi.org/10.1111/j.1365-2133.1990.tb08271.x
- Lucker GP, Steijlen PM. The Olmsted syndrome: mutilating palmoplantar and periorificial keratoderma. J Am Acad Dermatol 1994; 31:508-9; PMID:8077485; https://doi.org/10.1016/S0190-9622(09)80015-7
- Armstrong AP, Percival N. Olmsted's syndrome. J R Soc Med 1997; 90:81-2; PMID:9068437
- Larregue M, Callot V, Kanitakis J, Suau AM, Foret M. Olmsted syndrome: report of two new cases and literature review. J Dermatol 2000; 27:557-68; PMID:11052230; https://doi.org/10.1111/j.1346-8138.2000.tb02229.x
- Ogawa F, Udono M, Murota H, Shimizu K, Takahashi H, Ishida-Yamamoto A, Iizuka H, Katayama I. Olmsted syndrome with squamous cell carcinoma of extremities and adenocarcinoma of the lung: failure to detect loricrin gene mutation. Eur J Dermatol 2003; 13:524-8; PMID:14721769
- Elise Tonoli R, De Villa D, Hubner Frainer R, Pizzarro Meneghello L, Ricachnevsky N, de Quadros M. Olmsted syndrome. Case Rep Dermatol Med 2012; 2012:927305; PMID:23320205; https://doi.org/10.1155/2012/927305
- Danso-Abeam D, Zhang J, Dooley J, Staats KA, Van Eyck L, Van Brussel T, Zaman S, Hauben E, Van de Velde M, Morren MA, et al. Olmsted syndrome: exploration of the immunological phenotype. Orphanet J Rare Dis 2013; 8:79; PMID:23692804; https://doi.org/10.1186/1750-1172-8-79
- Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 2002; 418:186-90; PMID:12077606; https://doi.org/10.1038/nature00894
- Lin Z, Chen Q, Lee M, Cao X, Zhang J, Ma D, Chen L, Hu X, Wang H, Wang X, et al. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am J Hum Genet 2012; 90:558-64; PMID:22405088; https://doi.org/10.1016/j.ajhg.2012.02.006
- Lai-Cheong JE, Sethuraman G, Ramam M, Stone K, Simpson MA, McGrath JA. Recurrent heterozygous missense mutation, p.Gly573Ser, in the TRPV3 gene in an Indian boy with sporadic Olmsted syndrome. Br J Dermatol 2012; 167:440-2; PMID:22835024; https://doi.org/10.1111/j.1365-2133.2012.11115.x
- Nilius B, Biro T. TRPV3: a ‘more than skinny’ channel. Exp Dermatol 2013; 22:447-52; PMID:23800054; https://doi.org/10.1111/exd.12163
- Cheng W, Yang F, Takanishi CL, Zheng J. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J Gen Physiol 2007; 129:191-207; PMID:17325193; https://doi.org/10.1085/jgp.200709731
- Mari M, Bujny MV, Zeuschner D, Geerts WJ, Griffith J, Petersen CM, Cullen PJ, Klumperman J, Geuze HJ. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic 2008; 9:380-93; PMID:18088323; https://doi.org/10.1111/j.1600-0854.2007.00686.x
- Ishida-Yamamoto A, McGrath JA, Lam H, Iizuka H, Friedman RA, Christiano AM. The molecular pathology of progressive symmetric erythrokeratoderma: a frameshift mutation in the loricrin gene and perturbations in the cornified cell envelope. Am J Hum Genet 1997; 61:581-9; PMID:9326323; https://doi.org/10.1086/515518
- Ishida-Yamamoto A, Kato H, Kiyama H, Armstrong DK, Munro CS, Eady RA, Nakamura S, Kinouchi M, Takahashi H, Iizuka H. Mutant loricrin is not crosslinked into the cornified cell envelope but is translocated into the nucleus in loricrin keratoderma. J Invest Dermatol 2000; 115:1088-94; PMID:11121146; https://doi.org/10.1046/j.1523-1747.2000.00163.x
- Haghighi A, Scott CA, Poon DS, Yaghoobi R, Saleh-Gohari N, Plagnol V, Kelsell DP. A missense mutation in the MBTPS2 gene underlies the X-linked form of Olmsted syndrome. J Invest Dermatol 2013; 133:571-3; PMID:22931912; https://doi.org/10.1038/jid.2012.289
- Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, Ge P, Lilly J, Silos-Santiago I, Xie Y, et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 2002; 418:181-6; PMID:12077604; https://doi.org/10.1038/nature00882
- Sulk M, Seeliger S, Aubert J, Schwab VD, Cevikbas F, Rivier M, Nowak P, Voegel JJ, Buddenkotte J, Steinhoff M. Distribution and expression of non-neuronal transient receptor potential (TRPV) ion channels in rosacea. J Invest Dermatol 2012; 132:1253-62; PMID:22189789; https://doi.org/10.1038/jid.2011.424
- Majhi RK, Sahoo SS, Yadav M, Pratheek BM, Chattopadhyay S, Goswami C. Functional expression of TRPV channels in T cells and their implications in immune regulation. FEBS J 2015; 282:2661-81; PMID:25903376; https://doi.org/10.1111/febs.13306
- Asakawa M, Yoshioka T, Matsutani T, Hikita I, Suzuki M, Oshima I, Tsukahara K, Arimura A, Horikawa T, Hirasawa T, et al. Association of a mutation in TRPV3 with defective hair growth in rodents. J Invest Dermatol 2006; 126:2664-72; PMID:16858425; https://doi.org/10.1038/sj.jid.5700468
- Xiao R, Tian J, Tang J, Zhu MX. The TRPV3 mutation associated with the hairless phenotype in rodents is constitutively active. Cell Calcium 2008; 43:334-43; PMID:17706768; https://doi.org/10.1016/j.ceca.2007.06.004
- Cheng X, Jin J, Hu L, Shen D, Dong XP, Samie MA, Knoff J, Eisinger B, Liu ML, Huang SM, et al. TRP channel regulates EGFR signaling in hair morphogenesis and skin barrier formation. Cell 2010a; 141:331-43; https://doi.org/10.1016/j.cell.2010.03.013
- Li C, Ma W, Yin S, Liang X, Shu X, Pei D, Egan MT, Huang J, Pan A, Li Z. Sorting nexin 11 regulates lysosomal degradation of plasma membrane TRPV3. Traffic 2016; 17:500-14; PMID:26818531
- Cheng X, Shen D, Samie M, Xu H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett 2010b; 584:2013-21; https://doi.org/10.1016/j.febslet.2009.12.056
- Saito M, Hanson PI, Schlesinger P. Luminal chloride-dependent activation of endosome calcium channels: patch clamp study of enlarged endosomes. J Biol Chem 2007; 282:27327-33; PMID:17609211; https://doi.org/10.1074/jbc.M702557200
- Goswami C, Dreger M, Jahnel R, Bogen O, Gillen C, Hucho F. Identification and characterization of a Ca2+ -sensitive interaction of the vanilloid receptor TRPV1 with tubulin. J Neurochem 2004; 91:1092-103; PMID:15569253; https://doi.org/10.1111/j.1471-4159.2004.02795.x