
ABSTRACT
Burst-firing in thalamic neurons is known to play a key role in mediating thalamocortical (TC) oscillations that are associated with non-REM sleep and some types of epileptic seizure. Within the TC system the primary output of GABAergic neurons in the reticular thalamic nucleus (RTN) is thought to induce the de-inactivation of T-type calcium channels in thalamic relay (TR) neurons, promoting burst-firing drive to the cortex and the propagation of TC network activity. However, RTN neurons also project back onto other neurons within the RTN. The role of this putative negative feedback upon the RTN itself is less well understood, although is hypothesized to induce de-synchronization of RTN neuron firing leading to the suppression of TC oscillations. Here we tested two hypotheses concerning possible mechanisms underlying TC oscillation modulation. Firstly, we assessed the burst-firing behavior of RTN neurons in response to GABAB receptor activation using acute brain slices. The selective GABAB receptor agonist baclofen was found to induce suppression of burst-firing concurrent with effects on membrane input resistance. Secondly, RTN neurons express CaV3.2 and CaV3.3 T-type calcium channel isoforms known to contribute toward TC burst-firing and we examined the modulation of these channels by GABAB receptor activation. Utilizing exogenously expressed T-type channels we assessed whether GABAB receptor activation could directly alter T-type calcium channel properties. Overall, GABAB receptor activation had only modest effects on CaV3.2 and CaV3.3 isoforms. The only effect that could be predicted to suppress burst-firing was a hyperpolarized shift in the voltage-dependence of inactivation, potentially causing lower channel availability at membrane potentials critical for burst-firing. Conversely, other effects observed such as a hyperpolarized shift in the voltage-dependence of activation of both CaV3.2 and CaV3.3 as well as increased time constant of activation of the CaV3.3 isoform would be expected to enhance burst-firing. Together, we hypothesize that GABAB receptor activation mediates multiple downstream effectors that combined act to suppress burst-firing within the RTN. It appears unlikely that direct GABAB receptor-mediated modulation of T-type calcium channels is the major mechanistic contributor to this suppression.
Introduction
Thalamocortical (TC) oscillations play important roles in determining sleep and awake states, as well as contributing to the initiation and/or propagation of some types of epileptic seizure.Citation2,Citation12,Citation18 The relevant TC network comprises reticular thalamic nucleus (RTN) neurons, thalamic relay (TR) neurons and cortical layer V/VI neurons. While TR and cortical neurons send glutamatergic projections both to each other and to the RTN, the GABAergic RTN neurons project to TC neurons and also feedback to other neurons within the RTN. The GABAergic RTN innervation is believed to hyperpolarize TR neurons, driving the de-inactivation of low threshold T-type calcium channels which then promotes burst-firing either upon subsequent cessation of hyperpolarization (rebound burst-firing) or as a result of depolarizing drive from the cortex.Citation12 This burst-firing is believed to occur during drowsiness and non-REM sleep as well as in some types of epileptic seizure, such as absence seizures.Citation8 Alternatively, in the absence of hyperpolarization from the RTN TR neurons display a tonic firing pattern, believed to associate with the relay of sensory information during the awake state.Citation35
T-type calcium channels (CaV3.1, CaV3.2 and CaV3.3) underlie a low-threshold spike or calcium potential (LTCP), that activates around −60 mV, peaks at ∼ −40 mV and is 50 to 150 ms in duration.Citation7,Citation9,Citation19 It is this LTCP that triggers high frequency action potential firing to produce bursting.Citation7,Citation10,Citation11,Citation25 In RTN neurons mRNA expression analysis has identified the CaV3.2 and CaV3.3 T-type isoformsCitation38 with the relative contribution ratio estimated to be approximately 35:65 (CaV3.2:CaV3.3) as determined by pharmacological sensitivity,Citation21,Citation22,Citation23 and selective knockout in mice.Citation1 Of note, attenuation of the LTCP with high affinity T-type calcium channel antagonists both prevents RTN burst-firing in acute brain slices and prevents absence seizures in vivo.Citation40 Relevant to the underlying mechanism of epileptic activity, burst-firing in RTN neurons occurs in a time-locked manner with spike-wave discharges in both the GAERS and WAG/Rij rodent models of absence epilepsy and is thought to play a significant role in synchronization of the thalamocortical circuitry.Citation9,Citation20,Citation32,Citation33 Evidence suggests that burst-firing of RTN neurons promotes synchrony of intra-RTN, thalamic and thalamocortical rhythms via the intrinsic tendency of burst-firing to synchronize neuronal activity. Conversely, tonic firing is thought to lead to de-synchrony within the RTN, which would be expected to suppress thalamocortical oscillations.Citation39
Gamma-aminobutyric acid B (GABAB) receptors are metabotropic G protein-coupled receptors that couple to Gi/G0 to mediate slow inhibitory neurotransmission, typically via activation of potassium channels.Citation30,Citation31 These heterodimeric receptors have been implicated in several CNS disorders, for example, anxiety, depression and epilepsy and as such, have been proposed as promising therapeutic targets.Citation4,Citation24,Citation26,Citation37 The inhibition of GABAB receptors has been shown to induce seizure-like activity in thalamic slices and it has been proposed that intra-RTN inhibition reduces the number of RTN bursts.Citation14,Citation15,Citation36 The GABAB receptor agonist baclofen activates an outward conductance in RTN neuronsCitation41 which is believed to provide a sufficient time constant of hyperpolarization to allow for both the synchronization of burst-firing of RTN neurons and promote intrathalamic spindle oscillations.Citation16,Citation43 Given the feedback circuitry within the RTN it remains to be determined whether there are any effects of intra-RTN GABAB receptor activation on RTN ionic conductances and burst-firing.
Here we report that GABAB receptor activation inhibits RTN burst-firing in acute rat thalamic brain slices. This coincides with hyperpolarization of the neuronal resting membrane potential and decreased input resistance, both of which likely contribute toward the suppression of burst-firing. We further examine the effects of the direct modulation of T-type calcium channels by co-expression with heterodimeric GABAB1b and GABAB2 receptors. GABAB receptor activation is found to significantly shift the voltage-dependence of inactivation of both the CaV3.2 and CaV3.3 T-type isoforms which are hypothesized to contribute to the suppression of burst-firing in RTN neurons. However, additional effects such as a hyperpolarized shift in the voltage-dependence of activation of both CaV3.2 and CaV3.3 as well as an increase in the activation kinetics of the CaV3.3 isoform would be predicted to increase functional T-type currents and therefore increase burst-firing.
Results
Baclofen suppresses burst-firing in the RTN
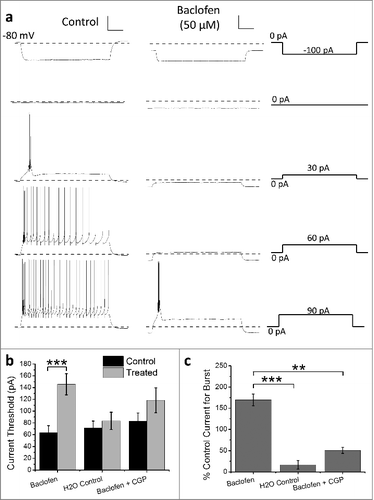
To examine burst-firing activity and passive membrane properties current-clamp recordings were performed in acute thalamic brain slices using 1000 ms hyperpolarizing and depolarizing current injections from their intrinsic resting membrane potential. RTN neurons responded to depolarizing current injection with burst-firing, often followed by tonic firing upon further depolarization (). In contrast, current injection causing hyperpolarization did not induce rebound burst-firing (). Application of baclofen (50 µM) significantly increased the depolarizing current injection required to induce burst-firing (control = 63.3 ± 12.02 pA, baclofen = 145.6 ± 18.0 pA (n = 8); p < 0.005) ( and ). Application of baclofen to cells pre-treated with the GABAB receptor antagonist CGP35348 (control + CGP = 82.9 ± 14.8 pA, baclofen + CGP = 118.3 ± 21.2 pA (n = 9)) or H20 control (control = 71.7 ± 11.4, H2O = 83.3 ± 14.8 (n = 6)) did not significantly affect the current threshold for burst-firing (). Comparing the percentage increase in current for the 3 treatment groups using multiple comparisons confirmed a significant increase in the current injection required to induce burst-firing in the baclofen versus CGP35348 pre-treated and H2O control application groups (; baclofen = 169.6 ± 30.7%, baclofen + CGP = 50.7 ± 7.2%, H2O = 16.8 ± 10.3%; [baclofen] vs [baclofen + CGP] p < 0.05, [baclofen] vs [H2O] p < 0.005 ANOVA with Tukey's post-test).
Figure 1. Baclofen suppresses burst-firing in RTN neurons. (a) Representative current-clamp recordings from neonatal RTN neurons in acute brain slices. Left and middle panels show control and baclofen-treated neuronal responses to the varying hyperpolarizing and depolarizing current injection protocols shown in right panels. (b) Histogram represents mean data for baclofen-treated neurons (n = 8) compared with the effects of H2O (n = 6) and baclofen perfusion with CGP35348 pretreatment (n = 9) on burst-firing threshold in control (black bars) and treated (gray bars) RTN neurons (Paired sample t-test). (c) Histogram represents the percentage change in current required to achieve threshold for burst-firing for data in (b) (ANOVA with Tukey's post-test). *p < 0.05, ***p < 0.005. Scale bars represent 20 mV and 200 ms.
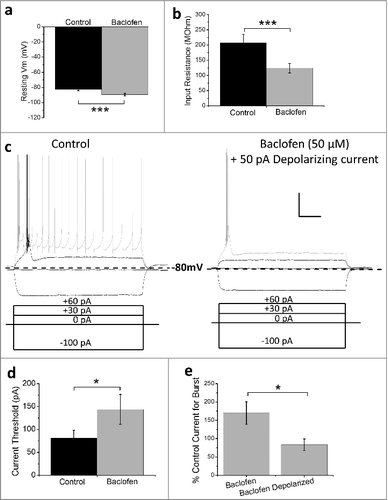
In addition to increasing the threshold for burst-firing, baclofen application also induced hyperpolarization of RTN neurons (), consistent with the activation of a GABAB receptor-mediated leak potassium conductance.Citation41 Further, we noted a corresponding significant decrease in input resistance in response to baclofen (). To assess the effect of baclofen on burst-firing without the compounding effects of hyperpolarization, constant depolarizing current was injected to “manually” return the resting membrane potential of RTN neurons to pre-baclofen levels. Under these conditions, baclofen still increased the current threshold required for burst-firing ( and ; control + current = 81.3 ± 16.8 pA, baclofen + current = 143.8 ± 32.7 pA; p < 0.05). Importantly, although burst-firing was still inhibited by baclofen after membrane potential had been returned to the pre-baclofen level, the percentage increase in current was significantly lower than when membrane potential was not corrected (; baclofen = 169.6 ± 30.7%, baclofen + depolarizing current = 83.3 ± 15.6%; P < 0.05). This indicates that hyperpolarization alone has a significant effect on the suppression of burst-firing following baclofen application.
Figure 2. Baclofen induces hyperpolarization and decreases input resistance in RTN neurons. Histograms representing mean resting membrane potential (a) and input resistance (b) of RTN neurons before (black columns) and after (gray columns) baclofen (50 uM) perfusion (n = 8). (c) Representative current-clamp recordings from RTN neurons under control conditions (left panel) and in the presence of baclofen with membrane potential corrected to pre-baclofen level with constant current injection (right panel). Recordings show response of RTN neurons to same current injection protocol (lower panels). (d) Histogram represents mean data from (c) for threshold current injection to achieve bursting in control conditions and with membrane-potential corrected, baclofen-treated neurons. (e) Histogram represents the percentage change in current required to achieve burst-firing for cells where depolarizing current is used to correct membrane potential vs. cells where membrane potential is not corrected following baclofen-mediated hyperpolarization. *p < 0.05, ***p < 0.005 Paired t-test. Scale bars represent 20 mV and 200 ms.
Taken together, these data demonstrate for the first time that GABAB receptor activation can suppress the activity of burst-firing neurons within the RTN itself.
GABAB receptor activation differentially affects T-type calcium channel isoforms
Given that T-type calcium channels play a key role in the generation of low threshold burst-firing (CaV3.2 and CaV3.3 in RTN neurons and CaV3.1 in TR neurons) we next sought to establish whether GABAB receptor activation can also modulate T-type calcium currents. Performing voltage-clamp in acute brain slices was associated with difficulties in space clamp and produced highly variable results, with 3 out of 8 cells showing a significant effect of baclofen application (∼10%, 25% and 80% inhibition in each affected cell), but with no effect on the remaining 5 cells. Notably, a large fraction of T-type calcium channels are located in dendrites and may have been lost in preparing dissociated neurons. As such, we opted to undertake a series of exogenous experiments using cloned GABAB1b and GABAB2 receptors co-transfected with individual CaV3.1, CaV3.2 or CaV3.3 T-type isoforms.
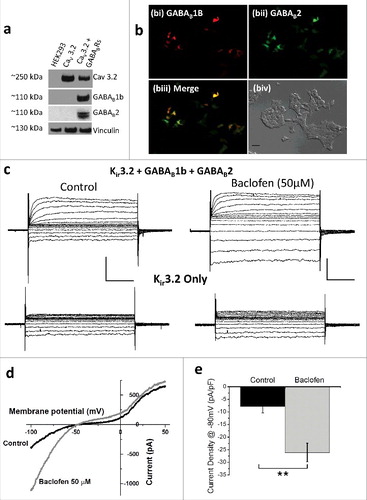
To confirm GABAB receptor expression, Western blotting was performed on lysates from HEK 293 cells stably expressing the CaV3.2 channel and co-transfected with GABAB1B and GABAB2 receptors (). Further, to validate GABAB receptor functional activity GABAB1B and GABAB2 receptors were co-transfected with Kir3.2 in HEK 293 cells. Application of baclofen resulted in potentiation of the Kir3.2 current in cells expressing GABAB receptors but not those expressing Kir3.2 alone ().
Figure 3. Expression and validation of GABAB receptors. (a) Western blot of lysates from untransfected, CaV3.2 transfected and CaV3.2 plus GABAB1b and GABAB2 transfected HEK 293 cells, exposed to CaV3.2, GABAB1b, GABAB2 and vinculin antibodies. (b) GFP (bi), RFP (bii) and merged GFP/RFP (biii) epi-fluorescence images with a corresponding transmitted light image (biv; scale bar = 50 µM) showing expression of GABAB1b (red) and GABAB2 (green) reporter genes transfected into HEK 293 cells. (c) Voltage-clamp traces showing response of Kir3.2 currents expressed in HEK 293 cells in control conditions (left panels) and following baclofen (50 µM) application (right panels) in cells expressing Kir3.2 co-transfected with GABAB1b and GABAB2 receptors (upper panels) or Kir3.2 alone (lower panels). (d) Representative Kir3.2 current in response to a 180 ms ramp depolarization from −100 to +50 mV under control conditions (black trace) and following baclofen (50 µM) application (gray trace). (e) Histogram represents mean current density data from ramp depolarizations in cells co-expressing Kir3.2 channels, plus GABAB1B and GABAB2 receptors under control conditions (black columns; n = 3) and following baclofen (50µM) application (gray column; n = 3). Scale bars represent 500 pA and 50 ms. **p < 0.01.
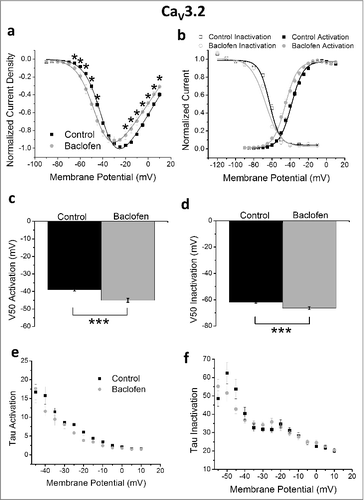
Cells co-expressing GABAB receptors with the CaV3.2 T-type channel resulted in several altered biophysical properties following baclofen application. An increase in CaV3.2 current density (normalized to pre-baclofen peak current density values to remove variability in current density magnitude between cells) was observed at test potentials between −65 and −45 mV with a decrease in current density at more depolarized test potentials (). These effects likely occur as a result of a hyperpolarizing shift in the voltage-dependence of activation ( and ; V50: control = −38.9 ± 0.9 mV, baclofen = −45.1 ± 1.1 mV; p < 0.005) shifting the current density voltage-relationship to the left (). The voltage-dependence of inactivation was also shifted toward more hyperpolarized potentials ( and ; V50: control = −38.9 ± 0.9 mV, baclofen = −45.1 ± 1.1 mV; p < 0.005). No significant effects were observed in the rates of activation or inactivation of CaV3.2 currents following GABAB receptor activation ( and ).
Figure 4. Mean data for HEK 293 cells expressing CaV3.2, GABAB1b and GABAB2 receptors. (a) Current density (n = 14), (b) current density normalized to peak current density of cell during control conditions (n = 14), (c) voltage-dependence of activation (n = 14), steady-state inactivation (n = 5), (d) tau activation (n = 14), (e) tau inactivation (n = 14). *p < 0.05.
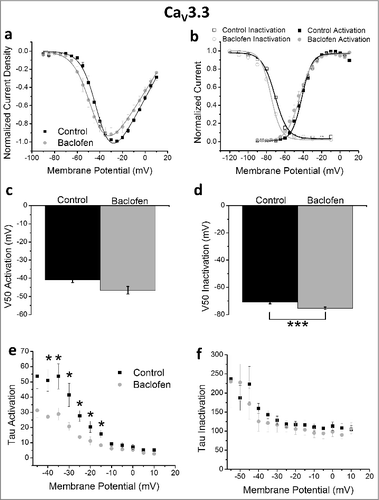
Distinct from the CaV3.2 isoform, in HEK 293 cells co-expressing GABAB receptors with the CaV3.3 T-subtype there was no significant effect of baclofen on current density (). There was also no significant effect observed in the voltage-dependence of activation ( and ). There was a significant shift in the voltage-dependence of inactivation following GABAB receptor activation ( and ; V50: control = −70.9 ± 1.2 mV, baclofen = −75.3 ± 0.8 mV; p < 0.05) (). A near significant increase was also observed in the slope factor potential following baclofen application (k: control = 5.86 ± 0.37, baclofen = 6.22 ± 0.27 mV; p = 0.07). Of note, the activation kinetics of CaV3.3 were faster following baclofen application, with the τact displaying a significantly reduced magnitude between −40 and −5 mV () and without any effect observed on inactivation kinetics ().
Figure 5. Mean data for HEK 293 cells expressing CaV3.3, GABAB1b and GABAB2 receptors. (a) Current density (n = 7), (b) current density normalized to peak current density of cell during control conditions (n = 7), (c) voltage-dependence of activation (n = 7), steady-state inactivation (n = 3), (d) tau activation (n = 7), (e) tau inactivation (n = 7). *p < 0.05.
Although the CaV3.1 T-type is not expressed in RTN cells, for completeness concerning the specificity of GABAB receptor-mediated modulation we also examined HEK 293 cells co-expressing GABAB receptor subunits with CaV3.1. However, baclofen had little effect on any of the biophysical parameters examined.
Discussion
GABA release within the RTN is believed to induce de-synchronization of thalamic spindle-like activity by preventing RTN neurons from burst-firing on each depolarization of an oscillation.Citation36 It is possible that desynchronization of the RTN contributes toward termination of both spindle oscillations and seizure activity within the thalamus and thalamocortical network, respectively. While the mechanism by which GABA receptors induce de-synchronization is not fully understood, it has been shown that selective GABAB receptor activation induces the activation of a leak potassium conductance in RTN neuronsCitation41 and that could be hypothesized to generate hyperpolarization. In the present study we examined the activity response of RTN neurons to GABAB receptor activation, confirming the induction of hyperpolarizationCitation41 and further that it occurs concurrently with a decrease in input resistance. Importantly, the amount of depolarizing current required to initiate burst-firing was decreased by GABAB receptor activation, even when the neuronal resting membrane potential was depolarized to control levels by constant current injection. This indicates that GABAB receptor-mediated membrane hyperpolarization alone is not sufficient to explain its effects on suppressing RTN burst-firing. Our data suggest that the underlying mechanism for suppressed burst-firing via GABAB receptors is the likely result of a shunt in excitability due to decreased input resistance. One possible candidate for the cause of this drop in input resistance, as well as the hyperpolarization of the membrane potential, is the activation of G protein-coupled Inwardly Rectifying Potassium Channels (GIRKS).Citation28 GIRKs are well-established effectors of GABAB receptors via direct activation by Gβγ subunits causing membrane hyperpolarization by increasing potassium efflux.Citation13,Citation34 A second possible candidate is activation of Two-Pore-Domain Potassium (K2P) currents, which have been described in thalamic relay neurons but are yet to be described in RTN neurons.Citation5,Citation27,Citation29 The K2P isoform, TREK-1 is potentiated by mGlu4 receptor activation via Gi/Go-mediated inhibition of PKA phosphorylation and as such it is possible that GABAB receptors may also enhance potassium conductance through this channel.Citation6
A significant hyperpolarizing shift in the V50 of activation was observed for the CaV3.2 T-type channel, resulting in increased current density at hyperpolarized membrane potentials and decreased current density at more depolarized membrane potentials. The increased current density at hyperpolarized potentials would be predicted to increase the LTCP magnitude, while the decreased current density at depolarized potentials may increase calcium conductance during action potentials.Citation7,Citation10,Citation25 Neither of these effects would be expected to contribute to the suppression of burst-firing observed with baclofen application. However, the noted hyperpolarizing shift in the V50 of steady-state inactivation would result in a significant reduction in the channel availability between −60 mV and −70 mV wherein T-type calcium channels critically contribute toward generating burst-firing. Parenthetically, we note that considering liquid junction potential correction this would equate to reduced channel availability between −67.8 mV and −77.8 mV; well within the realms of LTCP generation. As such, it is feasible that CaV3.2 T-type channel modulation contributes to the suppressed burst-firing observed with GABAB receptor activation.
The CaV3.3 T-type isoform displayed a distinct set of modulatory characteristics in response to baclofen application. In one aspect similar to CaV3.2 channels, GABAB receptor activation induced a hyperpolarizing shift in the V50 of CaV3.3 steady-state inactivation, resulting in a reduction in channel availability at membrane potentials between −70 mV and −80 mV (with liquid junction potential correction this would equate to between −77.8 mV and −87.8 mV). Distinct from that for CaV3.2 channels, GABAB receptor activation also increased the tau activation kinetics of CaV3.3. This modulatory effect would be expected to increase the rate of depolarization during LCTPs and therefore enhance burst-firing. It should also be considered that a neuronal milieu may be absent in HEK 293 cells, necessary for modulation of CaV3.2 and CaV3.3 channels by GABAB either directly by G proteins or via a secondary effector.
Little effect of GABAB receptor activation was observed on the CaV3.1 T-type channel isoform. This supports a previous study which demonstrated that baclofen (50 µM) does not affect the CaV3.1 T-type current expressed in dorsolateral lateral geniculate thalamocortical neurons.Citation17 Also, since CaV3.1 channels are not expressed in the RTN,Citation1,Citation23,Citation38 the lack of effect of GABAB receptor activation on CaV3.1 channels would not be expected to contribute to the observed effect of baclofen on burst-firing in the RTN.
The link between burst-firing, T-type calcium channels and synchronization has been extensively studied. Since burst-firing generally increases synchrony in neuronal networks, the de-synchronizing effect of intra-RTN GABAB-mediated signaling is predicted to result in an attenuation of burst-firing.Citation36,Citation41 Our results support the notion that GABAB receptor activation suppresses the ability of RTN neurons to burst-fire, which in turn may prevent incoming oscillatory volleys from maintaining RTN firing in a time-locked manner. This may have important consequences in thalamocortical system-mediated seizure generation, wherein a build-up of GABAB-mediated signaling throughout the duration of a seizure may lead to de-synchronous activity and seizure termination.
Methods
Acute brain slice preparation
P14-P22 Wistar rats (male and female; bred by the Zoology Department and Animal Resource Unit at The University of British Columbia, Canada) were used in acute brain slice current-clamp experiments. Neonatal rats were briefly anaesthetized using isoflurane, killed by cervical dislocation and the brains rapidly removed and placed in ice cold sucrose solution containing in mM: 234 sucrose, 24 NaHCO3, 1.25 NaH2PO4, 11 glucose, 2.5 KCl, 0.5 CaCl2, 6 MgCl2, bubbled with 95% O2:5% CO2. Brain tissue was glued to a cutting chamber, which was then filled with ice cold sucrose solution. Horizontal brain slices containing the whole thalamus (∼300–350 μm thick) were cut from the level of the ventral RTN and incubated for a minimum of one hour at 34°C in a current-clamp recording solution containing in mM: 126 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 2 MgCl2, 10 glucose, 1 kynurenic acid, 0.1 picrotoxin; bubbled with 95% O2:5% CO2.
Brain slice electrophysiology
Slices were transferred to the recording chamber superfused with current-clamp recording solution and maintained at 33–35°C. RTN neurons were visualized using a DIC microscope (Axioskop 2-FS Plus, Carl Zeiss) and infrared camera (IR-1000, DAGE MTI) and visually identified by their location, morphology and orientation. All recordings were undertaken using a Multiclamp 700B amplifier and pClamp software version 9 (Molecular Devices). The recording chamber was grounded with an Ag/AgCl pellet. Whole-cell current-clamp recordings were undertaken using fire polished borosilicate glass pipettes (4–6 MΩ) filled with the following solution containing in mM: 120 K-gluconate, 10 HEPES, 1 MgCl2, 1 CaCl2, 11 KCl, 11 EGTA, 4 MgATP, 0.5 NaGTP, pH adjusted to 7.2 using KOH, osmolarity adjusted to 290 mOsm/kg using D-mannitol. The liquid junction potential for current-clamp solutions was calculated as +13.3 mV and corrected off-line. To evaluate basic neuronal responses to hyperpolarization and depolarization, DC current was injected from −110 pA to +200 pA in 10 pA increments for a duration of 1.2 sec at the cell's intrinsic resting membrane potential. Neurons that did not exhibit burst-firing (as determined by a minimum of 3 action potentials within 100 ms during the current step) in response to depolarizing current steps were discarded. Voltage responses under current-clamp conditions were sampled at 50 kHz and filtered at 10 kHz. Bridge balance was monitored during recordings and any neurons displaying bridge balance values greater than 20 MΩ were discarded. Capacitance neutralization was performed between 3.8 and 4.2 pF. Data analysis was performed using Clampfit 9 (Axon Instruments Inc.) and software Origin version 8.5 (OriginLab Corp., Northampton, MA).
Cell culture
Human Embryonic Kidney (HEK 293) cell lines stably expressing human T-type calcium channels (hCav3.1, hCav3.2 and hCav3.3) were cultured in DMEM media containing 4 mM L-glutamine, supplemented with 10% heat inactivated FBS and 10 µl/ml of non-essential amino acids (Invitrogen 11140). Cells were grown and maintained at 37°C in a humidified atmosphere at 95% air and 5% CO2. Antibiotic selection was used to identify stable transfectants as described previously;Citation3 culture media was supplemented with 25 mg/ml zeocin (Invitrogen) for hCav3.1 and hCav3.2 or with 300 mg/ml hygromycin B (Invitrogen) for the hCav3.3-expressing cell line. For electrophysiological recordings, cells were plated on 35 mm Petri dishes with poly-D-Lysine-coated glass coverslips and transiently transfected after 24 hours with GABAB receptor subunits, hGABAB1b (in RFP expressing vector # QM512B-1, Systembio) and GABAB2 (in GFP expressing vector # CD111B-1, Systembio) using Lipofectamine 2000 Reagent (Invitrogen). HEK 293 cells were cotransfected with Kir 3.2 (in pcDNA3.1; 0.2 µg/dish), GABAB1b and hGABAB2 (0.25 µg/dish each) and whole-cell potassium currents were recorded 48 hours after transfection. Stable hCav3.x cell lines were cotransfected with hGABAB1b and hGABAB2 (0.25 – 0.5 µg/dish each) and the DNA-Lipofectamine complex was removed after 3 hours incubation and the selection agent immediately added to the culture media. T-type calcium currents were recorded 24–48 hours after transfection.
HEK 293 cell electrophysiology
Calcium currents were recorded at room temperature (22–24°C) using the whole-cell voltage-clamp technique with the following solutions: Internal (mM): 120 Cs-Methanesulphonate, 11 EGTA, 10 HEPES, 2 MgCl2, 5 MgATP and 0.3 NaGTP (pH 7.2 and osmolality corrected to 310 mM using D-mannitol); external (mM): 2 CaCl2, 1 MgCl2, 10 HEPES, 40 TEACl, 92 CsCl and 10 Glucose (pH 7.4). The liquid junction potential was calculated to be 7.8 mV but not corrected in data shown. Fire-polished patch pipettes (borosilicate glass) had typical resistances of 3 to 5 MΩ when containing internal solution. The recording chamber was grounded with a Ag/AgCl pellet. Whole-cell currents were recorded at room temperature using an Axopatch 200B amplifier (Axon instruments Inc., Union City, CA). Data were acquired with pClamp software package version 9 (Molecular Devices). Series resistance (Rs) was compensated by 65–75% and seals with Rs values higher than 20 MΩ or cells with peak current lower than 100 pA were discarded. Data analysis was performed using Clampfit 9 (Molecular Devices) and software Origin version 8.5 (OriginLab Corp., Northampton, MA).
Calcium current-voltage (I-V) relationships were obtained by depolarizing the membrane with 150 msec pulses from a holding potential of –110 mV (currents sampled at 10 kHz and filtered at 2 kHz). Test pulses from −90 to +10 mV were applied at 5 mV steps. Peak amplitude of calcium currents was plotted against test pulse potential and I-V curves were fitted using a modified Boltzmann equation: I = (Gmax*(Vm-Er))/(1+exp((V50-Vm)/k)), where Gmax is the maximum value of membrane conductance, Vm is the test potential, Er is the extrapolated reversal potential, V50 is the half-activation potential, and k reflects the voltage sensitivity. Activation curves were obtained by calculating conductance from the I-V curves and plotting the normalized conductance as a function of the membrane potential. The data was fitted with the Boltzmann equation: G/Gmax = A2 + (A1-A2)/(1 + exp((V50-Vm)/k)), where A1 is minimum normalized conductance, A2 is maximum normalized conductance, Vm is the test potential, V50 is the half-activation potential, and k value the slope of the activation curve (Slope constant).
Calcium current steady-state inactivation was studied using 90 msec test pulses at −30 mV applied after 2 sec conditioning pre-pulses ranging from −120 to −10 mV (currents sampled at 10k Hz and filtered at 2 kHz). The current magnitude obtained during each test pulse was normalized to the maximum at −120 mV and plotted as a function of the pre-pulse potential. The data was fitted with the Boltzmann equation: I/Imax = A2 + (A1-A2)/(1 + exp((Vm-V50)/k)), where A1 is minimum normalized current, A2 the maximum normalized current, Vm the test potential, V50 the half-inactivation potential, and k the slope of the inactivation curve (Slope constant). The time course for activation (τact) and inactivation (τinact) were analyzed by fitting current recordings obtained from the I-V protocol with a single exponential standard equation: I = Ae-t/τ, where A is the amplitude of the current, and τ is the time constant.
Inwardly-rectifying potassium currents were recorded using whole cell voltage-clamp with the following solutions: Internal (mM): 130 KCl, 20 NaCl, 5 EGTA, 5.5 MgCl2, 10 HEPES, 2.6 K-ATP, 0.3 Li-GTP (pH correct ed to 7.4 using KOH and osmolarity corrected to 317 mM using D-Mannitol); external (mM): 20 KCl, 140, NaCl, 0.5 CaCl2, 2 MgCl2, 10 HEPES (pH to 7.4 using NaOH and osmolarity corrected to 310 mM using D-Mannitol). I-V relationships were obtained by depolarizing the membrane with 100 msec pulses from a holding potential of –70 mV (currents sampled at 10 kHz and filtered at 2 kHz). Test pulses from −100 to +40 mV were applied at 10 mV steps. Current response to ramp depolarization was obtained by depolarizing the cell from −100 mV to +50 mV over a duration of 180 ms from a holding potential of −100 mV.
Western blotting
HEK 293 cell extracts were homogenized using Laemmli buffer and liquid nitrogen. Sample lysate concentration was determined using the Pierce 660nm Protein Assay (Fisher # PI22660) with the addition of the IDCR reagent (Fisher # PI22663) to compensate for the use of Laemmli buffer in the sample preparation. All samples were run on NuPAGE® Novex® 3–8% Tris-Acetate Midi Gels, from Invitrogen, using NuPAGE Tris-Acetate SDS Running Buffer. Samples for CaV3.2 detection were loaded with 5 μg per lane. Samples for GABAB1b and GABAB2 detection were loaded with 10 μg per lane. Samples for Vinculin detection were loaded with 5 μg per lane. The samples were blotted overnight at 4°C, running at 75 mA per gel. The transfer buffer used contained (in mM) Tris-HCl (40), Sodium Acetate (20), EDTA (2), 20% Methanol and 10% SDS.Citation42 Samples were blotted on to Nitrocellulose membrane Amersham Hybond ECL 0.45 um (Fisher # 45000929). After transfer, the membranes were equilibrated for 10 minutes in TBS + 0.1% Tween-20, then blocked for 1 hour at room temperature in TBS-T + 2% non-fat milk powder. The primary antibodies were diluted in TBS-T + 2% milk and incubated for 2 hours at room temperature. The membranes were washed 3 times for 20 minutes in 2% milk buffer, followed by incubation for one hour at room temperature with the secondary antibodies diluted in 2% milk buffer. After the secondary incubation, the membranes were washed 3 times for 10 minutes in TBS + 0.05% Tween-20. The ECL reagent was then added, and the membranes exposed to Amersham Hyperfilm ECL (Fisher # 45001505). Antibodies: Anti-CaV3.2 mouse monoclonal antibody at 0.5 μg/ml (Neuromab #75–095), anti-GABAB1b mouse monoclonal at 1 μg/ml (Santa Cruz # sc-166408), anti-GABAB2 rabbit polyclonal at 0.2 μg/ml (Santa Cruz #sc-28792), and anti-vinculin mouse monoclonal at 1/10,000 (Sigma #V9131), secondary goat anti-mouse HRP at 1/5000 for GABAB1b and CaV3.2, and 1/10 000 for vinculin (Pierce # PI32230), and secondary goat anti-Rabbit Poly-HRP at 1/5000 (Pierce cat# PI32260). The densitometric results were analyzed using Image Studio Lite software (LICOR).
Statistics
All data plotted as mean ± standard error mean. Data followed a normal distribution and was analyzed by Paired Sample t-test or by One-way ANOVA with Tukey's post-test where appropriate. Significance was considered where p < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
T.P. Snutch is supported by an operating grant from the Canadian Institutes of Health Research (#10677) and the Canada Research Chair in Biotechnology and Genomics-Neurobiology. S.M. Cain was supported by a postdoctoral fellowship from the B.C. Epilepsy Society and the Michael Smith Foundation for Health Research, a research grant from the BC Epilepsy Society and is currently funded by the CURE – Taking Flight Award. We thank Dr. Harley Kurata (University of British Columbia) for his generous gift of the Kir3.2 cDNA.
References
- Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Lüthi A. The CaV3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A. 2011;108:13823-8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21808016 [Accessed August 17, 2011]. https://doi.org/10.1073/pnas.1105115108. PMID:21808016.
- Beierlein M, Fall CP, Rinzel J, Yuste R. Thalamocortical bursts trigger recurrent activity in neocortical networks: Layer 4 as a frequency-dependent gate. J Neurosci. 2002;22:9885-94. https://doi.org/10.1089/adt.2009.191. PMID:12427845.
- Belardetti F, Tringham E, Eduljee C, Jiang X, Dong H, Hendricson A, Shimizu Y, Janke DL, Parker D, Mezeyova J, et al. A fluorescence-based high-throughput screening assay for the identification of T-type calcium channel blockers. Assay Drug Dev Technol. 2009;7:266-80. PMID:19530894.
- Benke D. GABAB receptor trafficking and interacting proteins: Targets for the development of highly specific therapeutic strategies to treat neurological disorders? Biochem Pharmacol. 2013;86:1525-30. https://doi.org/10.1016/j.bcp.2013.09.016. PMID:24084431.
- Bista P, Meuth SG, Kanyshkova T, Cerina M, Pawlowski M, Ehling P, Landgraf P, Borsotto M, Heurteaux C, Pape H-C, et al. Identification of the muscarinic pathway underlying cessation of sleep-related burst activity in rat thalamocortical relay neurons. Pflüg Arch. 2012;463:89-102. https://doi.org/10.1007/s00424-011-1056-9. PMID:22083644.
- Cain SM, Meadows HJ, Dunlop J, Bushell TJ. mGlu4 potentiation of K(2P)2.1 is dependant on C-terminal dephosphorylation. Mol Cell Neurosci. 2008;37:32-39. https://doi.org/10.1016/j.mcn.2007.08.009. PMID:17916432.
- Cain SM, Snutch TP. Contributions of T-type calcium channel isoforms to neuronal firing. Channels. 2010;4:44-51. https://doi.org/10.4161/chan.4.6.14106.
- Cain SM, Snutch TP. Voltage-gated calcium channels in epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta A, editors. Jasper's basic mechanisms of the epilepsies. 4th Edition. Bethesda (U.S.A.): Oxford University Press; 2012a. p. 66-84.
- Cain SM, Snutch TP. T-type calcium channels in burst-firing, network synchrony, and epilepsy. Biochim Biophys Acta. 2012b;1828:1572-8. https://doi.org/10.1016/j.bbamem.2012.07.028.
- Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P. Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol. 2002;540:3-14. https://doi.org/10.1113/jphysiol.2001.013269. PMID:11927664.
- Chorev E, Manor Y, Yarom Y. Density is destiny - On the relation between quantity of T-type Ca2+ channels and neuronal electrical behavior. CNS Neurol Disord Drug Targets. 2006;5:655-62. https://doi.org/10.2174/187152706779025517. PMID:17168749.
- Contreras D. The role of T-channels in the generation of thalamocortical rhythms. CNS Neurol Disord Drug Targets.2006;5:571-85. https://doi.org/10.2174/187152706779025526. PMID:17168743.
- Dascal N. Signalling via the G protein-activated K+ channels. Cell Signal. 1997;9:551-73. https://doi.org/10.1016/S0898-6568(97)00095-8. PMID:9429760.
- Destexhe A. Spike-and-wave oscillations based on the properties of GABAB receptors. J Neurosci. 1998;18:9099-111. https://doi.org/10.1113/jphysiol.1995.sp020710. PMID:9787013.
- Destexhe A, Bal T, McCormick DA, Sejnowski TJ. Ionic mechanisms underlying synchronized oscillations and propagating waves in a model of ferret thalamic slices. J Neurophysiol. 1996;76:2049-70. PMID:8890314.
- Golomb D, Wang XJ, Rinzel J. Synchronization properties of spindle oscillations in a thalamic reticular nucleus model. J Neurophysiol. 1994;72:1109-26. PMID:7807198.
- Guyon A, Leresche N. Modulation by different GABAB receptor types of voltage-activated calcium currents in rat thalamocortical neurones. J Physiol. 1995;485(Pt 1):29-42. PMID:7658381.
- Huguenard JR. Neuronal circuitry of thalamocortical epilepsy and mechanisms of antiabsence drug action. Adv Neurol. 1999;79:991-9. https://doi.org/10.1016/0006-8993(93)91641-5. PMID:10514881.
- Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804-17. PMID:1403085.
- Inoue M, Duysens J, Vossen JM, Coenen AM. Thalamic multiple-unit activity underlying spike-wave discharges in anesthetized rats. Brain Res. 1993;612:35-40. PMID:8330210.
- Joksovic PM, Bayliss DA, Todorovic SM. Different kinetic properties of two T-type Ca2+ currents of rat reticular thalamic neurones and their modulation by enflurane. J Physiol. 2005;566:125-42. https://doi.org/10.1113/jphysiol.2005.086579. PMID:15845580.
- Joksovic PM, Doctor A, Gaston B, Todorovic SM. Functional regulation of T-type calcium channels by s-nitrosothiols in the rat thalamus. J Neurophysiol. 2007;97:2712-21. https://doi.org/10.1152/jn.00926.2006. PMID:17287440.
- Joksovic PM, Nelson MT, Jevtovic-Todorovic V, Patel MK, Perez-Reyes E, Campbell KP, Chen CC, Todorovic SM. CaV3.2 is the major molecular substrate for redox regulation of T-type Ca2+ channels in the rat and mouse thalamus. J Physiol. 2006;574:415-30. https://doi.org/10.1113/jphysiol.2006.110395. PMID:16644797.
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca2+ channels. Neuron. 2001;31:35-45. https://doi.org/10.1016/S0896-6273(01)00343-9. PMID:11498049.
- Kozlov AS, McKenna F, Lee JH, Cribbs LL, Perez-Reyes E, Feltz A, Lambert RC. Distinct kinetics of cloned T-type Ca2 + channels lead to differential Ca2 + entry and frequency-dependence during mock action potentials. Eur J Neurosci. 1999;11:4149-58. https://doi.org/10.1046/j.1460-9568.1999.00841.x. PMID:10594640.
- Kumar K, Sharma S, Kumar P, Deshmukh R. Therapeutic potential of GABA(B) receptor ligands in drug addiction, anxiety, depression and other CNS disorders. Pharmacol Biochem Behav. 2013;110:174-84. https://doi.org/10.1016/j.pbb.2013.07.003. PMID:23872369.
- Meuth SG, Aller MI, Munsch T, Schuhmacher T, Seidenbecher T, Meuth P, Kleinschnitz C, Pape H-C, Wiendl H, Wisden W, et al. The contribution of TWIK-related acid-sensitive K+-containing channels to the function of dorsal lateral geniculate thalamocortical relay neurons. Mol Pharmacol. 2006;69:1468-76. https://doi.org/10.1124/mol.105.020594. PMID:16424077.
- Misgeld U, Bijak M, Jarolimek W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol. 1995;46:423-62. https://doi.org/10.1016/0301-0082(95)00012-K. PMID:8532848.
- Musset B, Meuth SG, Liu GX, Derst C, Wegner S, Pape H-C, Budde T, Preisig-Müller R, Daut J. Effects of divalent cations and spermine on the K+ channel TASK-3 and on the outward current in thalamic neurons. J Physiol. 2006;572:639-57. https://doi.org/10.1113/jphysiol.2006.106898. PMID:16513667.
- Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123-47. https://doi.org/10.1016/S1054-3589(10)58006-2. PMID:20655481.
- Pinard A, Seddik R, Bettler B. GABAB receptors: Physiological functions and mechanisms of diversity. Adv Pharmacol. 2010;58:231-55. https://doi.org/10.1016/S1054-3589(10)58010-4. PMID:20655485.
- Pinault D. The thalamic reticular nucleus: Structure, function and concept. Brain Res Brain Res Rev. 2004;46:1-31 https://doi.org/10.1016/j.brainresrev.2004.04.008. PMID:15297152.
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol. 1998;509(Pt 2):449-56. https://doi.org/10.1111/j.1469-7793.1998.449bn.x. PMID:9575294.
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iñiguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 1994;370:143-6. https://doi.org/10.1038/370143a0. PMID:8022483.
- Sherman SM. Tonic and burst firing: Dual modes of thalamocortical relay. Trends Neurosci. 2001;24:122-6. https://doi.org/10.1016/S0166-2236(00)01714-8. PMID:11164943.
- Sohal VS, Huguenard JR. Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci. 2003;23:8978-88. https://doi.org/10.1371/journal.pone.0024933. PMID:14523100.
- Tadavarty R, Rajput PS, Wong JM, Kumar U, Sastry BR. Sleep-deprivation induces changes in GABA(B) and mGlu receptor expression and has consequences for synaptic long-term depression. PloS One. 2011;6:e24933. PMID:21980366.
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895-911. https://doi.org/10.1097/00001756-199803090-00024. PMID:10066243.
- Thomas E, Lytton WW. Computer model of antiepileptic effects mediated by alterations in GABA(A)-mediated inhibition. Neuroreport. 1998;9:691-6. PMID:9559940.
- Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, Eduljee C, Jiang X, Smith P, Morrison J-L, et al. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med. 2012;4:121ra19. https://doi.org/10.1126/scitranslmed.3003120. PMID:22344687.
- Ulrich D, Huguenard JR. GABAB receptor-mediated responses in GABAergic projection neurones of rat nucleus reticularis thalami in vitro. J Physiol. 1996;493(Pt 3):845-54. https://doi.org/10.1113/jphysiol.1996.sp021427. PMID:8799904.
- Wang K, Fanger BO, Guyer CA, Staros JV. Electrophoretic transfer of high-molecular-weight proteins for immunostaining. Methods Enzymol. 1989;172:687-96. https://doi.org/10.1016/0306-4522(93)90474-T. PMID:2747545.
- Wang XJ, Rinzel J. Spindle rhythmicity in the reticularis thalami nucleus: Synchronization among mutually inhibitory neurons. Neuroscience. 1993;53:899-904. PMID:8389430.