
ABSTRACT
In the last 5 years inhibitors of the potassium channel KV1.3 have been shown to reduce neuroinflammation in rodent models of ischemic stroke, Alzheimer’s disease, Parkinson’s disease and traumatic brain injury. At the systemic level these beneficial actions are mediated by a reduction in microglia activation and a suppression of pro-inflammatory cytokine and nitric oxide production. However, the molecular mechanisms for the suppressive action of KV1.3 blockers on pro-inflammatory microglia functions was not known until our group recently demonstrated that KV1.3 channels not only regulate membrane potential, as would be expected of a voltage-gated potassium channel, but also play a crucial role in enabling microglia to resist depolarizations produced by the danger signal ATP thus regulating calcium influx through P2X4 receptors. We here review the role of KV1.3 in microglial signaling and show that, similarly to their role in T cells, KV1.3 channels also regulated store-operated calcium influx in microglia.
Introduction
The voltage-gated potassium channel KV1.3 was originally described in T cells [Citation1] and subsequently pursued as a target for treating T-cell mediated autoimmune diseases such as multiple sclerosis, rheumatoid arthritis and psoriasis [Citation2–4]. More recently KV1.3 has emerged as an attractive pharmacological target for reducing so-called “neuroinflammation” by modulating microglia activation. In both mouse and rat models of ischemic stroke KV1.3 blockers have been shown by us and others to reduce infarct area and improve neurological deficit [Citation5,Citation6]. In transgenic mouse models of Alzheimer’s disease KV1.3 blockade decreased cerebral amyloid load, enhanced hippocampal neuronal plasticity, and improved behavioral deficits [Citation7]. Similarly, in multiple animal models of Parkinson’s disease administration of the small molecule KV1.3 blocker PAP-1 inhibited degeneration of dopaminergic neurons and improved behavioral outcomes [Citation8]. KV1.3 inhibition or genetic deletion has further been shown to ameliorate white matter pathology after traumatic brain injury [Citation9], postoperative cognitive decline [Citation10], and radiation induced brain injury [Citation11]. In all these animal models of neurological disease increased KV1.3 expression was observed on pathology-associated microglia and treatment with KV1.3 blockers resulted in reduced microglia activation and reduced inflammatory cytokine levels in the brains of the animals.
The disease-associated increased KV1.3 expression seems to translate to humans since strong KV1.3 expression has also been found in postmortem human brains on microglia in infarcted areas following ischemic stroke [Citation5,Citation12], on microglia surrounding amyloid plaques in Alzheimer’s disease [Citation7,Citation13], and on microglia in the prefrontal cortex and substantia nigra in Parkinson’s disease [Citation8]. Taken together, these encouraging findings in humans and animal models strongly suggest microglial KV1.3 channels as a therapeutic target for reducing detrimental inflammatory responses in the brain. But how do KV1.3 blockers work? How does blocking a voltage-gated potassium channel in microglia, and presumably also brain infiltrating monocyte derived macrophages reduce inflammatory cytokine production and inflammation mediated tissue damage?
Effects of KV1.3 blockers on microglia function
Cultured neonatal mouse and rat microglia, as well as acutely isolated microglia from mouse brain or tissue printed microglia from rat brain have been shown to express KV1.3 currents by whole-cell patch-clamp that increase substantially following in vitro stimulation or in vivo intracerebroventricular injection with the TLR-4 ligand lipopolysaccharide (LPS) [Citation14–18]. Stimulation with the gram-negative cell wall component LPS is widely used to induce a “classically” activated “M1-like” state of microglia simulating general inflammation like would be associated with a bacterial infection [Citation19,Citation20]. In the field of neuroimmunology LPS stimulated microglia should ideally be called M(LPS) microglia based on the stimulus used to induce polarization to avoid using the M1/M2 terminology, which is regarded as oversimplified [Citation21], but nevertheless widely used because of its therapeutic attractiveness. Following stimulation with LPS increased KV1.3 expression can be observed at the mRNA level and at the protein level by Western blotting, immunohistochemistry, and as increased current density measured by whole-cell patch-clamp [Citation15–18]. A similar, but slightly less intense increase in Kv1.3 expression occurs when cultured neonatal microglia are stimulated with oligomeric amyloid-β (AβO) or α-synuclein [Citation7,Citation8], two proteins that are misfolded and aggregated in Alzheimer’s or Parkinson’s disease, and which are often used to create an in vitro model of microglia responses associated with the respective disease.
In these types of in vitro systems KV1.3 inhibition or genetic deletion reduces the expression and production of multiple inflammatory mediators measured at 24- or 48-hours following microglia activation. About 20 years ago the laboratory of Lyanne Schlichter reported that KV1.3 inhibitors suppress phorbol ester stimulated respiratory burst [Citation22] and NO production in cultured neonatal rat microglia, thus reducing microglia mediated neuronal killing in a transwell culture system [Citation16]. In a more recent study from our own laboratory [Citation17], which compared the effects of KV1.3 and KCa3.1 blockers with the widely used microglia inhibitor minocycline on differentially polarized neonatal mouse microglia, we observed that Kv1.3 inhibition reduced expression and production of the pro-inflammatory cytokines IL-1β and TNF-α at 24 and 28 hours and prevented upregulation of the inflammation associated enzymes cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS). The later effect was probably responsible for the reduced NO production observed in microglia treated with KV1.3 inhibitors at 24 and 48 hours after LPS stimulation. Similar to the earlier work, we also observed that KV1.3 inhibitors prevent microglia mediated neuronal killing in organotypic hippocampal slice cultures 3 days after microglia were activated either through 1-hour of hypoxia/aglycemia to simulate ischemic stroke [Citation5] or by treatment with oligomeric amyloid-β to simulate microglia activation in AD [Citation7]. In addition to reducing AβO induced microglia activation, a phenomenon which can be clearly shown by staining microglia for CD11b or Iba-1 and evaluating fluorescence intensity, the KV1.3 inhibitor PAP-1 has also been found to prevent AβO-induced, microglia-mediated impairment of long-term potentiation (LPT) in hippocampal slices and to reduce AβO-induced production of IL-1β, TNF-α and NO in cultured mouse microglia [Citation7], very similar to what had been found for KV1.3 inhibitors with LPS stimulated microglia [Citation17,Citation18]. In experiments where cultured neonatal mouse microglia from wild-type and KV1.3−/- mice were stimulated with aggregate α-synuclein (αSynAgg), which activates microglia through binding to the pattern recognition receptors TLR2 and CD36 [Citation23], both pharmacological KV1.3 inhibition with PAP-1 and genetic deletion of the channel resulted in reduced IL-1β, IL-6, IL-12 and TNF-α production and prevented NLRP3 inflammasome protein upregulation at 24 hours after microglia stimulation [Citation8]. Conversely, overexpression of KV1.3 in a microglial cell line increased αSynAgg-induced expression or secretion of IL-1β, TNF-α, IL-6 and IL-12 at 24 hours after stimulation [Citation8]
Various studies by other groups confirmed that KV1.3 blockers reduce pro-inflammatory microglia functions when using similar stimuli or stimuli associated with other inflammatory pathologies. For example, in cultured rat microglia activated with the HIV Tat protein or with glycoprotein 120 (gp120) to simulate HIV-1-associated neurocognitive disorder, KV1.3 inhibition or knockdown with siRNA was found to reduce IL-1β, TNF-α and NO production and suppressed neurotoxicity [Citation24,Citation25]. Using a model of ischemic stroke and oxygen/glucose deprivation of cultured microglia Ma et al. reported that KV1.3 inhibition shifted the microglial phenotype from a pro-inflammatory M1-like to an M2-like response and reduced NLRP3 inflammasome activation [Citation5,Citation6], similar to the above described studies in models of Parkinson’s disease [Citation8].
Using freshly isolated CD11b+ cells, which consist of both CNS infiltrating macrophages and brain resident microglia from the brains of mice that had been systemically treated with LPS in the presence and absence of the KV1.3 blocking peptide ShK-223, Rangaraju et al. confirmed that KV1.3 inhibition reduces expression of inflammatory cytokines like IL-1β but showed that KV1.3 inhibition also reduced expression of TAP1 and prevented upregulation of EHD1 in CD11b+CD45low microglia as measured by flow cytometry [Citation26]. Both TAP1 and EDH1 are proteins that are involved in MHC class-I assembly and trafficking to the cell surface. The authors therefore next investigated the effect of ShK-223 treatment on MHC class-1 expression on acutely isolated microglia, and, based on reduced MHC expression, hypothesized that LPS-induced increases in the antigen presenting capacity of microglia are KV1.3-dependent processes, suggesting that KV1.3 inhibition could reduce antigen presentation to CD8+ cytotoxic T cells. In a subsequent weighted co-expression network analysis applied to microglial transcriptomic datasets from neuroinflammatory and neurodegenerative disease mouse models the same group identified KV1.3 as part of a pro-inflammatory gene signature that also included Tlr2, Ptgs2, Il12b, Il1b, CD44, NFκB, Stat1, and RelA [Citation27]. KV1.3 blockade with ShK-223 could shift this pro-inflammatory signature to one with increased expression of anti-inflammatory genes in disease associated microglia. What is interesting in this context, is that Kv1.3 inhibitors have been reported in other studies to further increase the migration of cultured microglia stimulated with the anti-inflammatory cytokines IL-4 and IL-10 [Citation28] and to not affect microglial phagocytosis of microbeads [Citation26] or to even increase phagocytosis of amyloid-β [Citation7].
Taken together, there is thus ample evidence that microglia express KV1.3 and that KV1.3 blockers suppress pro-inflammatory, neurotoxic microglia functions and might even shift microglia toward a more neuroprotective phenotype. We here mostly concentrated on summarizing data obtained with cultured microglial or slice cultures because in these reductionist systems there is no doubt that knockout, knockdown or pharmacological blockade exerts its effect directly through inhibition of microglial KV1.3 channels and not indirectly through affecting KV1.3 channels in T cells, B cells, dendritic cells or monocytes, which all express KV1.3 [Citation29], and could thus contribute to the in vivo effects of KV1.3 blockers or to findings made with KV1.3−/- mice. However, we would like to point out here that in addition to numerous studies showing KV1.3 expression on microglia in rodents and humans by immunohistochemistry, the Kettenmann laboratory and our own group have repeatedly shown that increased KV1.3 expression on activated microglia is not just a tissue culture phenomenon and that Kv1.3 currents can be recorded from microglia in acute slices [Citation30,Citation31] and from acutely isolated microglia from the brains of 5xFAD mice [Citation7] or from the infarcted area after middle cerebral artery occlusion [Citation5,Citation12], a model for ischemic stroke.
Signaling pathways affected by KV1.3 inhibition or modulating Kv1.3 expression
Studies probing into signaling pathways () that are affected by KV1.3 blockers to produce these beneficial reductions in pro-inflammatory microglia functions 24 or 48 hours later have rendered somewhat conflicting results, suggesting that the effects of KV1.3 blockers are dependent on the stimulus and maybe the species. While Fordyce et al. reported that KV1.3 blockers do not affect p38MAPK phosphorylation in LPS treated neonatal rat microglia [Citation16], several other studies using mouse microglia have found that KV1.3 blockers strongly reduce p38MAPK phosphorylation as well as activation of NF-κB following stimulation with LPS, AβO, αSynAgg or HIV-1 glycoprotein 120 [Citation7,Citation8,Citation24]. In the microglial cell line BV2 KV1.3 has also been shown to regulate S727 phosphorylation of STAT1 [Citation26]. Interestingly, whether KV1.3 blockers inhibit NFAT translocation, one of the best studied effects of KV1.3 blockers in T cells [Citation2,Citation32,Citation33] has not been investigated in microglia so far.
Figure 1. Cartoon showing the involvement of KV1.3 and a selected number of other ion channels and receptors in microglia signaling. Abbreviations: COX, cyclooxygenase; DAG, diacylglycerol; Fyn, Src-family of kinase; IL, interleukin; iNOS, inducible nitric oxide synthase; KV, voltage-gated K+ channel; Kir, inward-rectifier K+ channel; KCa, calcium-activated K+ channel; IP3, ionositol-triphosphate; NF-κB, Nuclear factor kappa B; NLRP, nod-like receptor protein; Orai1, calcium release-activated calcium channel protein 1; P2YR, metabotropic purinergic receptor; P2X, ionotropic purinergic receptor; p38-MAPK, p38 mitogen-activated protein kinase; SERCA, sarco/endoplasmic reticulum Ca-ATPase; STIM, stromal interaction molecule; TLR, toll-like receptor; TNFα, tumor necrosis factor alpha; TRP, transient receptor potential channel
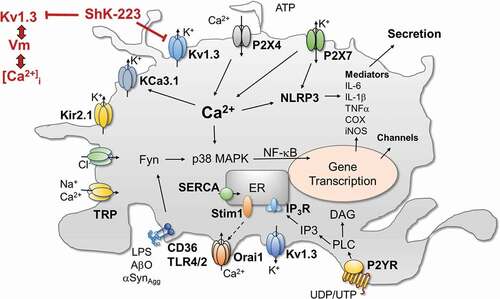
The increases in KV1.3 expression following stimulation of microglia with LPS, AβO or αSynAgg seem to involve many of the same signaling cascades that are affected by KV1.3 inhibition, and KV1.3 blockers typically prevent increases in Kv1.3 expression. The KV1.3 promoter contains multiple NF-κB and SP1 binding sites and inhibitors of NF-κB or p38MAPK independently reduce αSynAgg stimulated Kv1.3 up-regulation in microglia [Citation8], demonstrating that both the p38MAPK and the classical NF-κB pathways mediate the transcriptional regulation of KV1.3 (). The Src family kinase Fyn works upstream of the NF-κB transcription factor by regulating the nuclear translocation of its component p65 [Citation34] and by phosphorylating the PKC isoform PKCδ. Sarkar et al. reported that Fyn knockout reduces both LPS and αSynAgg induced increases in KV1.3 current density in cultured microglia, an effect that can be mimicked by treatment of microglia with the Fyn inhibitor saracatinib or by deficiency for PKCδ, suggesting that the Fyn/PKCδ kinase signaling cascade is involved in mediating KV1.3 upregulation in response to inflammatory stimuli [Citation8]. As demonstrated in a Duolink proximity ligation assay, Fyn is directly interacting with Kv1.3 and is phosphorylating the channel at Y135 to increase its activity.
Kv1.3 regulates membrane potential and resists depolarization
Despite the above described well documented ameliorative effects of KV1.3 blockers on neuroinflammation in vivo and on microglial signaling cascades and inflammatory mediator expression and secretion in vitro, the fundamental molecular mechanisms of how KV1.3 blockers affect microglial functions was long unclear. Many investigators, our own laboratory included, simply assumed that KV1.3 was serving the same function in microglia that it was fulfilling in T cells, namely regulating membrane potential and calcium influx [Citation29,Citation32,Citation33]. In T cells, KV1.3 inhibition leads to membrane depolarization and reduces calcium influx through calcium release-activated calcium channels (CRAC) following T cell receptor stimulation [Citation33,Citation35,Citation36]. Calcium influx via CRAC channels is critically required for dephosphorylation and subsequent nuclear translocation of the calcium/calmodulin-dependent transcription factor NFAT [Citation29], which triggers transcription of the cytokine IL-2 [Citation29,Citation33]. IL-2 is released from activated T cells upon synthesis and stimulates T cell clonal expansion in an autocrine and a paracrine manner. By inhibiting the counterbalancing potassium efflux that is necessary to sustain calcium influx through CRAC channels KV1.3 blockers disrupt T cells activation, prevent NFAT dephosphorylation and nuclear translocation [Citation32] leading to reduced T cell proliferation and reduced inflammatory cytokine production and secretion at 48 hours [Citation5,Citation33,Citation35,Citation36]. However, it had never been tested in microglia, which unlike T cells also express the inward rectifier Kir2.1 [Citation17,Citation37,Citation38] and the two-pore potassium channel THIK-1 (K2P13.1) [Citation39], whether KV1.3 blockers would have the same molecular effects or if the role of KV1.3 was taken over by these other potassium channels. In a recent study our group accordingly addressed this question by first reexamining the fundamental significance of KV1.3 in maintaining resting membrane potential and resisting depolarization [Citation40]. As expected, overexpression of KV1.3 in CHO cells lowered the typically very depolarized resting membrane potential of these cells from −20 mV to −50 mV, while KV1.3 inhibition with ShK-223 depolarized microglia, which inherently maintain a more hyperpolarized resting membrane potential, which in our hands averaged around −90 mV in undifferentiated microglia predominantly expressing Kir2.1 and around −65 mV in LPS stimulated microglia expressing higher KV1.3 and lower Kir2.1 current densities [Citation40].
While KV1.3 thus certainly contributes to regulating resting membrane potential in microglia (), we believe, its more important role, and the reason for why Kv1.3 expression increases in activated microglia, is the channel’s ability to offset extreme and prolonged membrane depolarization that potentially can negatively impact microglial health and physiology as revealed by current injection experiments. While current injections of 25 pA are not able to depolarize KV1.3 expressing microglia beyond −30 mV in the absence of blockers, microglia pretreated with KV1.3 blockers depolarize to potentials of +50 mV in response to the same 25 pA current injections [Citation40], suggesting that KV1.3 is able to “clamp” membrane potential close to the channel’s activation threshold potential to prevent excessive membrane depolarizations (). The situation in microglial physiology where this capacity is important is when microglia are exposed to extracellular ATP, a danger signal that can reach concentrations in the high micromolar range due to release of cytosolic ATP from damaged neurons or ATP-containing synaptic vesicles [Citation41], which activate P2X receptor channels leading to intense depolarizations [Citation42]. In our own hands, current clamp experiments performed on undifferentiated and LPS stimulated microglia showed that acute exposure to 100 μM ATP, which out of the P2X receptors expressed in microglia primarily activates P2X4, lead to more pronounced depolarizations in undifferentiated microglia, despite their more hyperpolarized resting membrane potential than in KV1.3high LPS-stimulated microglia, which did not depolarize above −40 mV. KV1.3 blockers impaired this ability to resist ATP-induced depolarizations and microglia depolarized in their presence [Citation40].
Figure 2. KV1.3 regulates membrane potential and ATP-induced P2X4 receptor-mediated calcium entry. KV1.3 enables microglia to resist depolarizing current injections in whole-cell patch-clamp experiments and to sustain larger ATP induced calcium signals in calcium imaging experiments. Both effects were inhibited or reduced by the KV1.3 blocking peptide ShK-223. For details see [Citation40]
![Figure 2. KV1.3 regulates membrane potential and ATP-induced P2X4 receptor-mediated calcium entry. KV1.3 enables microglia to resist depolarizing current injections in whole-cell patch-clamp experiments and to sustain larger ATP induced calcium signals in calcium imaging experiments. Both effects were inhibited or reduced by the KV1.3 blocking peptide ShK-223. For details see [Citation40]](/cms/asset/3dd2ba01-2ea9-4616-8e6f-9a0c85cc7c6a/kchl_a_1853943_f0002_oc.jpg)
KV1.3 modulates P2X4 receptor mediated calcium influx in microglia
Calcium imaging experiments revealed that this ability of KV1.3 channels to resist ATP induced P2X4-mediated depolarizations enhances Ca2+ entry into microglia through P2X4 receptors stimulated with 100 μM of ATP [Citation40]. For example, we observed that the KV1.3 blockers ShK-223 and PAP-1 reduced ATP-induced Ca2+ influx through P2X4 receptors in LPS-differentiated microglia which express high KV1.3 current densities, much more strongly than in unstimulated microglia despite the fact that LPS stimulation downregulates P2X4 receptor expression in cultured microglia. In contrast, KV1.3 blockade had no effect on ATP-induced calcium transients in IL-4 stimulated microglia, which express very low levels of KV1.3. Interestingly, while LPS also increases KV1.3 and reduces P2X4 expression on microglia in vivo, both KV1.3 and P2X4 expression increase in parallel in the setting of ischemic stroke as shown by our laboratory on acutely isolated microglia from the infarcted brain hemisphere of mice subjected to ischemic stroke and by other groups on rodent and human microglia by immunohistochemistry or qPCR [Citation5,Citation12,Citation43–46]. This suggests that following ischemic stroke KV1.3 might be regulating the more complex purinergic microglial calcium signaling initiated by the danger signal ATP more strongly than in vitro. ATP and other neuronal debris in the infarcted area of the brain activate microglia, which then continuously produce and release more inflammatory mediators within 24 to 72 hours and thus damage and kill more neurons in the penumbra leading to an inflammatory expansion of the infarct. The increased KV1.3 expression that is observed on microglia in the infarct border within 2–5 days [Citation5] presumably enables activated microglia to sustain stronger calcium signals through the increased number of P2X4 receptors and thus to produce more inflammatory mediators, damaging more neurons and activating more microglia. This hypothesis would be in line with the finding that the KV1.3 blocker PAP-1 reduced microglia activation and brain levels of IL‐1β and IFN‐γ, leading to roughly 40% smaller infarct areas and improved neurological deficit scores in both mouse and rat models of ischemic stroke [Citation5]. By depolarizing microglia Kv1.3 blockers reduce calcium influx through P2X4 receptors triggered by ATP released from damaged neurons and thus dampen further calcium-dependent activation of the NLRP3 inflammasome, NF-κB and other transcription factors leading to reduced inflammatory mediator production () and reduced pathology.
What other calcium influx pathways in microglia are regulated by KV1.3?
While KV1.3 blockade clearly reduced ATP-induced calcium influx through P2X4 receptors [Citation40] and thus constitutes an important regulator of this calcium influx pathway in microglia (), this clearly is not the only KV1.3 or ATP dependent calcium signaling process in microglia. In addition to P2X receptor channels microglia also express metabotropic P2Y purinergic receptors which following activation by ATP or UTP can induce calcium release from internal stores [Citation47] and activate store-operated Ca2+ entry (SOCE) following depletion of the relatively small intracellular calcium stores in microglia [Citation38]. It therefore is certainly possible that additional SOCE contributed to the ATP induced Ca2+ signals we had imaged and that some of the KV1.3 blocker effects were mediated by additionally reducing SOCE. This is a very likely hypothesis, since the “classical” molecular mechanism for KV1.3 blockers in T cells is reduction of SOCE through CRAC channels following T cell receptor activation and subsequently reduced transcription factor activation and cytokine production and release [Citation29,Citation33,Citation36,Citation48]. Rat and mouse microglia have been repeatedly reported to exhibit currents with biophysical properties typical for CRAC currents [Citation49,Citation50] and to express the molecular components of CRAC channels, Orai and STIM [Citation51,Citation52]. There further is evidence that genetic deletion of Orai1, STIM1, and STIM2 reduces CRAC current and/or SOCE and inhibits migration and phagocytosis in cultured mouse microglia [Citation52], while the small molecule CRAC channel blocker CM-EX-137 has recently been reported to decrease microglia activation and lesion size in a mouse model of traumatic brain injury [Citation53].
Blockade of KV1.3 channels attenuates store-operated calcium entry in LPS stimulated microglia
As a follow-up experiment to our previous paper on the role of KV1.3 in regulating P2X4 mediated calcium influx, we here specifically asked the question if the KV1.3 blocker ShK-223 would also reduce SOCE in LPS differentiated microglia, the same stimulation condition, where we had observed the highest KV1.3 current densities and the largest effects of KV1.3 inhibition on ATP induced calcium signaling [Citation40]. Using the ratiometric calcium indicator Fura-2 to track changes in the free intracellular calcium concentration ([Ca2+]i) we found that LPS differentiated microglia had a resting cytosolic calcium concentration [Ca2+]i that varied between 60 nM and 370 nM, averaging 150 ± 8.8 nM (n = 50; ), which is consistent with a previous report [Citation54]. To elicit SOCE, intracellular calcium stores were first depleted with thapsigargin (Tg), a blocker of the endoplasmic reticulum Ca2+-ATPase, applied in a nominally Ca2+-free bath solution. Removal of extracellular calcium produced a roughly 25 nM decrease in [Ca2+]i () indicating the presence of a calcium influx at rest. Subsequent application of Tg (1 μM) elicited small amplitude (> 25 nM) asynchronous elevations in [Ca2+]i due to passive calcium mobilization from stores. Re-addition of calcium into the bath solution elicited a rapid elevation in [Ca2+]i above the resting levels caused by SOCE. The magnitude of the SOCE-induced [Ca2+]i transients (“peak SOCE”) measured as the difference between [Ca2+]i level in the Ca2+-free bath solution and maximal [Ca2+]i elevation following calcium re-addition varied significantly among individual cells with peak SOCE distribution displaying a maximum at 120 nM and a tail up to 1800 nM (). This heterogeneity in the strength of SOCE is likely caused by variations in resting membrane potential, which provides the driving force for calcium entry during SOCE. The resting membrane potential of LPS-differentiated microglia has been found to vary between −90 mV and −40 mV in individual cells [Citation40,Citation55] and thus could account for the observed differences in SOCE.
Figure 3. The KV1.3 blocker ShK-223 reduces SOCE in LPS differentiated neonatal microglia. Primary neonatal microglia cultures were prepared from 3 independent litters and stimulated with LPS as described in our publication investigating the effect of KV1.3 blockers on P2X4 mediated Ca2+ influx in microglia [Citation40]. Loading microglia with Fura-2 was performed as previously described [Citation56]. Fura-2 fluorescence signals evoked by 340- and 380-nm excitation (F340 and F380, respectively) were acquired every 12 s. Data acquisition was performed using MetaFluor version 7.0 software (Molecular Devices). Estimated [Ca2+]i was calculated from background-subtracted F340/F380 ratio values and Fura-2 calibration as previously described [Citation57]. Differences between data sets were analyzed using one-way analysis of variance (one-way ANOVA) with Bonferroni correction (Origin 7 software)
![Figure 3. The KV1.3 blocker ShK-223 reduces SOCE in LPS differentiated neonatal microglia. Primary neonatal microglia cultures were prepared from 3 independent litters and stimulated with LPS as described in our publication investigating the effect of KV1.3 blockers on P2X4 mediated Ca2+ influx in microglia [Citation40]. Loading microglia with Fura-2 was performed as previously described [Citation56]. Fura-2 fluorescence signals evoked by 340- and 380-nm excitation (F340 and F380, respectively) were acquired every 12 s. Data acquisition was performed using MetaFluor version 7.0 software (Molecular Devices). Estimated [Ca2+]i was calculated from background-subtracted F340/F380 ratio values and Fura-2 calibration as previously described [Citation57]. Differences between data sets were analyzed using one-way analysis of variance (one-way ANOVA) with Bonferroni correction (Origin 7 software)](/cms/asset/df7a4ccc-01a3-429e-b693-8a73fd29cd8c/kchl_a_1853943_f0003_b.gif)
A previous study reported that elevation in resting [Ca2+]i was associated with a reduced ability of external stimuli such as the P2Y receptor agonist UTP or complement factor 5a to elicit [Ca2+]i transients in LPS-activated microglia [Citation54]. Because in our study both resting [Ca2+]i levels and peak SOCE values varied among individual cells, we explored whether variation in resting [Ca2+]i may account for heterogeneity in peak SOCE values. Plotting peak SOCE values versus resting [Ca2+]i, revealed a positive correlation between peak SOCE and resting [Ca2+]i (). When we split the data into two groups with a cutoff value of 600 nM for peak SOCE, the relationships between resting [Ca2+]i and peak SOCE in each group were stronger than in the whole population, indicating the presence of at least two cell populations, which differ in both magnitude of SOCE and strength of correlation between resting [Ca2+]i and peak SOCE. Thus, within the population of LPS-treated cells, an elevation in resting [Ca2+]i is not causally linked to the reduced ability of activated microglia to respond to external stimuli with a [Ca 2+]i transient, as was previously suggested by Hoffman et al. Our data suggest that a common influence, such as the level of membrane potential, which is controlled by a variety of channels, including KV1.3, Kir2.1 and THIK-1, may be responsible for the positive correlation between resting [Ca2+]i and peak SOCE observed in our study. For example, overexpression or functional upregulation of hyperpolarizing channels could hyperpolarize the membrane and increase the driving force for both calcium entry via constitutively active calcium permeable channels such as metabolically activated transient receptor potential (TRP) channels and store-operated channels activated following store depletion such as CRAC channels.
Pre-incubation of cells with the KV1.3 inhibitor ShK-223 did not significantly affect resting [Ca2+]i (), but eliminated peak SOCE responses larger than 300 nM and shifted the maximum of the distribution of peak SOCE values from 130 nM (, control) to 70 nM (, ShK-223). The difference between the values of peak SOCE in control cells and cells preincubated with ShK-223 was significant (). The relationship between resting [Ca2+]i and peak SOCE still displayed a mild positive correlation (). Taken together, our data indicate that similarly to T lymphocytes, blockade of KV1.3 channels significantly reduces SOCE in LPS differentiated microglia. The extent of the inhibition varied among cells with a greater effect on cells expressing large magnitude (> 300 nM) SOCE. The specific characteristics of the cells with high and low peak SOCE remain to be determined.
Under physiological conditions, SOCE can be triggered subsequently to store depletion following activation of G-protein coupled P2Y receptors and contributes to the combined [Ca2+]i elevation during activation of P2Y and P2X receptors elicited by common purinergic agonists, such as ATP. The magnitude of the SOCE varies significantly among LPS differentiated cells, which may be a function of the unique combination of differential expression of store-operated channels and channels controlling membrane potential, such as KV1.3. Reduction in peak SOCE by ShK-223 indicates that KV1.3 channels are potent regulators of SOCE and uncover an additional mechanism by which KV1.3 blockers attenuate calcium signaling and subsequent functional responses (e.g. cytokine production) following calcium dependent transcription factor activation in microglia.
Conclusions
While it is certainly challenging to sort out the contributions effects on cells of the peripheral immune system, particularly on memory T cells and monocyte derived macrophages, make to the beneficial effects of KV1.3 blockers on inflammation in the CNS, it is clear that KV1.3 plays important roles in regulating membrane potential, preventing depolarization and controlling calcium signaling events that subsequently lead to microglia activation and inflammatory mediator production. We would like to suggest that from a therapeutic perspective it is probably desirable to inhibit KV1.3 in multiple immune system cell types. However, it would be intellectually very satisfying and helpful for therapy development to clarify what the major cellular target of KV1.3 inhibitors is and whether therapeutics need to be brain penetrant in order to exert optimal ameliorating effects in CNS diseases. These questions should ideally be addressed both pharmacologically by testing brain penetrant small molecules side by side with peripheral restricted petidic KV1.3 inhibitors or anti-KV1.3 antibodies [Citation58,Citation59] in animal models of Alzheimer’s and Parkinson’s disease. It would further be highly informative to compare the effects of conditional KV1.3 deletion in microglia, monocyte derived macrophages and different T cell subsets to the effects of drug treatment to gain a more detailed understanding of the role of KV1.3 in neuroinflammation and the cellular target of KV1.3 inhibitors. However, we believe that at the cellular level the fundamental role of KV1.3 will always be what was first described in T cells, namely regulation of membrane potential and calcium influx and subsequent activation of calcium dependent transcription factors. What sometimes does not get stressed enough in this respect, is probably that KV1.3’s biophysical properties like its half-activation voltage and its relatively slow inactivation [Citation60,Citation61], perfectly “qualify” the channel to resist membrane depolarizations and thus enable KV1.3 to not only regulate SOCE, as was described 30 years ago in T cells, but also P2X receptor mediated calcium influx.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- DeCoursey TE, Chandy KG, Gupta S, et al. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307(5950):465–468.
- Beeton C, Wulff H, Standifer NE, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci USA. 2006;103(46):17414–17419.
- Tarcha EJ, Chi V, Munoz-Elias EJ, et al. Durable pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease. J Pharmacol Exp Ther. 2012;342(3):642–653.
- Chandy KG, Norton RS. Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Curr Opin Chem Biol. 2017;38:97–107.
- Chen YJ, Nguyen HM, Maezawa I, et al. Inhibition of the potassium channel Kv1.3 reduces infarction and inflammation in ischemic stroke. Ann Clin Transl Neurol. 2018;5(2):147–161.
- Ma DC, Zhang NN, Zhang YN, et al. Kv1.3 channel blockade alleviates cerebral ischemia/reperfusion injury by reshaping M1/M2 phenotypes and compromising the activation of NLRP3 inflammasome in microglia. Exp Neurol. 2020;332:113399.
- Maezawa I, Nguyen HM, Di Lucente J, et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: preclinical proof of concept. Brain. 2018;141(2):596–612.
- Sarkar S, Nguyen HM, Malovic E, et al. Kv1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J Clin Invest. 2020;130(8):4195–4212.
- Reeves TM, Trimmer PA, Colley BS, et al. Targeting Kv1.3 channels to reduce white matter pathology after traumatic brain injury. Exp Neurol. 2016;283(PtA):188–203.
- Lai IK, Valdearcos M, Morioka K, et al. Blocking Kv1.3 potassium channels prevents postoperative neuroinflammation and cognitive decline without impairing wound healing in mice. Br J Anaesth. 2020;125(3):298–307.
- Peng Y, Lu K, Li Z, et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol. 2014;16(4):528–539.
- Chen YJ, Nguyen HM, Maezawa I, et al. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2016;36(12):2146–2161.
- Rangaraju S, Gearing M, Jin LW, et al. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer’s disease. J Alzheimers Dis. 2015;44(3):797–808.
- Norenberg W, Gebicke-Haerter PJ, Illes P. Inflammatory stimuli induce a new K+ outward current in cultured rat microglia. Neurosci Lett. 1992;147(2):171–174.
- Kotecha SA, Schlichter LC. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci. 1999;19(24):10680–10693.
- Fordyce CB, Jagasia R, Zhu X, et al. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25(31):7139–7149.
- Nguyen HM, Grossinger EM, Horiuchi M, et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia. 2017;65(1):106–121.
- Di Lucente J, Nguyen HM, Wulff H, et al. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia. 2018;66(9):1881–1895.
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201.
- Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. 2015;131:65–86.
- Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20.
- Khanna R, Roy L, Zhu X, et al. K+ channels and the microglial respiratory burst. Am J Physiol Cell Physiol. 2001;280(4):C796–806.
- Kim C, Ho DH, Suk JE, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.
- Liu J, Xu C, Chen L, et al. Involvement of Kv1.3 and p38 MAPK signaling in HIV-1 glycoprotein 120-induced microglia neurotoxicity. Cell Death Dis. 2012;3:e254.
- Liu J, Xu P, Collins C, et al. HIV-1 Tat protein increases microglial outward K+ current and resultant neurotoxic activity. PLoS One. 2013;8(5):e64904.
- Rangaraju S, Raza SA, Pennati A, et al. A systems pharmacology-based approach to identify novel Kv1.3 channel-dependent mechanisms in microglial activation. J Neuroinflammation. 2017;14(1):128.
- Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):24.
- Lam D, Lively S, Schlichter LC. Responses of rat and mouse primary microglia to pro- and anti-inflammatory stimuli: molecular profiles, K+ channels and migration. J Neuroinflammation. 2017;14(1):166.
- Feske S, Wulff H, Skolnik EY. Ion Channels in Innate and Adaptive Immunity. Annu Rev Immunol. 2015;33:291–353.
- Lyons SA, Pastor A, Ohlemeyer C, et al. Distinct physiologic properties of microglia and blood-borne cells in rat brain slices after permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2000;20(11):1537–1549.
- Boucsein C, Zacharias R, Farber K, et al. Purinergic receptors on microglial cells: functional expression in acute brain slices and modulation of microglial activation in vitro. Eur J Neurosci. 2003;17(11):2267–2276.
- Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci U S A. 1994;91(7):2873–2877.
- Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. 2009;231(1):59–87.
- Panicker N, Saminathan H, Jin H, et al. Fyn kinase regulates microglial neuroinflammatory responses in cell culture and animal models of Parkinson’s disease. J Neurosci. 2015;35(27):10058–10077.
- Lewis RS, Cahalan MD. Potassium and calcium channels in lymphocytes. Annu Rev Immunol. 1995;13:623–653.
- Leonard RJ, Garcia ML, Slaughter RS, et al. Selective blockers of voltage-gated K+ channels depolarize human T lymphocytes: mechanism of the antiproliferative effect of charybdotoxin. Proc Natl Acad Sci U S A. 1992;89(21):10094–10098.
- Eder C. Ion channels in microglia (brain macrophages). Am J Physiol. 1998;275(2):C327–42.
- Kettenmann H, Hanisch UK, Noda M, et al. Physiology of microglia. Physiol Rev. 2011;91(2):461–553.
- Madry C, Kyrargyri V, Arancibia-Carcamo IL, et al. Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK-1. Neuron. 2018;97(2):299–312 e6.
- Nguyen HM, Di Lucente J, Chen YJ, et al. Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia. 2020;68(11):2377–2394.
- Pankratov Y, Lalo U, Verkhratsky A, et al. Vesicular release of ATP at central synapses. Pflugers Arch. 2006;452(5):589–597.
- Visentin S, Renzi M, Frank C, et al. Two different ionotropic receptors are activated by ATP in rat microglia. J Physiol. 1999;519(Pt 3):723–736.
- Wixey JA, Reinebrant HE, Carty ML, et al. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J Neuroimmunol. 2009;212(1–2):35–43.
- Cavaliere F, Florenzano F, Amadio S, et al. Up-regulation of P2X2, P2X4 receptor and ischemic cell death: prevention by P2 antagonists. Neuroscience. 2003;120(1):85–98.
- Li F, Wang L, Li JW, et al. Hypoxia induced amoeboid microglial cell activation in postnatal rat brain is mediated by ATP receptor P2X4. BMC Neurosci. 2011;12:111.
- Gao T, Raza SA, Ramesha S, et al. Temporal profiling of Kv1.3 channel expression in brain mononuclear phagocytes following ischemic stroke. J Neuroinflammation. 2019;16(1):116.
- McLarnon JG. Purinergic mediated changes in Ca2+ mobilization and functional responses in microglia: effects of low levels of ATP. J Neurosci Res. 2005;81(3):349–356.
- Verheugen JA, Vijverberg HP, Oortgiesen M, et al. Voltage-gated and Ca2+-activated K+ channels in intact human T lymphocytes. Noninvasive measurements of membrane currents, membrane potential, and intracellular calcium. J Gen Physiol. 1995;105(6):765–794.
- Hahn J, Jung W, Kim N, et al. Characterization and regulation of rat microglial Ca2+ release-activated Ca2+ (CRAC) channel by protein kinases. Glia. 2000;31(2):118–124.
- Beck A, Penner R, Fleig A. Lipopolysaccharide-induced down-regulation of Ca2+ release-activated Ca2+ currents (I CRAC) but not Ca2+-activated TRPM4-like currents (I CAN) in cultured mouse microglial cells. J Physiol. 2008;586(2):427–439.
- Ohana L, Newell EW, Stanley EF, et al. The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels (Austin). 2009;3(2):129–139.
- Michaelis M, Nieswandt B, Stegner D, et al. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia. 2015;63(4):652–663.
- Mizuma A, Kim JY, Kacimi R, et al. Microglial calcium release-activated calcium channel inhibition improves outcome from experimental traumatic brain injury and microglia-induced neuronal death. J Neurotrauma. 2019;36(7):996–1007.
- Hoffmann A, Kann O, Ohlemeyer C, et al. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci. 2003;23(11):4410–4419.
- Chung S, Lee MY, Soh H, et al. Modulation of membrane potential by extracellular pH in activated microglia in rats. Neurosci Lett. 1998;249(2–3):139–142.
- Dadsetan S, Zakharova L, Molinski TF, et al. Store-operated Ca2+ influx causes Ca2+ release from the intracellular Ca2+ channels that is required for T cell activation. J Biol Chem. 2008;283(18):12512–12519.
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450.
- Wang RE, Wang Y, Zhang Y, et al. Rational design of a Kv1.3 channel-blocking antibody as a selective immunosuppressant. Proc Natl Acad Sci USA. 2016;113(41):11501–11506.
- Bednenko J, Harriman R, Marien L, et al. A multiplatform strategy for the discovery of conventional monoclonal antibodies that inhibit the voltage-gated potassium channel Kv1.3. MAbs. 2018;10(4):636–650.
- Grissmer S, Dethlefs B, Wasmuth JJ, et al. Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc Natl Acad Sci U S A. 1990;87(23):9411–9415.
- Gutman GA, Chandy KG, Grissmer S, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508.