
ABSTRACT
The cardiac late sodium current (INa,late) is the small sustained component of the sodium current active during the plateau phase of the action potential. Several studies demonstrated that augmentation of the current can lead to cardiac arrhythmias; therefore, INa,late is considered as a promising antiarrhythmic target. Fundamentally, enlarged INa,late increases Na+ influx into the cell, which, in turn, is converted to elevated intracellular Ca2+ concentration through the Na+/Ca2+ exchanger. The excessive Ca2+ load is known to be proarrhythmic. This review describes the behavior of the voltage-gated Na+ channels generating INa,late in health and disease and aims to discuss the physiology and pathophysiology of Na+ and Ca2+ homeostasis in context with the enhanced INa,late demonstrating also the currently accessible antiarrhythmic choices.
Introduction
The cardiac action potential (AP) is composed of several ion currents. During the initial depolarization, voltage-gated Na+ channels open to further depolarize the membrane. This is followed by a tightly regulated process, in which L-type Ca2+ channels open to let the Ca2+ ions flow into the cell (L-type Ca2+ current, ICa,L), so that the contraction can occur in the process called Ca2+-induced Ca2+ release (CICR) by opening the ryanodine receptors (RyR). Besides the depolarizing inward currents, a number of outwardly driven K+ currents repolarize the membrane. During repolarization, Ca2+ is removed from the cytoplasm; therefore, complete relaxation of the cell occurs while the membrane potential returns to its resting value so that the next AP can be elicited [Citation1–3].
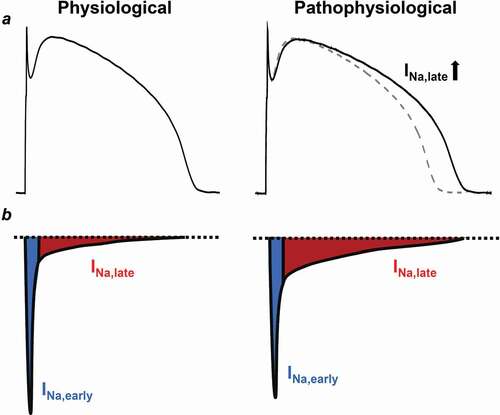
The behavior of Na+ current is not monotonic in time. Once the membrane potential reaches the threshold level for the voltage-gated Na+ channels, a significant Na+ influx depolarizes the membrane and creates the upstroke of the AP. However, this fast, early peak Na+ current (INa,early) is rapidly inactivated causing the fast decay of the INa,early [Citation4,Citation5]. Under certain conditions, Na+ channels might recover from inactivation and reopen during the plateau phase of the AP, bringing a further depolarizing Na+ influx, termed as the late Na+ current (INa,late) () [Citation6]. As INa,early increases the intracellular Na+ concentration [Na+]i at the upstroke of the AP, the Na+/Ca2+ exchange (NCX) switches to its reverse mode and removes Na+ from the cell at the cost of intracellular Ca2+ load. This reverse mode persists for only a very short period of time and NCX works in its forward mode at the rest of the AP, underlying the vast majority of sarcolemmal Ca2+ extrusion [Citation1,Citation7]. INa,late is a minute, but persistent inward current which is much smaller in amplitude than INa,early in healthy myocytes. However, under certain pathophysiological conditions, INa,late can become much larger and might cause Na+ (and Ca2+) overload leading to arrhythmogenesis (). In the present review we aim to discuss the arrhythmogenic role of INa,late in context with the intracellular Na+ and Ca2+ overload.
Figure 1. Comparison of the physiological and the pathological INa,late in ventricular cardiomyocytes. (a) Ventricular action potentials recorded from healthy and diseased hearts. Diseased INa,late is increased causing a longer action potential. Dashed line shows the control action potential. (b) Representative electrophysiological recordings of the INa in normal and diseased myocytes. Blue shows the early, peak component of the INa (INa,early), while red shows the sustained, late component of the current (INa,late)
Voltage-gated sodium channels
Since the original observations by Hodgkin and Huxley on squid giant axon, voltage-gated Na+ channels are known to be regulated by changes in the actual membrane potential [Citation8]. If the membrane voltage is favorable for channel opening, the movement of ions is determined by the electrochemical gradient of the ion. Voltage-gated Na+ channels consist of a large pore-forming pseudotetrameric α subunit, accessory β subunits and scaffolding proteins ()). To date there are 9 different α subunits (Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.5, Nav1.6, Nav1.7, Nav1.8, Nav1.9) encoded by 9 different genes (SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN8A, SCN9A, SCN10A, SCN11A) of which the Nav1.5 is considered to be the dominant cardiac subtype [Citation9,Citation10]. Nav1.5 is relatively insensitive to the Na+ channel blocker neurotoxin, tetrodotoxin (TTX) [Citation9,Citation11–13]. However, other TTX sensitive subtypes – such as Nav1.1, Nav1.2, Nav1.3, Nav1.4 and Nav1.6 – are also reported to be expressed in the heart [Citation14–18]. An α subunit encoded by a specific gene determines not only the channel subtype itself, but also the receptor population of the particular channel. The 6 possible β subunits (β1, β1A, β1B, β2, β3, β4) are encoded by four genes (SCN1B, SCN2B, SCN3B, SCN4B) [Citation19–22]. The α subunit alone is sufficient to form a functional channel; however, the auxiliary β subunits are required for regular channel kinetics and cell surface expression [Citation20]. In fact, as the β subunits modulate the number of the available channels on the cell surface, they play a role in regulating peak current density [Citation20]. Furthermore, β subunits control activation, inactivation, and recovery from inactivation by altering their voltage range [Citation23,Citation24].
Figure 2. Structure of the cardiac Nav1.5 α subunit. (a) Alpha and beta subunits of the cardiac voltage-gated Na+ channel isoform showing the four domains of the alpha subunit (DI-DIV) and the six transmembrane segments (S1-S6) in each domain and the auxiliary beta subunits. Grey zone shows the pore-forming domain. Red shows the inactivation gate between the DIII and DIV (IDIII/IV). (b) Side and (c) top (intracellular) view of the cryo-EM structure of the rat Nav1.5 α subunit displaying the domains in different colors (PDB ID: 6UZ3) and the channel pore (c) generated by PyMol Software.[Citation182]
![Figure 2. Structure of the cardiac Nav1.5 α subunit. (a) Alpha and beta subunits of the cardiac voltage-gated Na+ channel isoform showing the four domains of the alpha subunit (DI-DIV) and the six transmembrane segments (S1-S6) in each domain and the auxiliary beta subunits. Grey zone shows the pore-forming domain. Red shows the inactivation gate between the DIII and DIV (IDIII/IV). (b) Side and (c) top (intracellular) view of the cryo-EM structure of the rat Nav1.5 α subunit displaying the domains in different colors (PDB ID: 6UZ3) and the channel pore (c) generated by PyMol Software.[Citation182]](/cms/asset/659ca5f9-9f67-4e5a-b633-1ee5b888dad4/kchl_a_1854986_f0002_oc.jpg)
Voltage-gated Na+ channel α subunits consist of approximately 2000 amino acid residues, creating four domains (DI-DII-DIII-DIV) ()). Each domain is formed by six transmembrane segments (S1-S6). Segments 1–4 (S1-S4) function as the voltage sensor domain of the channel. This domain senses membrane depolarization leading to channel activation. The S1-S4 connects to the channel’s pore-forming domain S5-S6 via an intracellular linker. This structure encompasses the central aqueous pore domain ()). The selectivity filter is also located in the pore domain, recognizing the charge and radius of the ion.
At membrane potentials negative to the threshold of the Na+ channel the channel’s open probability is low. Upon depolarization, however, the α subunit undergoes a conformational change, the voltage sensor activates, the activation gate – and therefore the Na+ channel – quickly opens, thereby conducting Na+ current and resulting in the upstroke of the cardiac AP. A few milliseconds later the channel inactivates quickly as the inactivation gate closes into the channel’s pore domain yielding a nonconducting state [Citation25]. The homologous domains are connected by intracellular interdomain loops (IDI/II, IDII/III and IDIII/IV). Inactivation gate incorporates the smallest interdomain cytoplasmic loop (IDIII/IV) and functions as a lid that locks the pore during inactivation ()) [Citation25–29]. Normal inactivation is needed to prevent excess depolarization and to ensure timely repolarization. It has also been proposed that the inactivation gate is formed and stabilized as a molecular complex, formed by the IDIII/IV and the C-terminal loop of the α subunit [Citation30]. After depolarization and inactivation, during the repolarization phase, Na+ channels recover from inactivation ready to be activated again [Citation31].
Late sodium current
INa,late is normally a small but persistent current (). It is active during the plateau phase of the cardiac AP; therefore, the current can play a significant role in determining the duration and the shape of the AP ()) [Citation32–34]. A possible explanation of this discrepancy (i.e., a tiny current causing large effects on the AP) is given by the role of net membrane current. During the plateau phase of the AP the impedance of the cell membrane is high [Citation35] and according to Ohm’s law, at this stage, small changes in net membrane current lead to relatively large changes in the membrane potential, and consequently, in AP duration (APD) [Citation36,Citation37].
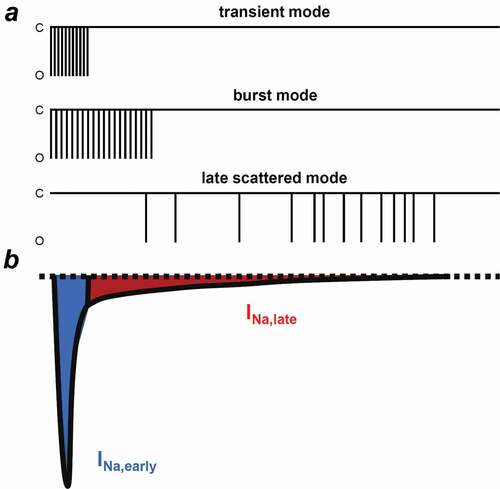
Three different gating modes of Nav1.5 have been described in ventricular cells () [Citation38]. The transient mode is the main gating mode for the INa,early. Burst mode and late scattered mode are responsible for INa,late; however, burst mode openings decline quickly, leaving the late scattered mode to be the main gating for INa,late during the plateau phase. Furthermore, several inactivation processes have been proposed, each governing APD, Na+ channel steady-state inactivation and Na+ flux balance of the cell [Citation39,Citation40]. Fast inactivation takes place only in the first milliseconds and the channel recovers rapidly at negative membrane potentials. This is followed by the intermediate inactivation which recovers slowly compared to the fast inactivation. Slow inactivation from the open state occurs over hundreds of milliseconds and finally, ultraslow inactivation can take seconds.
Figure 3. Different gating modes determining the INa,early and INa,late. (a) Schematic illustrations of the three Nav1.5 gating modes. INa,early is determined by the transient mode. Burst mode and late scattered mode are responsible for INa,late, however, the late scattered mode is the main gating for INa,late. (b) Representative electrophysiological recording of the INa. Blue shows the early, peak component of the INa (INa,early), while red shows the sustained, late component of the current (INa,late). C, closed and O, opened
The window Na+ current is a well-known phenomenon characterizing Na+ channels. Due to an overlap between steady-state activation and inactivation curves (“window of potentials”), a fraction of Na+ channels can recover from inactivation and might reopen. However, considering the voltage range of this window current (approximately – 70 mV), it is far below the physiological plateau potential of the AP, so it is unlikely to play a major role in the INa,late [Citation34,Citation41–43].
There are marked interspecies differences in the profiles of the INa,late. One practical difference is the shape and duration of the AP, resulting in distinct INa,late profiles. Our group has recently demonstrated that INa,late in human and canine ventricular myocytes is markedly disparate from cells isolated from guinea pig hearts [Citation44] or some other mammals including rabbits and pigs [Citation45–47]. The greatest difference was in the time course and profile of the INa,late. In human and canine cells, the amplitude of the INa,late monotonically decreases during the time course of the AP. On the contrary, guinea pig cells show a different current profile, namely, the current amplitude increases during the plateau phase to only decline during the terminal repolarization. In addition, Horváth et al. and Hegyi et al. reported that the density of the INa,late is comparable to the major repolarizing K+ currents in guinea pig and rabbit myocytes [Citation45,Citation47]. Also in rabbits, the atrial density of INa,late was greater than measured in the ventricles [Citation48].
INa,late shows reverse rate-dependent properties, that is, the higher the pacing frequency (i.e. heart rate), the lower the density of the current [Citation37,Citation49,Citation50]. However, the early and late components behave differently. The higher [Na+]i, observed at high frequency, is mainly determined by the early component as INa,late recovers slowly from inactivation at rapid pacing [Citation51]. Additionally, at higher frequencies, APD is usually shortened, allowing less time for activation of INa,late. Therefore the shorter the APD the smaller the INa,late and Na+ influx. In contrast, during bradycardia APD is longer and shows greater beat-to-beat variability [Citation52]. Therefore, bradycardia, associated with enhanced INa,late, may strongly be proarrhythmic [Citation45,Citation53,Citation54]. As there are marked interspecies differences in heart rate and APD, species having long APs (e.g. human, canine, guinea pig) are expected to manifest larger INa,late and Na+ influx than species with fundamentally shorter APs (e.g. rat, mouse).
Late sodium current in disease
In normal, healthy myocytes, the amplitude of INa,late is much smaller, less than 0.1% of the peak INa,early [Citation55,Citation56]. However, as stated before, the current is persistent, lasting for 100–400 ms; therefore, the inward charge carried by INa,late is comparable to INa,early mediated within 1–2 ms [Citation57,Citation58]. Some papers in the literature refer to this as endogenous INa,late and is thought to be without any arrhythmic properties.
On the other hand, the density of INa,late can be increased under many pathophysiological conditions, such as heart failure (HF) [Citation53,Citation59], hypertrophic cardiomyopathy, inherited long QT syndrome 3 (LQTS-3) [Citation16,Citation34,Citation53,Citation60,Citation61], oxidative stress, or atrial fibrillation (AF) with intracellular Ca2+ handling abnormalities [Citation62]. Moreover, even a low heart rate or pharmacological interventions can elevate INa,late [Citation63]. INa,late is also augmented in myocardial ischemia/reperfusion injury [Citation18,Citation62] and in the presence of characteristic components of ischemia (e.g. hypoxia, ischemic metabolites, hydrogen peroxide) as documented in voltage clamp experiments [Citation64–67].
The AP lengthening effect of the augmented INa,late can also be observed in HF [Citation68]. The increased INa,late results in a Na+ overload, which, in turn, leads to elevation of intracellular Ca2+ concentration [Ca2+]i. The concomitant abnormal conduction can cause sudden death in HF patients. Conduction velocity is determined also by the Na+ channel function [Citation69]. In the ventricular conductive system (Purkinje fibers) – in contrast to ventricular myocardium – slow pacing generates a higher, while fast pacing results in a significantly lower INa,late. This transmural inhomogeneity may be a trigger for cardiac arrhythmias [Citation70]. All these mechanisms may lead to complex pathological electrical and mechanical performance, such as contractile dysfunction [Citation71], disturbed myocardial energetics [Citation72] and arrhythmias [Citation73]. Increased INa,late is most arrhythmogenic in those cases, where the repolarization reserve is already compromised, such as during treatment with IKr inhibitors [Citation74], or in the remodeled myocardium.
In the above mentioned diseases several pathways can play a role in the alteration of INa,late. INa,late can be elevated by reactive oxygen species (ROS), H2O2 [Citation60,Citation66,Citation75], acidosis [Citation76,Citation77], hypoxia [Citation78,Citation79], or nitric oxide (NO) [Citation80]. Furthermore, INa,late is also altered by transcriptional regulation [Citation81], N-glycosylation [Citation82,Citation83], phosphorylation on tyrosine residues [Citation84] or arginine methylation [Citation85]. Modulation of channel function can also be achieved by mechanosensitivity [Citation86,Citation87], β-adrenergic stimulation [Citation47], or CaMKII [Citation88–90].
Calcium and sodium homeostasis
In the case of facilitated sarcolemmal Na+ entry to the cytoplasm, [Na+]i is going to increase with a concomitant rise in the [Ca2+]i, which is considered to be arrhythmogenic [Citation3]. Furthermore, high Ca2+ via the Ca2+/calmodulin-dependent protein kinase II (CaMKII) – protein kinase C (PKC) pathway can further increase INa,late thereby initiating a vicious circle, leading to spatial heterogeneity of Ca2+ transients and triggered activities [Citation90–92]. Therefore, a better understanding of the effects of the elevated INa,late on Na+ and Ca2+ homeostasis is critically important.
The Na+ balance of a healthy myocyte consists of influx and efflux of Na+. The main sources for Na+ influx from the extracellular compartment are the Na+ channels, the NCX and the Na+/H+ exchanger (NHX). Na+ leaves the cell via the Na+/K+ pump (NKP) and NCX operating in its reverse mode. Approximately 25% of the Na+ entry is produced by the Na+ channels, equally distributing between INa,early and INa,late, while NCX provides about 60% of total Na+ influx [Citation93]. Additionally, other routes for Na+ fluxes may contribute to a minor extent. The Na+ and Ca2+ homeostasis are strictly coupled processes [Citation93,Citation94]. Beyond the conversion of the elevated intracellular Na+ to Ca2+ by the NCX, it is easy to consider that a sustained depolarization above −40 mV, due to the augmented INa,late, may increase the open probability of L-type Ca2+ channels. In other words, a longer AP causes higher Ca2+ influx and Ca2+ load [Citation95–99].
Enhancement of the INa,late can be achieved through the Ca2+ – calmodulin (CaM) – CaMKII pathway. CaM and CaMKII can regulate the channel individually and cooperatively as well [Citation90,Citation100,Citation101]. CaM modulates Na+ channel function by binding to an IQ domain of the channel protein at the C-terminus and enhances slow inactivation () [Citation102–104]. CaM decreases the sustained INa,late during depolarization, therefore reduces the risk of arrhythmias [Citation105]. Until recently, understanding of the association of Na+ channels and CaM was limited, as most of the studies applied the Ca2+-free CaM, apocalmodulin (apoCAM). The binding site for both Ca2+-free and Ca2+-occupied CaM is the IQ motif [Citation106]. Wang et al., however, recently demonstrated that Ca2+ induces a conformational switch in the CaM, in which the N-lobe of the CaM contacts with the distal IQ motif of the C-terminal domain of the Na+ channel, while the C-lobe of the CaM (Ca2+ free) remains anchored to the IQ motif and this action is isoform-specific [Citation107]. There are controversial studies on whether Ca2+ alone can regulate Na+ channels [Citation102,Citation105,Citation108]. Gardill et al. concluded that the position of the EF-hand domain regulates Ca2+-dependent inactivation [Citation106]. Anomalous diffraction studies, on the other hand, proposed a Ca2+-sensor role for CaM rather than the EF-hand of the Na+ channel C-terminal domain [Citation109]. CaMKIIδ – the predominant cardiac isoform – may also alter the inactivation properties of Na+ channels. The Ca2+-CaM complex activates CaMKII which, in turn, phosphorylates the Na+ channel and enhances INa,late [Citation88–90,Citation110]. CaMKII increases intermediate inactivation and slows recovery, but slows the open state inactivation of INa,early and increases INa,late, increasing ultimately [Na+]i [Citation110–112]. Na+ channel regulation by CaMKII can also take place by association with the channel and by phosphorylation of the channel proteins [Citation111]. In rabbits, phosphorylation of Na+ channels by endogenous CaMKII occurs even at physiological Ca2+ levels [Citation111]. In addition to the effects on Na+ channels, phosphorylation by CaMKII enhances protein kinase A (PKA), ICa,L and sarcoendoplasmic reticulum Ca-ATPase (SERCA) and activates RyR () [Citation113–118]. Sustained depolarization by the augmented INa,late also contributes to cell Ca2+gain. These altogether increase the sarcoplasmic reticulum (SR) Ca2+ content and the open probability of RyR, therefore giving substrate for spontaneous Ca2+ release events [Citation112,Citation119].
Figure 4. Connection of the Ca2+/CaM complex to the Na+ channel. Crystal structure shows the human Nav – Ca2+/CaM complex (PBD ID: 6MUD [Citation106]) designed by PyMol Software [Citation106]. Blue shows the C-terminal region of the Nav1.5 α subunit, while red shows the CaM with 4 Ca2+ ions bound (cyan spheres). CaM, calmodulin
![Figure 4. Connection of the Ca2+/CaM complex to the Na+ channel. Crystal structure shows the human Nav – Ca2+/CaM complex (PBD ID: 6MUD [Citation106]) designed by PyMol Software [Citation106]. Blue shows the C-terminal region of the Nav1.5 α subunit, while red shows the CaM with 4 Ca2+ ions bound (cyan spheres). CaM, calmodulin](/cms/asset/dbc167d5-0630-407b-be89-581bb72134a0/kchl_a_1854986_f0004_oc.jpg)
In HF (both human HF and animal model of HF) expression and activity of CaMKII are increased, which may be proarrhythmogenic [Citation120–122]. Furthermore, it has been shown that transgenic overexpression of cytosolic CaMKII can induce HF [Citation117,Citation118]. Acute overexpression of CaMKII enhances INa,late and increases [Na+]i, slows inactivation of INa,early and recovery from inactivation, while shifting steady-state inactivation to more negative membrane potential in a Ca2+-dependent manner [Citation111]. All these effects of acute overexpression of CaMKII can be hindered by CaMKII inhibition. In undiseased ventricular cells, it has been shown that a fourfold increase in Na+ current density was required to achieve a significant increase in [Na+]i [Citation61,Citation123]. Wei et al. demonstrated that phosphorylation of CaMKII and the expression of Nav1.5 channel protein has been significantly elevated in the left ventricle upon treatment with the Ca2+ channel activator Bay K 8644 and the Na+ channel activator sea anemone toxin II (ATX-II). These effects were readily reversible by the application of TTX [Citation124]. Bay K 8644 and ATX-II increased the APD more powerfully when applied simultaneously and caused ventricular tachycardia with high incidence. This synergistic connection between high [Ca2+]i and high [Na+]i potentiates their arrhythmogenic activities.
Mitochondria are important Ca2+ buffering stores [Citation125,Citation126]. They contribute to Ca2+ homeostasis by taking up cytosolic Ca2+ via the mitochondrial Ca2+ uniporter (MCU) or releasing Ca2+ through the mitochondrial NCX (mNCX), the latter being an [Na+]i sensitive transporter [Citation67,Citation127,Citation128]. Under conditions of Ca2+ overload, as suggested by Ronchi et al. in a simulated ischemia protocol in rat ventricular myocytes, blockade of the sarcolemmal NCX turned mitochondria into a Ca2+ source from being a Ca2+ sink. It was concluded that during Ca2+ overload mitochondria may play a role in providing extra cytosolic Ca2+ and may be responsible for the INa,late mediated perturbation of the intracellular milieu [Citation129].
Elevated [Na+]i, when exceeding the functional reserve of the NKP, increases [Ca2+]i by switching NCX to a reverse mode operation with a consequent loading of the SR Ca2+ stores (). Even a relatively small, a few millimolar increase in [Na+]i slows Ca2+ extrusion by NCX [Citation93]. In addition, CaMKII phosphorylates the SERCA regulatory protein phospholamban (PLN), thereby augmenting SERCA activity and further gaining SR Ca2+ content [Citation118]. This predisposes SR and RyR to spontaneous Ca2+ releases which lead to the development of delayed afterdepolarizations (DAD). DADs occur in diastole after full repolarization and are usually the results of intracellular Ca2+ overload and spontaneous SR Ca2+ release ()). The abnormal Ca2+ release generates a depolarizing current by activating the forward mode of NCX [Citation130]. The development of DADs has clinical importance as they generate triggered activity which contributes to arrhythmogenesis in certain diseases, such as catecholaminergic polymorphic ventricular tachycardia (CPVT), HF or AF [Citation3].
Figure 5. Schematic illustration of the physiological and pathophysiological processes leading to arrhythmias upon increased INa,late. (a) In a healthy myocyte, excitation-contraction coupling controls contraction by periodically increasing and decreasing the intracellular Ca2+ concentration. (b) If INa,late is elevated, as in the case of many diseases, intracellular Na+ and a concomitant Ca2+ overload may lead to arrhythmias. High intracellular Na+ concentration can activate the reverse mode NCX to further load the cell with Ca2+. Ca2+ overload and the longer action potential duration predispose the cell to proarrhythmic events. Red arrows show Ca2+ related, while blue arrows show Na+ related processes. Dashed lines indicate the phosphorylation targets of the Ca2+ – CaM – CaMKII pathway. APD, action potential duration; CaM, calmodulin; CaMKII, Ca/calmodulin-dependent protein kinase II; DAD, delayed afterdepolarization; EAD, early afterdepolarization; ICa,L, L-type Ca2+ current; INa,early, the fast, early component of the Na+ current; INa,late, the persistent, late component of the Na+ current; NCX, Na+/Ca2+ exchange; NHX, Na+/H+ exchanger; NKP, Na+/K+ pump; PLN, phospholamban; PMCA, plasma membrane Ca2+-ATPase; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+-ATPase; SV, short term beat-to-beat variability of action potential duration
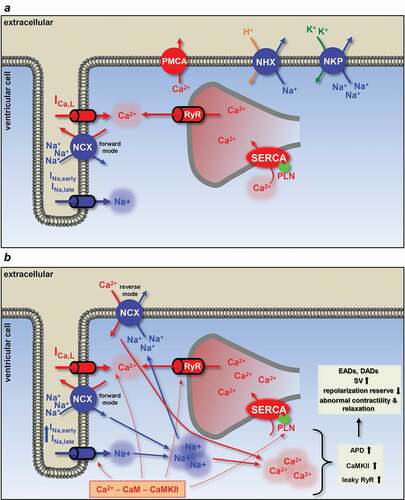
Figure 6. Basic mechanisms for early and delayed afterdepolarizations. (a) Factors involved in the generation of delayed afterdepolarizations (DAD). Increased [Na+]i elevates [Ca2+]i (and SR Ca2+ content) by switching NCX to reverse mode if the functional reserve of the NKP is reached. CaMKII phosphorylates phospholamban, also increasing SR Ca2+ content. High SR Ca2+ causes spontaneous Ca2+ release via the ryanodine receptors. This abnormal Ca2+ signaling switches NCX to forward mode, generating the transient inward current and this membrane depolarization can lead to triggered activity. Usually happens at high frequency, during diastole. Membrane potential recording shows a typical DAD. (b) Early afterdepolarization (EAD) occurs when the outward currents are reduced (reduced repolarization reserve) and/or the inward currents are enhanced. INa,late promotes EAD generation by the reactivation of ICa,L during the plateau phase, NCX activation and SR Ca2+ overload. Membrane potential recording shows a typical phase EAD. EAD, early afterdepolarization; DAD, delayed afterdepolarization; ICa,L, L-type Ca2+ current; IK1, inward rectifier K+ current; IKr, rapid component of delayed rectifier K+ current; IKs, slow component of delayed rectifier K+ current; INa, Na+ current; INCX, Na+/Ca2+ exchange; Iti, transient outward current; NKP, Na+/K+ pump; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; SERCA, sarcoplasmic reticulum Ca2+-ATPase; TA, triggered activity
![Figure 6. Basic mechanisms for early and delayed afterdepolarizations. (a) Factors involved in the generation of delayed afterdepolarizations (DAD). Increased [Na+]i elevates [Ca2+]i (and SR Ca2+ content) by switching NCX to reverse mode if the functional reserve of the NKP is reached. CaMKII phosphorylates phospholamban, also increasing SR Ca2+ content. High SR Ca2+ causes spontaneous Ca2+ release via the ryanodine receptors. This abnormal Ca2+ signaling switches NCX to forward mode, generating the transient inward current and this membrane depolarization can lead to triggered activity. Usually happens at high frequency, during diastole. Membrane potential recording shows a typical DAD. (b) Early afterdepolarization (EAD) occurs when the outward currents are reduced (reduced repolarization reserve) and/or the inward currents are enhanced. INa,late promotes EAD generation by the reactivation of ICa,L during the plateau phase, NCX activation and SR Ca2+ overload. Membrane potential recording shows a typical phase EAD. EAD, early afterdepolarization; DAD, delayed afterdepolarization; ICa,L, L-type Ca2+ current; IK1, inward rectifier K+ current; IKr, rapid component of delayed rectifier K+ current; IKs, slow component of delayed rectifier K+ current; INa, Na+ current; INCX, Na+/Ca2+ exchange; Iti, transient outward current; NKP, Na+/K+ pump; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; SERCA, sarcoplasmic reticulum Ca2+-ATPase; TA, triggered activity](/cms/asset/0e15793e-598f-4a95-9394-2ad06f53767a/kchl_a_1854986_f0006_oc.jpg)
Besides DADs, early afterdepolarizations (EAD) can be generated in the case of abnormal Na+ channel function ()). APD is lengthened upon slower INa,early inactivation predisposing the cell to the generation of EADs. There are several subtypes of EADs (phase 2, phase 3 and late phase 3 EAD) but, in general, they occur before the terminal repolarization. In most cases, a longer AP (except for the late phase 3 EAD) allows ICa,L to recover from inactivation generating a positive feedback loop triggering further APs [Citation3]. It is important to note that activation of CaMKII itself may also contribute to the facilitation and reactivation of ICa,L [Citation131,Citation132]. It has been shown, however, that both the SR Ca2+ load with the concomitant spontaneous Ca2+ release and the inward depolarizing current delivered by the NCX and the reactivated INa are also accountable for the generation of EADs [Citation133–135]. Two possible mechanisms have been proposed to explain the EAD generation by INa,late; SR Ca2+ overload and the reactivation of ICa,L during the plateau phase of the AP [Citation63]. Our experiments in guinea pig myocytes showed that INa,late-induced EADs are mediated by spontaneous SR Ca2+ release as the first occurrence of EAD precedes – i.e. occurs at more positive membrane potential – the window current voltage range of ICa,L, therefore making ICa,L reactivation as a key feature less likely [Citation45].
It has been demonstrated in LQTS-3 patients, that a gain-of-function mutation of SCN5A resulted in an enlargement of INa,late [Citation136]. Besides increasing INa,late, the mutation also caused elevation of [Na+]i and [Ca2+]i which was associated with a reduction in the forward mode and an increase in the reverse mode activity of NCX [Citation123,Citation137–139]. Other studies have shown high SR Ca2+ content and spontaneous diastolic Ca2+ transients in isolated cells from LQTS-2 mutant mice [Citation140,Citation141].
Previously, we established the concept of relative short-term beat-to-beat variability of APD (RSV), which might be a novel approach for predicting arrhythmias [Citation52,Citation142]. In those settings, higher RSV is considered to be more arrhythmogenic by increasing the dispersion of refractoriness. Those experiments showed that higher Na+ current causes higher variations in the APD, which is in agreement with the proarrhythmic role of INa,late. In our experiments, Na+ current was inhibited by TTX and lidocaine, or alternatively, activated by veratridine. Similar increase in beat-to-beat variability was observed under Ca2+ overload conditions [Citation143] and in situations where the repolarization reserve has been compromised [Citation52].
INa,late can be directly or indirectly regulated by Ca2+, CaM, and CaMKII. In general, the higher [Ca2+]i shifts the steady-state inactivation curve of the Na+ current to more positive voltages and increases the availability of the channels at more positive potentials [Citation45,Citation108,Citation144]. Consequently, the buffering of [Ca2+]i should decrease INa,late. Our experiments, however, showed that INa,late is rather influenced by the shape and voltage profile of the AP than by Ca2+ itself [Citation45]. On the other hand, inhibition of CaMKII successfully prevented the catecholamine-induced spontaneous Ca2+ waves, DADs and EADs while improving contractile function [Citation145–148]. Unfortunately, targeting CaMKII as an antiarrhythmic option is rather difficult considering its immensely complex signaling network.
It has recently been shown that inhibition of the exchange protein directly activated by cAMP (Epac) can induce EADs [Citation149]. The mechanism involves oxidative activation of CaMKII by an increase in cellular reactive oxygen species, ROS an increase in INa,late and prolongation of APD. ROS activation of CaMKII phosphorylates RyR and Nav1.5, leading to SR Ca2+ leak through RyR and enhanced INa,late [Citation150]. Application of ranolazine prevented the proarrhythmic effects: decreased APD and abolished EADs, i.e. the impaired Epac signaling induced arrhythmias.
For a detailed review about the role of Ca2+ in arrhythmogenesis see a recent review of Kistamás et al [Citation3].
Antiarrhythmic drug development
It became clear that elevation of INa,late, [Na+]i, and [Ca2+]i is arrhythmogenic; therefore, an effective antiarrhythmic treatment is necessary under these conditions. An obvious objective is the inhibition of voltage-gated Na+ channels. However, the disappointing results of the CAST and SWORD studies clarified that blocking a single specific ion channel alone can lead to unexpected adverse effects. Recently, a need for selective inhibitors that are able to distinguish between INa,early and INa,late is rather emerging. The selectivity here is critically important since blocking the early component of the current can lead to a decrease in conduction velocity and might lead to conduction block and reentrant arrhythmias [Citation3,Citation151,Citation152].
A number of inhibitors have been developed to date including ranolazine, eleclazine (GS-6615), lidocaine, GS-458967, GS-462808, and F15845, however, mainly ranolazine was used for excessive experimental and clinical studies () [Citation153]. The main issue with most of these inhibitors is that they function in a voltage-dependent manner and exert their INa,late selective effects mainly at lower than physiological membrane potentials. Coming closer to the physiologically relevant membrane voltage range, these inhibitors tend to block INa,early more and more, thus losing their selectivity [Citation153]. Secondly, their blocking effect is rate-dependent, in other words, the inhibition of Na+ current increases with the pacing frequency, exerting thus a lesser impact on INa,late in bradycardia, when INa,late is thought to be significantly greater [Citation154].
Table 1. Summary of the effects of INa,late inhibitors
GS-458967, like other Na+ channel blockers including ranolazine and mexiletine, reduces APD and suppresses EAD or DAD formation and generation of Torsade de Pointes (TdP) type ventricular tachyarrhythmias [Citation18,Citation155–159]. F15845, an anti-ischemic drug, was also shown to inhibit INa,late and prevent ventricular tachycardia and fibrillation [Citation155]. The latest promising inhibitor compound was the eleclazine (GS-6615) which has undergone clinical trials. Eleclazine was demonstrated to bind to the Na+ channels with rapid kinetics and block INa,late with minimal effects on other ion currents and without adverse side effects [Citation160]. Eleclazine shortened APD and the QT-interval, decreased spatiotemporal dispersion of repolarization, and suppressed the epinephrine induced ventricular tachycardias. Despite the encouraging results with eleclazine, the drug and the clinical trials were suspended as the number of implantable cardioverter defibrillator (ICD) shocks was higher in the eleclazine-treated group. The use of amiodarone, a blocker with mixed effects, seemed to be a promising drug in HF, but there was a higher incidence of QT prolongation and bradycardia or pulmonary fibrosis, hepatotoxicity, and thyrotoxicity [Citation161].
The most extensively studied selective INa,late inhibitor is the antiischemic ranolazine. Ranolazine reduces Na+ dependent Ca2+ overload by inhibiting INa,late [Citation162]. This compound also inhibits the rapid component of the delayed rectifier K+ current (IKr) [Citation163], ICa,L [Citation73] and reverse mode NCX [Citation164]; however, effects on ICa,L and NCX are mainly out of the therapeutic concentration range of the drug. Ranolazine is also a weak β-adrenergic agonist [Citation165], while it has minimal effects on blood pressure or heart rate [Citation166]. Furthermore, ranolazine reduced the beat-to-beat variability of APD [Citation167]. Unfortunately, ranolazine shares the same disadvantages, namely, the enhancement of INa,early inhibition in the case of partially depolarized membrane observed in diseased hearts or at higher activation rates [Citation168,Citation169]. At low pacing rates inhibition of INa,late successfully decreased the arrhythmic events, such as EADs, DADs or TdP [Citation45,Citation53,Citation75,Citation170–172]. Ranolazine reduced the dispersion of repolarization [Citation173] the occurrence of EADs and TdP [Citation75,Citation174]. Dispersion of repolarization is caused by the shortening of the APD of midmyocardial cells, where INa,late is the most prominent. In LQTS-3 patients ranolazine decreased [Citation175], while in a different study it increased the QT interval, due to IKr blockade [Citation176].
It has been demonstrated that suppression of INa,late hinders Ca2+ overload [Citation167,Citation177–179]. The hallmark of INa,late inhibition is the suppression of Ca2+-dependent triggered activities, by reducing [Na+]i and [Ca2+]i. A second feature of the INa,late blockade is the normalization of repolarization, allowing restoring the repolarization reserve [Citation55].
Characterization of the true gating mechanism of INa,late (the potential gating mechanisms were discussed previously) may also bring a better therapeutic protocol closer, since each gating mechanism has its own characteristic drug affinity and sensitivity profile [Citation164,Citation180,Citation181].
Conclusions
In summary, we have reviewed the arrhythmogenic behavior of the augmented late Na+ current and the concomitant elevation of intracellular Na+ and Ca2+ concentrations. In spite of the tremendous work that had been done on understanding the background of the Ca2+ related arrhythmias, there is still a need for more research to better design antiarrhythmic treatments and drugs. It became clear that more attention has to be paid to INa,late in patients with Ca2+-dependent arrhythmias, especially in the case of bradycardia.
Various processes can lead to Ca2+ overload and the therapeutic options are rather complex if we take into account the augmented INa,late. Hence, a selective INa,late inhibitor may only treat a part of the issue. To date, the most selective drug to INa,late in the market is ranolazine; however, it exerts electrophysiological effects on other ion currents as well. Therefore, more work is necessary to gain a better understanding of the role of INa,late and Ca2+ handling in cardiac arrhythmias and to develop novel antiarrhythmic therapies with a focus on translational aspects.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205.
- Eisner DA, Caldwell JL, Kistamas K, et al. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121:181–195.
- Kistamas K, Veress R, Horvath B, et al. Calcium handling defects and cardiac arrhythmia syndromes. Front Pharmacol. 2020;11:72.
- Mitsuiye T, Noma A. Inactivation of cardiac Na+ channel simply through open states as revealed by single-channel analysis in guinea pig ventricular myocytes. Jpn J Physiol. 2002;52:457–469.
- Scanley BE, Hanck DA, Chay T, et al. Kinetic analysis of single sodium channels from canine cardiac Purkinje cells. J Gen Physiol. 1990;95:411–437.
- Zaza A, Rocchetti M. The late Na+ current–origin and pathophysiological relevance. Cardiovasc Drugs Ther. 2013 ;27(1):61–68.
- Sipido KR, Maes M, Van de Werf F. Low efficiency of Ca2+ entry through the Na(+)-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na(+)-Ca2+ exchange. Circ Res. 1997;81:1034–1044.
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544.
- Catterall WA, Goldin AL, Waxman SG. International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409.
- de Lera Ruiz M, Kraus RL. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem. 2015;58:7093–7118.
- Gellens ME, George AL Jr., Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:554–558.
- Maier SK, Westenbroek RE, Yamanushi TT, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci U S A. 2003;100:3507–3512.
- Maier SK, Westenbroek RE, Schenkman KA, et al. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078.
- Yang T, Atack TC, Stroud DM, et al. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–332.
- Biet M, Barajas-Martinez H, Ton AT, et al. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J Mol Cell Cardiol. 2012;53:593–598.
- Valdivia CR, Chu WW, Pu J, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483.
- Haufe V, Cordeiro JM, Zimmer T, et al. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–127.
- Maier LS, Sossalla S. The late Na current as a therapeutic target: where are we? J Mol Cell Cardiol. 2013;61:44–50.
- Cusdin FS, Clare JJ, Jackson AP. Trafficking and cellular distribution of voltage-gated sodium channels. Traffic. 2008;9:17–26.
- Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. 2011;2:53.
- Kazen-Gillespie KA, Ragsdale DS, D’Andrea MR, et al. Cloning, localization, and functional expression of sodium channel beta1A subunits. J Biol Chem. 2000;275:1079–1088.
- Qin N, D’Andrea MR, Lubin ML, et al. Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur J Biochem. 2003;270:4762–4770.
- Chen C, Cannon SC. Modulation of Na+ channel inactivation by the beta 1 subunit: a deletion analysis. Pflugers Arch. 1995;431:186–195.
- Yu EJ, Ko SH, Lenkowski PW, et al. Distinct domains of the sodium channel beta3-subunit modulate channel-gating kinetics and subcellular location. Biochem J. 2005;392:519–526.
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25.
- Kellenberger S, Scheuer T, Catterall WA. Movement of the Na+ channel inactivation gate during inactivation. J Biol Chem. 1996;271:30971–30979.
- West JW, Patton DE, Scheuer T, et al. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–10914.
- Patton DE, West JW, Catterall WA, et al. Amino acid residues required for fast Na(+)-channel inactivation: charge neutralizations and deletions in the III-IV linker. Proc Natl Acad Sci U S A. 1992;89:10905–10909.
- Stuhmer W, Conti F, Suzuki H, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603.
- Motoike HK, Liu H, Glaaser IW, et al. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165.
- Armstrong CM. Na channel inactivation from open and closed states. Proc Natl Acad Sci U S A. 2006;103:17991–17996.
- Kiyosue T, Arita M. Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ Res. 1989;64:389–397.
- Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. A J Physiol. 1979;236:H561–7.
- Maltsev VA, Sabbah HN, Higgins RS, et al. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552.
- Zaniboni M, Pollard AE, Yang L, et al. Beat-to-beat repolarization variability in ventricular myocytes and its suppression by electrical coupling. Am J Physiol Heart Circ Physiol. 2000;278:H677–87.
- Barandi L, Virag L, Jost N, et al. Reverse rate-dependent changes are determined by baseline action potential duration in mammalian and human ventricular preparations. Basic Res Cardiol. 2010;105:315–323.
- Banyasz T, Horvath B, Virag L, et al. Reverse rate dependency is an intrinsic property of canine cardiac preparations. Cardiovasc Res. 2009;84:237–244.
- Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127.
- Mangold KE, Brumback BD, Angsutararux P, et al. Mechanisms and models of cardiac sodium channel inactivation. Channels (Austin). 2017;11:517–533.
- Bers DM. Excitation-contraction coupling and cardiac contractile force. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001.
- Liu H, Sun HY, Lau CP, et al. Regulation of voltage-gated cardiac sodium current by epidermal growth factor receptor kinase in guinea pig ventricular myocytes. J Mol Cell Cardiol. 2007;42:760–768.
- Wang DW, Viswanathan PC, Balser JR, et al. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation. 2002;105:341–346.
- Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92(Suppl 4):iv1–iv5.
- Horvath B, Hezso T, Szentandrassy N, et al. Late sodium current in human, canine and guinea pig ventricular myocardium. J Mol Cell Cardiol. 2020;139:14–23.
- Horvath B, Banyasz T, Jian Z, et al. Dynamics of the late Na(+) current during cardiac action potential and its contribution to afterdepolarizations. J Mol Cell Cardiol. 2013;64:59–68.
- Hegyi B, Bossuyt J, Griffiths LG, et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc Natl Acad Sci U S A. 2018;115:E3036–E44.
- Hegyi B, Banyasz T, Izu LT, et al. beta-adrenergic regulation of late Na(+) current during cardiac action potential is mediated by both PKA and CaMKII. J Mol Cell Cardiol. 2018;123:168–179.
- Luo A, Ma J, Song Y, et al. Larger late sodium current density as well as greater sensitivities to ATX II and ranolazine in rabbit left atrial than left ventricular myocytes. Am J Physiol Heart Circ Physiol. 2014;306:H455–61.
- Wu L, Ma J, Li H, et al. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation. 2011;123:1713–1720.
- Nagatomo T, January CT, Ye B, et al. Rate-dependent QT shortening mechanism for the LQT3 deltaKPQ mutant. Cardiovasc Res. 2002;54:624–629.
- Carmeliet E. Action potential duration, rate of stimulation, and intracellular sodium. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S2–S7.
- Szentandrassy N, Kistamas K, Hegyi B, et al. Contribution of ion currents to beat-to-beat variability of action potential duration in canine ventricular myocytes. Pflugers Arch. 2015;467:1431–1443.
- Coppini R, Ferrantini C, Yao L, et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–584.
- Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–9.
- Burashnikov A. Late INa inhibition as an antiarrhythmic strategy. J Cardiovasc Pharmacol. 2017;70:159–167.
- Clancy CE, Tateyama M, Kass RS. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest. 2002;110:1251–1262.
- Shryock JC, Song Y, Rajamani S, et al. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res. 2013;99:600–611.
- Undrovinas A, Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–359.
- Pourrier M, Williams S, McAfee D, et al. CrossTalk proposal: the late sodium current is an important player in the development of diastolic heart failure (heart failure with a preserved ejection fraction). J Physiol. 2014;592:411–414.
- Sossalla S, Wagner S, Rasenack EC, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43.
- Yao L, Fan P, Jiang Z, et al. Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am J Physiol Cell Physiol. 2011;301:C577–86.
- Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. 2014;63:2335–2345.
- Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol Ther. 2008;119:326–339.
- Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241.
- Tang Q, Ma J, Zhang P, et al. Persistent sodium current and Na+/H+ exchange contributes to the augmentation of the reverse Na+/Ca2+ exchange during hypoxia or acute ischemia in ventricular myocytes. Pflugers Arch. 2012;463:513–522.
- Song Y, Shryock JC, Wagner S, et al. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222.
- Ma J, Song Y, Shryock JC, et al. Ranolazine attenuates hypoxia- and hydrogen peroxide-induced increases in sodium channel late openings in ventricular myocytes. J Cardiovasc Pharmacol. 2014;64:60–68.
- Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505.
- Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763.
- Li W, Yu Y, Hou JW, et al. Larger rate dependence of late sodium current in cardiac Purkinje cells: A potential link to arrhythmogenesis. Heart Rhythm. 2017;14:422–431.
- Sossalla S, Maurer U, Schotola H, et al. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res Cardiol. 2011;106:263–272.
- Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288.
- Antzelevitch C, Nesterenko V, Shryock JC, et al. The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol. 2014;221:137–168.
- Wu L, Rajamani S, Li H, et al. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na(+) current in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1048–57.
- Song Y, Shryock JC, Wu L, et al. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199.
- Murphy L, Renodin D, Antzelevitch C, et al. Extracellular proton depression of peak and late Na current in the canine left ventricle. Am J Physiol Heart Circ Physiol. 2011;301:H936–44.
- Jones DK, Claydon TW, Ruben PC. Extracellular protons inhibit charge immobilization in the cardiac voltage-gated sodium channel. Biophys J. 2013;105:101–107.
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999;79:917–1017.
- Shimoda LA, Polak J. Hypoxia. 4. Hypoxia and ion channel function. Am J Physiol Cell Physiol. 2011;300:C951–67.
- Ahern GP, Hsu SF, Klyachko VA, et al. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem. 2000;275:28810–28815.
- Yang P, Kupershmidt S, Roden DM. Cloning and initial characterization of the human cardiac sodium channel (SCN5A) promoter. Cardiovasc Res. 2004;61:56–65.
- Zhang Y, Hartmann HA, Satin J. Glycosylation influences voltage-dependent gating of cardiac and skeletal muscle sodium channels. J Membr Biol. 1999;171:195–207.
- Cortada E, Brugada R, Verges M. N-Glycosylation of the voltage-gated sodium channel beta2 subunit is required for efficient trafficking of NaV1.5/beta2 to the plasma membrane. J Biol Chem. 2019;294:16123–16140.
- Sarhan MF, Van Petegem F, Ahern CA. A double tyrosine motif in the cardiac sodium channel domain III-IV linker couples calcium-dependent calmodulin binding to inactivation gating. J Biol Chem. 2009;284:33265–33274.
- Beltran-Alvarez P, Pagans S, Brugada R. The cardiac sodium channel is post-translationally modified by arginine methylation. J Proteome Res. 2011;10:3712–3719.
- Beyder A, Strege PR, Reyes S, et al. Ranolazine decreases mechanosensitivity of the voltage-gated sodium ion channel Na(v)1.5: a novel mechanism of drug action. Circulation. 2012;125:2698–2706.
- Beyder A, Rae JL, Bernard C, et al. Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J Physiol. 2010;588:4969–4985.
- Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther. 2005;106:39–55.
- Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63:476–486.
- Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–187.
- Maltsev VA, Reznikov V, Undrovinas NA, et al. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–608.
- Maltsev VA, Kyle JW, Undrovinas A. Late Na+ current produced by human cardiac Na+ channel isoform Nav1.5 is modulated by its beta1 subunit. J Physiol Sci. 2009;59: 217–225. .
- Despa S, Bers DM. Na(+) transport in the normal and failing heart - remember the balance. J Mol Cell Cardiol. 2013;61:2–10.
- Clancy CE, Chen-Izu Y, Bers DM, et al. Deranged sodium to sudden death. J Physiol. 2015;593:1331–1345.
- Banyasz T, Horvath B, Jian Z, et al. Profile of L-type Ca(2+) current and Na(+)/Ca(2+) exchange current during cardiac action potential in ventricular myocytes. Heart Rhythm. 2012;9:134–142.
- Banyasz T, Fulop L, Magyar J, et al. Endocardial versus epicardial differences in L-type calcium current in canine ventricular myocytes studied by action potential voltage clamp. Cardiovasc Res. 2003;58:66–75.
- Fulop L, Banyasz T, Magyar J, et al. Reopening of L-type calcium channels in human ventricular myocytes during applied epicardial action potentials. Acta Physiol Scand. 2004;180:39–47.
- Linz KW, Meyer R. Profile and kinetics of L-type calcium current during the cardiac ventricular action potential compared in guinea-pigs, rats and rabbits. Pflugers Arch. 2000;439:588–599.
- Linz KW, Meyer R. Control of L-type calcium current during the action potential of guinea-pig ventricular myocytes. J Physiol. 1998;513(Pt 2):425–442.
- Maier LS. CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm. 2011;8:474–477.
- Scheuer T. Regulation of sodium channel activity by phosphorylation. Semin Cell Dev Biol. 2011;22:160–165.
- Tan HL, Kupershmidt S, Zhang R, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447.
- Wang DW, Makita N, Kitabatake A, et al. Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circ Res. 2000;87:E37–43.
- Veldkamp MW, Viswanathan PC, Bezzina C, et al. Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ Res. 2000;86:E91–7.
- Kim J, Ghosh S, Liu H, et al. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012.
- Gardill BR, Rivera-Acevedo RE, Tung CC, et al. Crystal structures of Ca(2+)-calmodulin bound to NaV C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc Natl Acad Sci U S A. 2019;116:10763–10772.
- Wang C, Chung BC, Yan H, et al. Structural analyses of Ca(2)(+)/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat Commun. 2014;5:4896.
- Wingo TL, Shah VN, Anderson ME, et al. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219–225.
- Wang C, Chung BC, Yan H, et al. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012;20:1167–1176.
- Grandi E, Herren AW. CaMKII-dependent regulation of cardiac Na(+) homeostasis. Front Pharmacol. 2014;5:41.
- Wagner S, Dybkova N, Rasenack EC, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138.
- Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. 2019;127:246–259.
- Liu N, Ruan Y, Denegri M, et al. Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C±) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011;50:214–222.
- Kashimura T, Briston SJ, Trafford AW, et al. In the RyR2(R4496C) mouse model of CPVT, beta-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res. 2010;107:1483–1489.
- Venetucci L, Denegri M, Napolitano C, et al. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575.
- Guo T, Zhang T, Mestril R, et al. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406.
- Maier LS, Zhang T, Chen L, et al. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911.
- Zhang T, Maier LS, Dalton ND, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919.
- Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–640.
- Ai X, Curran JW, Shannon TR, et al. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322.
- Hoch B, Meyer R, Hetzer R, et al. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–721.
- Kirchhefer U, Schmitz W, Scholz H, et al. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–261.
- Kornyeyev D, El-Bizri N, Hirakawa R, et al. Contribution of the late sodium current to intracellular sodium and calcium overload in rabbit ventricular myocytes treated by anemone toxin. Am J Physiol Heart Circ Physiol. 2016;310:H426–35.
- Wei XH, Yu SD, Ren L, et al. Inhibition of late sodium current suppresses calcium-related ventricular arrhythmias by reducing the phosphorylation of CaMK-II and sodium channel expressions. Sci Rep. 2017;7:981.
- Smith GL, Eisner DA. Calcium buffering in the heart in health and disease. Circulation. 2019;139:2358–2371.
- Eisner DA, Caldwell JL, Trafford AW, et al. The control of diastolic calcium in the heart: basic mechanisms and functional implications. Circ Res. 2020;126:395–412.
- Boyman L, Williams GS, Khananshvili D, et al. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–213.
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364.
- Ronchi C, Torre E, Rizzetto R, et al. Late sodium current and intracellular ionic homeostasis in acute ischemia. Basic Res Cardiol. 2017;112:12.
- Mechmann S, Pott L. Identification of Na-Ca exchange current in single cardiac myocytes. Nature. 1986;319:597–599.
- Kohlhaas M, Zhang T, Seidler T, et al. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244.
- Wu Y, Temple J, Zhang R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293.
- Volders PG, Kulcsar A, Vos MA, et al. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997;34:348–359.
- Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. A J Physiol. 1990;258:H1796–805.
- Szabo B, Sweidan R, Rajagopalan CV, et al. Role of Na+:Ca2+ exchange current in Cs(+)-induced early afterdepolarizations in Purkinje fibers. J Cardiovasc Electrophysiol. 1994;5:933–944.
- Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28:287–294.
- Belardinelli L, Giles WR, Rajamani S, et al. Cardiac late Na(+) current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm. 2015;12:440–448.
- Larbig R, Torres N, Bridge JH, et al. Activation of reverse Na+-Ca2+ exchange by the Na+ current augments the cardiac Ca2+ transient: evidence from NCX knockout mice. J Physiol. 2010;588:3267–3276.
- Baartscheer A, Schumacher CA, Belterman CN, et al. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57:986–995.
- Lindegger N, Hagen BM, Marks AR, et al. Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J Mol Cell Cardiol. 2009;47:326–334.
- Fredj S, Lindegger N, Sampson KJ, et al. Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome type 3 myocytes. Circ Res. 2006;99:1225–1232.
- Nanasi PP, Magyar J, Varro A, et al. Beat-to-beat variability of cardiac action potential duration: underlying mechanism and clinical implications. Can J Physiol Pharmacol. 2017;95:1230–1235.
- Kistamas K, Szentandrassy N, Hegyi B, et al. Changes in intracellular calcium concentration influence beat-to-beat variability of action potential duration in canine ventricular myocytes. J Physiol Pharmacol. 2015;66:73–81.
- Sarhan MF, Tung CC, Van Petegem F, et al. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci U S A. 2012;109:3558–3563.
- Sag CM, Wadsack DP, Khabbazzadeh S, et al. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. 2009;2:664–675.
- Sossalla S, Kallmeyer B, Wagner S, et al. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–2342.
- Curran J, Brown KH, Santiago DJ, et al. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32.
- Anderson ME, Braun AP, Wu Y, et al. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006.
- Yang Z, Kirton HM, Al-Owais M, et al. Epac2-rap1 signaling regulates reactive oxygen species production and susceptibility to cardiac arrhythmias. Antioxid Redox Signal. 2017;27:117–132.
- Rokita AG, Anderson ME. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. 2012;126:2125–2139.
- Moreno JD, Zhu ZI, Yang PC, et al. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci Transl Med. 2011;3:98ra83.
- Echt DS, Liebson PR, Mitchell LB, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The cardiac arrhythmia suppression trial. N Engl J Med. 1991;324:781–788.
- Horvath B, Hezso T, Kiss D, et al. Late sodium current inhibitors as potential antiarrhythmic agents. Front Pharmacol. 2020;11:413.
- Pless SA, Galpin JD, Frankel A, et al. Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat Commun. 2011;2:351.
- Carneiro JS, Bento AS, Bacic D, et al. The selective cardiac late sodium current inhibitor GS-458967 suppresses autonomically triggered atrial fibrillation in an intact porcine model. J Cardiovasc Electrophysiol. 2015;26:1364–1369.
- Bonatti R, Silva AF, Batatinha JA, et al. Selective late sodium current blockade with GS-458967 markedly reduces ischemia-induced atrial and ventricular repolarization alternans and ECG heterogeneity. Heart Rhythm. 2014;11:1827–1835.
- Pezhouman A, Madahian S, Stepanyan H, et al. Selective inhibition of late sodium current suppresses ventricular tachycardia and fibrillation in intact rat hearts. Heart Rhythm. 2014;11(3):492–501. .
- Hasenfuss G, Maier LS. Mechanism of action of the new anti-ischemia drug ranolazine. Clin Res Cardiol. 2008;97:222–226.
- Trenor B, Gomis-Tena J, Cardona K, et al. In silico assessment of drug safety in human heart applied to late sodium current blockers. Channels (Austin). 2013;7:249–262.
- Bacic D, Carneiro JS, Bento AA, et al. Eleclazine, an inhibitor of the cardiac late sodium current, is superior to flecainide in suppressing catecholamine-induced ventricular tachycardia and T-wave alternans in an intact porcine model. Heart Rhythm. 2017;14:448–454.
- Burton L, Knollan B. Goodman and Gilman’s the pharmacological basis of therapeutics. 12 ed. McGraw-Hill, NY, USA; 2010.
- Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation. 2006;113:2462–2472.
- Rajamani S, Shryock JC, Belardinelli L. Rapid kinetic interactions of ranolazine with HERG K+ current. J Cardiovasc Pharmacol. 2008;51:581–589.
- Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl 4):iv6–iv14.
- Letienne R, Vie B, Puech A, et al. Evidence that ranolazine behaves as a weak beta1- and beta2-adrenoceptor antagonist in the rat [correction of cat] cardiovascular system. Naunyn-Schmiedeberg’s Arch Pharmacol. 2001;363:464–471.
- Doshi D, Morrow JP. Potential application of late sodium current blockade in the treatment of heart failure and atrial fibrillation. Rev Cardiovasc Med. 2009;10(Suppl 1):S46–52.
- Undrovinas AI, Belardinelli L, Undrovinas NA, et al. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S169–S77.
- Zygmunt AC, Nesterenko VV, Rajamani S, et al. Mechanisms of atrial-selective block of Na(+) channels by ranolazine: I. Experimental analysis of the use-dependent block. Am J Physiol Heart Circ Physiol. 2011;301:H1606–14.
- Szel T, Koncz I, Jost N, et al. Class I/B antiarrhythmic property of ranolazine, a novel antianginal agent, in dog and human cardiac preparations. Eur J Pharmacol. 2011;662:31–39.
- Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047.
- Belardinelli L, Liu G, Smith-Maxwell C, et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther. 2013;344:23–32.
- Rajamani S, Liu G, El-Bizri N, et al. The novel late Na(+) current inhibitor, GS-6615 (eleclazine) and its anti-arrhythmic effects in rabbit isolated heart preparations. Br J Pharmacol. 2016;173:3088–3098.
- Zygmunt AC, Eddlestone GT, Thomas GP, et al. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol. 2001;281:H689–97.
- Baartscheer A, Schumacher CA, van Borren MM, et al. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024.
- Moss AJ, Zareba W, Schwarz KQ, et al. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–1293.
- Wilson SR, Scirica BM, Braunwald E, et al. Efficacy of ranolazine in patients with chronic angina observations from the randomized, double-blind, placebo-controlled MERLIN-TIMI (Metabolic Efficiency with ranolazine for less ischemia in non-ST-segment elevation acute coronary syndromes) 36 Trial. J Am Coll Cardiol. 2009;53:1510–1516.
- Hale SL, Leeka JA, Kloner RA. Improved left ventricular function and reduced necrosis after myocardial ischemia/reperfusion in rabbits treated with ranolazine, an inhibitor of the late sodium channel. J Pharmacol Exp Ther. 2006;318:418–423.
- Calderon-Sanchez EM, Dominguez-Rodriguez A, Lopez-Haldon J, et al. Cardioprotective effect of ranolazine in the process of ischemia-reperfusion in adult rat cardiomyocytes. Rev Esp Cardiologia. 2016;69:45–53.
- Aldakkak M, Camara AK, Heisner JS, et al. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res. 2011;64:381–392.
- Morita N, Lee JH, Xie Y, et al. Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. 2011;57:366–375.
- Hoyer K, Song Y, Wang D, et al. Reducing the late sodium current improves cardiac function during sodium pump inhibition by ouabain. J Pharmacol Exp Ther. 2011;337:513–523.
- Jiang D, Shi H, Tonggu L, et al. Structure of the cardiac sodium channel. Cell. 2020;180:122–34 e10.
- Hale SL, Shryock JC, Belardinelli L, et al. Late sodium current inhibition as a new cardioprotective approach. J Mol Cell Cardiol. 2008;44:954–967.
- Sicouri S, Glass A, Belardinelli L, et al. Antiarrhythmic effects of ranolazine in canine pulmonary vein sleeve preparations. Heart Rhythm. 2008;5:1019–1026.
- Gralinski MR, Black SC, Kilgore KS, et al. Cardioprotective effects of ranolazine (RS-43285) in the isolated perfused rabbit heart. Cardiovasc Res. 1994;28:1231–1237.
- Haigney MC, Lakatta EG, Stern MD, et al. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399.
- Antoons G, Oros A, Beekman JD, et al. Late na(+) current inhibition by ranolazine reduces torsades de pointes in the chronic atrioventricular block dog model. J Am Coll Cardiol. 2010;55:801–809.
- Alves Bento AS, Bacic D, Saran Carneiro J, et al. Selective late INa inhibition by GS-458967 exerts parallel suppression of catecholamine-induced hemodynamically significant ventricular tachycardia and T-wave alternans in an intact porcine model. Heart Rhythm. 2015;12:2508–2514.
- Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910.
- Bossu A, Houtman MJC, Meijborg VMF, et al. Selective late sodium current inhibitor GS-458967 suppresses Torsades de Pointes by mostly affecting perpetuation but not initiation of the arrhythmia. Br J Pharmacol. 2018;175:2470–2482.
- Sicouri S, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of the highly selective late sodium channel current blocker GS-458967. Heart Rhythm. 2013;10:1036–1043.
- Azam MA, Zamiri N, Masse S, et al. Effects of late sodium current blockade on ventricular refibrillation in a rabbit model. Circ Arrhythm Electrophysiol 2017; 10:e004331.
- Badri M, Patel A, Patel C, et al. Mexiletine prevents recurrent torsades de pointes in acquired long QT syndrome refractory to conventional measures. JACC Clin Electrophysiol. 2015;1:315–322.
- Frommeyer G, Garthmann J, Ellermann C, et al. Broad antiarrhythmic effect of mexiletine in different arrhythmia models. Europace. 2018;20:1375–1381.
- Funasako M, Aiba T, Ishibashi K, et al. Pronounced shortening of QT interval with mexiletine infusion test in patients with type 3 congenital long QT syndrome. Circ J. 2016;80:340–345.
- Bos JM, Crotti L, Rohatgi RK, et al. Mexiletine shortens the QT interval in patients with potassium channel-mediated type 2 long QT syndrome. Circ Arrhythm Electrophysiol. 2019;12:e007280.
- Fujisawa T, Aizawa Y, Katsumata Y, et al. Mexiletine shortens the QT interval in a pedigree of KCNH2 related long QT syndrome. J Arrhythm. 2020;36:193–196.
- Pignier C, Rougier JS, Vie B, et al. Selective inhibition of persistent sodium current by F 15845 prevents ischaemia-induced arrhythmias. Br J Pharmacol. 2010;161:79–91.
- Vacher B, Pignier C, Letienne R, et al. 15845 inhibits persistent sodium current in the heart and prevents angina in animal models. Br J Pharmacol. 2009;156:214–225.