
ABSTRACT
Voltage-gated calcium channels (VGCCs) represent key regulators of the calcium influx through the plasma membrane of excitable cells, like neurons. Activated by the depolarization of the membrane, the opening of VGCCs induces very transient and local changes in the intracellular calcium concentration, known as calcium nanodomains, that in turn trigger calcium-dependent signaling cascades and the release of chemical neurotransmitters. Based on their central importance as concierges of excitation-secretion coupling and therefore neuronal communication, VGCCs have been studied in multiple aspects of neuronal function and malfunction. However, studies on molecular interaction partners and recent progress in omics technologies have extended the actual concept of these molecules. With this review, we want to illustrate some new perspectives of VGCCs reaching beyond their function as calcium-permeable pores in the plasma membrane. Therefore, we will discuss the relevance of VGCCs as voltage sensors in functional complexes with ryanodine receptors, channel-independent actions of auxiliary VGCC subunits, and provide an insight into how VGCCs even directly participate in gene regulation. Furthermore, we will illustrate how structural changes in the intracellular C-terminus of VGCCs generated by alternative splicing events might not only affect the biophysical channel characteristics but rather determine their molecular environment and downstream signaling pathways.
Introduction
Transient changes of the intracellular calcium concentration are a major trigger for many signaling cascades and cellular processes. Besides being a key parameter for neuronal communication, intracellular calcium levels control the entire cell life from fertilization to programmed cell death and regulate, inter alia, gene expression, heart and muscle contraction, as well as enzyme activity within subcellular compartments [Citation1–3]. In excitable cells like neurons, voltage-gated calcium channels (VGCCs) are important regulators of the calcium concentration by controlling the influx of calcium ions (Ca2+) across the plasma membrane [Citation4–6]. The huge signaling power of Ca2+ , which is the most widely used and at the same time most strictly controlled second messenger molecule [Citation1,Citation7], might account for the fact that mutations in VGCCs have been reported in the context of severe disorders reaching from cardiovascular channelopathies to neurological and psychiatric conditions such as ataxic and epileptic phenotypes, chronic pain, autism, schizophrenia, and depression [Citation8–10]. However, there is a growing body of evidence suggesting that the functional relevance of VGCCs goes beyond their central role as Ca2+ -conducting elements. One structural feature that might facilitate such a many-sided picture of VGCCs is their design as multi-subunit complexes. The basic Ca2+ -conducting pore is formed by the α1 subunit, a 190–270 kDa membrane-spanning protein. Today, ten variants of the pore forming α1 calcium channel subunits have been described and grouped into three families, termed CaV1, CaV2 and CaV3, based on their biophysical kinetics and pharmacological properties [Citation4,Citation11]. This diversity of calcium channel phenotypes is strongly increased by the association of the auxiliary subunits β, α2δ, and γ as well as many other regulatory proteins that interact with specific binding domains located in the intracellular loops of α1[Citation9,Citation12]. Historically, the functional importance of auxiliary VGCC subunits was considered primarily in association with the α1 pore of CaV1 and CaV2 high-voltage-activated (HVA) VGCCs. From this point of view, β and α2δ isoforms were extensively shown to promote the trafficking of the channel to the membrane, as well as to significantly modulate the biophysical properties of the multi-subunit channel complex [Citation13,Citation14]. However, several reports published over the last two decades demonstrate that VGCC auxiliary subunits can serve additional functions, which do not necessarily involve or require a direct interaction with the channel pore. Furthermore, there are studies suggesting that the pore-forming subunit of VGCCs, besides being the traditional source of Ca2+ from the extracellular space, has some side-functions, e.g. as voltage sensor and interaction partner for signaling complexes as well as in gene expression. These extended roles of VGCCs will be discussed in the following to illustrate some possibilities of how VGCCs might participate in neuronal network development, maintenance, and plasticity.
VGCCs: Not only voltage-gating but voltage-sensing
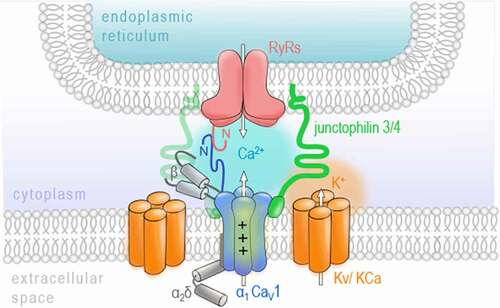
As already mentioned above, VGCCs are key regulators of the Ca2+ influx across the plasma membrane of excitable cells. In neurons, they have been widely described to shape neuronal communication by initializing the release of neurotransmitter molecules. This process mainly involves the transient influx of Ca2+ in response to the depolarization of the plasma membrane triggered by an arriving action potential and is therefore known as excitation-secretion coupling [Citation4–6]. However, besides the Ca2+-conducting aspect, a second key feature of VGCCs is their ability to sense membrane depolarization to initialize channel gating. The role VGCCs as voltage sensors was firstly described in skeletal muscle. Here, the excitation-contraction coupling does not require the influx of extracellular Ca2+ via VGCCs but rather depends on their voltage-sensing properties to trigger the release of Ca2+ from intracellular stores of the sarcoplasmic reticulum [Citation15–17]. This functional coupling involves the physical interaction of CaV1.1, a member of the CaV1 family, and calcium release channels in the sarcoplasmic reticulum called ryanodine receptors (RyR), especially the isoform 1, called RyR1. When binding to the skeletal RyR1, CaV1.1 transduces the sarcolemma depolarization to directly induce a mechanical gating of RyR1 by conformational interaction resulting in calcium release from intracellular stores [Citation18,Citation19]. Interestingly, the molecular basics and, more importantly, a similar process of voltage-induced calcium release (also called depolarization-induced calcium release) has been documented in the brain and spinal cord [Citation20–23]. Mouton and colleagues have found RyR1 channels, although poorly expressed in the brain when compared to the other RyR isoforms 2 and 3 [Citation23–27], in a complex with the pore-forming subunits of the CaV1 family members CaV1.2 and CaV1.3 in the rat brain [Citation22]. The mechanical coupling between RyR1 and CaV1 channels was later shown to be specific for CaV1.2, while RyR2 was associated with CaV1.3 in spinal cord dorsal columns and whole brain [Citation20]. A study by Kim et al. has further characterized the specific interaction of CaV1.3 and RyR2 in the rat hippocampus and demonstrated the importance of both N termini of CaV1.3 and RyR2 for their functional coupling [Citation23]. Notably, also the activation of RyR2 in hippocampal neurons seems to be independent of the Ca2+ influx through CaV1.3 since RyR2 opening was also triggered in Ca2+ -free extracellular solution [Citation23]. This highlights the function of CaV1.3 as a voltage sensor in neurons, which seems to differ from its classical role in the cardiac muscle where CaV1.3 opening has been widely reported to trigger calcium-induced calcium release [Citation28]. Although the co-localization of Cav1 isoforms and RyRs in neurons was confirmed using immunocytochemistry and super-resolution microscopy [Citation23,Citation29], it has been noted that only a few of the CaV1-RyR complexes appear co-localized and are sparsely distributed along axon cylinders [Citation20]. More recently, studies have revealed additional interaction partners, including potassium channels and junctophilin proteins, participating in a multi-protein complex on the junction between the outer plasma membrane and the ER in mammalian neurons () [Citation29,Citation30]. However, further investigation is necessary to evaluate the stability of these complexes and their functional implication for, e.g. synaptic plasticity. Suggestions have been made on a role in activity-dependent transport of signaling molecules [Citation20], neuronal excitability [Citation29], and a possible link to transmit membrane activity to gene expression in the nucleus [Citation22]. From a pathophysiological perspective, it has been shown that RyR1 mediates the release of damaging quantities of Ca2+ from the ER when triggered by ischemic depolarizations sensed by CaV1.2 in rat dorsal columns [Citation20]. Therefore, the voltage-sensing contribution of VGCCs in the release of Ca2+ from intracellular stores should be considered when studying pathophysiological elevations of calcium levels involved in, e.g. axonal damage [Citation31]. Although a participation of the Ca2+ influx through VGCCs cannot be ruled out completely, especially for CaV1.3-RyR2-complexes, this functional coupling is a first example of how VGCCs, as calcium sensors and interaction partners, can contribute to more complex signaling mechanisms.
Figure 1. VGCCs as voltage sensors
C-terminal fragments of the VGCC pore and CaVβ subunits participate in gene regulation
Since intracellular Ca2+ acts as a second messenger molecule in a plethora of signaling pathways [Citation1], it is evident that VGCCs, beeing main plasma membrane Ca2+ sources in excitable cells, contribute to transcriptional regulation processes [Citation32]. This excitation-transcription coupling allows the conversion of activity-induced, very local calcium transients into long-term effects on gene regulation pathways with distinct transcription factors. Early studies have already demonstrated that the genes regulated via VGCC-induced Ca2+ influx differ from those targeted by other Ca2+ origins, for example, receptor-activated Ca2+ channels like NMDA receptors, store-operated Ca2+ channels like ORAI and TRP members, or by Ca2+ release from intracellular stores [Citation33,Citation34]. To date, many pathways orchestrating neuronal development, survival, and communication have been identified that specifically involve VGCCs' Ca2+ signaling to activate CREB [Citation35,Citation36], NFAT [Citation37] or downstream regulator element antagonist modulator (DREAM) transcription factors [Citation38], to name only a few [Citation32]. Even if the specific contribution of VGCCs has been reported to initialize these pathways, it is the transcriptional signaling power of Ca2+ in combination with accessory Ca2+-binding proteins that allows for the nuclear forwarding and transcriptional action described above. However, several observations have been made, underpinning the idea that VGCC subunits and intracellular domains might also directly act as gene regulators ().
Figure 2. VGCCs participate in gene regulation
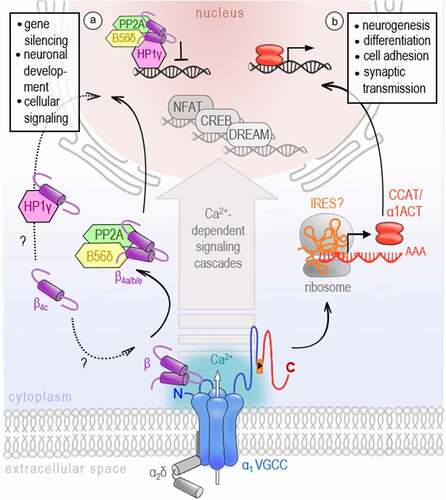
In 2006, two independent studies have reported C-terminal fragments of CaV1.2 and CaV2.1 that translocate to the nucleus of neurons in vitro and in vivo [Citation39,Citation40]. Gomez-Ospina et al. describe a so-called calcium channel associated transcriptional regulator (CCAT), a C-terminal fragment of the CaV1.2 channel subunit. Initially, it was assumed that CCAT is proteolytically processed from the full-length CaV1.2. However, later studies have shown that an internal promoter located in exon 46 of the CACNA1C gene might drive CCAT’s expression independently of the CaV1.2 channel protein [Citation41]. CCAT was verified in many different neuronal cell types throughout the brain, with a strong nuclear expression in GAD65-positive inhibitory neurons [Citation39,Citation41]. Notably, its nuclear localization is developmentally regulated and controlled by changes in intracellular calcium levels [Citation39]. In the nucleus, CCAT associates with the transcriptional regulator p54(nrb)/NonO [Citation39] and can activate transcription reporters and endogenous genes affecting, for example, cell excitability, neurite extension, and neural differentiation [Citation42]. A similar observation has been made by Kordasiewicz and colleagues, who have found a C-terminal fragment of the neuronal CaV2.1 VGCC, which they have termed α1ACT, enriched in nuclei of cerebellar neurons [Citation40]. The expression of α1ACT was proven to involve the cap-independent translation as a second gene product via an internal ribosomal entry site (IRES) located in the C-terminus of the CACNA1A gene [Citation43]. Using chromatin immunoprecipitation-based sequencing and high-throughput RNA sequencing (RNA-seq), α1ACT was shown to orchestrate a complex network of neuronal genes associated with neurogenesis (Dusp4, Efnb2, Fgfr3, Gfra2, Ntn1, Ptger3, Penk, and Odc1), synaptic transmission (Hcn4, Slc18A3, and Syn2), and cell adhesion (L1cam) essential for early cerebellar development and neonatal survival [Citation43,Citation44]. Tet-off transgenic reintroduction of α1ACT in Purkinje cells of knockin-knockout compound heterozygote mice (KIKO), which exhibit a marked reduction of the full-length CaV2.1 mRNA isoform (+exon47), improved mouse survival and early motor development demonstrating a highly age-dependent operation window of α1ACT in early life [Citation44]. Importantly, α1ACT was further shown to partially rescue the CACNA1A knockout phenotype associated with seizures, dystonia, ataxia, and death by postnatal days P18–P21 [Citation45], at behavioral, histological, and electrophysiological levels [Citation44]. These results demonstrate that both gene products, the ion channel and a nuclear protein α1ACT, play an important role in neuronal development and that in the case of perinatally decreased CACNA1A gene expression, the reintroduction of α1ACT might be a potential early intervention therapy [Citation44]. Taken together with the results from CaV1.2 and recent reports on C-terminal proteins identified across all functional VGCC classes (also CaV1.3 [Citation46] and CaV3.2 [Citation44]), it seems likely that the bicistronic expression of calcium channel proteins might be conserved across the gene family, even if a nuclear protein has not been documented for all channel isoforms, yet, and the nature of its expression mechanism is currently not fully elucidated.
Notably, not only intracellular domains of the VGCC pore have been identified to participate in transcriptional regulation, but also some isoforms of the auxiliary CaVβ subunit of the VGCC complex have been found in the nucleus. This intracellular family of VGCC subunits is widely known to support the trafficking and expression of functional VGCCs. Four isoforms of the CaVβ subunit are described (β1-β4), each having different splice variants that might interact with any α1 channel isoform in a tissue-dependent manner [Citation11,Citation47]. In addition to their essential effect on the forward trafficking of VGCCs [Citation48,Citation49], β subunits can also modulate the channel’s kinetics, reported as a shift of the activation potential and raise of the opening probability of HVA channels resulting in larger current densities [Citation14,Citation50,Citation51]. More surprisingly, several studies uncovered a calcium channel-independent function of β4 which has been shown to translocate to the nucleus and might be directly involved in activity-dependent gene regulation. At first, Hibino and colleagues have described an atypical short splice variant of the β4 subunit, namely β4C, to directly interact with the chromo shadow domain of chromobox protein 2/heterochromatin protein 1γ (CHCB2/HP1γ), a nuclear protein involved in the epigenetic control of gene regulation and gene silencing. While having only slight effects on channel activation and inactivation kinetics, the co-expression of β4C with CHCB2/HP1γ fosters the recruitment of β4C to the nuclei of mammalian cells and significantly reduces its transcriptional repression activity [Citation52]. Despite some controversial results about the molecular underpinnings, a number of studies have now confirmed the nuclear targeting of various β4 variants [Citation52–55] and their direct involvement in gene regulation via interactions with proteins of the epigenetic machinery such as HP1s [Citation52,Citation56] or the regulatory subunit of protein phosphatases-2A [Citation57,Citation58]. Notably, the subcellular localization and thus the function of β4, either as a VGCC channel subunit or transcription regulator, was shown to be under the control of electrical activity and Ca2+ influx [Citation54,Citation57]. Subramanyam et al. further interpret this activity-dependent shuttling of β4b into and out of the nucleus, and probably its switch between two independent physiological functions, as a possible mechanism of VGCCs to communicate their state of activity to the nucleus. Reconstitution experiments performed on cultured hippocampal neurons and cerebellar granule cells prepared from E17 lethargic (β4-null) mice have shown that the extent of β4 nuclear targeting significantly varied for the tested β4 splice variants which localized in neuronal nuclei with a rank order of β4b > β4a > β4e [Citation55]. The differential subcellular distribution of β4 splice variants suggests that they might regulate distinct genetic programs. Indeed, Etemad and colleagues report that the gene regulatory power of β4 splice variants correlates with their nuclear-targeting properties. However, they further point out that the nature of regulated genes, which are mainly implicated in cellular signaling, membrane/vesicle transport, and neuronal development, is quite similar for the tested β4 splice variants [Citation55]. This could indeed indicate an identical gene regulatory mechanism but remains to be explored in more detail.
α2δ subunits are many-sided extracellular interaction partners involved in synaptogenesis
In the extracellular space, VGCCs are represented by the auxiliary α2δ subunits that are attached to the outer loops of the α1 subunits. To date, four α2δ isoforms (α2δ1-α2δ4), encoded by the CACNA2D1-CACNA2D4 genes, have been identified [Citation13,Citation14]. Being transcribed from a single gene [Citation59], the α2δ protein undergoes post-translational proteolytic cleavage into α2 and δ polypeptides [Citation60] that remain linked via disulfide bonds [Citation61]. Although it is generally assumed that every α2δ isoform can associate with any HVA α1 pore-forming subunit, recent findings suggest distinct α1-preferences for some α2δ isoforms [Citation62–65]. Hence, the observed differences in the distribution and expression levels of the individual α2δ isoforms across tissues and brain regions [Citation66–71] might reflect such preferential interaction between α2δ and α1 subunits. Experimental and clinical data on α2δ knockouts or mutations in α2δ genes revealed their important role for the development of neuronal networks and establishment of excitation-to-inhibition balance. In particular, genetic aberrations in CACNA2D1 and CACNA2D2 in humans are associated with a developmental delay, mental disability and symptomatic epilepsy [Citation72,Citation73]. In several studies of normotypic and autistic individuals, mutations in CACNA2D3 were consistently identified as a risk factor for autism spectrum disorders [Citation74]. In the last few years, several new interaction partners of α2δ subunits were identified, including thrombospondin [Citation75], prion protein [Citation76], LRP1 [Citation77], BK channels [Citation78], NMDA receptors [Citation79], and α-neurexin [Citation62,Citation63]. Given the wide range of possible interactions, the idea that α2δ subunits can exert calcium channel-independent functions is increasingly gaining favor ()
Figure 3. Channel-independent functions of the auxiliary ɑ2 δ subunit
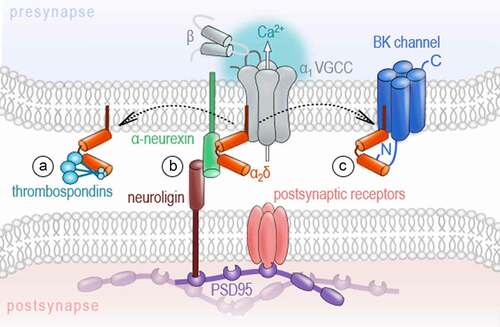
One of the most intriguing aspects related to such autonomous functions of α2δ subunits is their ability to trigger synaptogenesis in developing neurons. The α2δ1 subunit was shown to induce glutamatergic synapse formation in murine neurons through a process requiring interaction with thrombospondins, extracellular matrix proteins released by young astrocytes [Citation75]. Similarly, straightjacket (the ortholog of α2δ3) was reported to be essential for the development of excitatory synaptic boutons in Drosophila, with the extracellular α2 peptide chain being necessary and sufficient for bouton formation [Citation80]. The finding of Kurshan and colleagues that the synapse establishment was significantly impaired upon deletion of straightjacket, but was not affected by the knockout of the pore-forming subunit cacophony [Citation80], provided one of the first pieces of evidence that synaptogenic function of α2δ subunits is independent of the α1 subunit. A more recent report provided further compelling evidence for the channel-independent action of α2δ subunits on the formation of presynaptic release machinery and transsynaptic organization. In cultured hippocampal neurons and at the calyx of Held, triple knockout of CaV2.1, CaV2.2, and CaV2.3 strongly impaired evoked vesicle release but did not alter the structure of presynaptic terminals or transsynaptic organization [Citation81]. Importantly, the localization of the α2δ1 in nerve terminals was not affected by the knockout of CaV2 channels as compared to control neurons, hence demonstrating that the synaptic localization of the α2δ1 is independent of the channel pore [Citation81]. Furthermore, the total deletion of α2δ subunits in hippocampal neurons supported their relevance as a nucleation point for the formation of glutamatergic synapses [Citation82]. Consistent with the role of α2δ1 subunits in glutamatergic synaptogenesis, the overexpression of α2δ1 in adult mice leads to cortical hyperexcitability, epileptiform activity, and an increased glutamatergic synaptic density [Citation83], while a significant decrease in the number of excitatory synapses was shown for cortical neurons of α2δ1 knockout mice [Citation84]. We have recently demonstrated that α2δ subunits can trigger the formation of not only excitatory, but also inhibitory synapses in an isoform-specific manner [Citation64]. While α2δ1 subunits selectively improved neurotransmitter release in glutamatergic synapses, the upregulation of the α2δ3, but not α2δ1, resulted in a significantly higher density of GABAergic synapses and facilitated both the axonal growth in GAD67-positive interneurons and spontaneous GABA release during early development [Citation64]. Although modulatory effects on the channel pore cannot be completely ruled out, the expression, surface delivery, and major current properties of CaV2.1 and CaV2.2 channels were not affected by association with either α2δ1 or α2δ3 in heterologous expression systems [Citation64].
A strong argument for the autonomous function of α2δ subunits is the plurality of their additional interaction partners. At the nanoscale organization of a synapse, their interaction of α2δ subunits with the synaptic cell adhesion molecule α-neurexin might be particularly interesting in the context of transcellular units, so-called nanocolumns [Citation85,Citation86]. These structures have been recently reported to align the presynaptic nerve terminal, synaptic cleft, and postsynaptic compartment fostering efficient neurotransmission [Citation85,Citation86]. Similarly to α2δ subunits, α-neurexins exhibit a wide array of potential binding partners, including the postsynaptic neuroligin [Citation87,Citation88], neurexophilin [Citation89], dystroglycan [Citation90], LRRTM proteins [Citation91,Citation92], and cerebellin [Citation93]. The complex formed by neurexin isoforms and neuroligins, for example, has been described as a transsynaptic assembly found at excitatory and inhibitory synapses that is involved in synapse specification, establishment, maturation, and plasticity [Citation94]. Notably, Tong and colleagues have reported a selective interaction of the α2δ3 protein with neurexin1α[Citation62]. Since the confirmation of a specific association between α2δ isoforms and α-neurexin failed in overexpression experiments [Citation63], the physiological interaction could be regulated rather via a cell type- or synapse-specific expression of these molecules than selective binding mechanisms. It is also conceivable that slight differences in the binding affinities can dynamically influence these molecular interactions. This could transiently modulate the calcium channel properties and, in turn, synaptic transmission and plasticity in an activity- or synapse-specific manner. Indeed, other partners, besides α-neurexin, e.g. BK potassium channels [Citation80], have been also described to compete with the channel pore for the α2δ subunits [Citation78]. Moreover, the channel-to-α2δ coupling is expected to be rather loose [Citation95–97] and the analysis of surface dynamics of the α2δ1 subunits and various α1 proteins further revealed, apart from a pool of α2δ1 bound to the channel, two subpopulations of the α2δ1 and α1 subunits that are not interacting with each other [Citation98]. Whether the dissociation of the α2δ subunit from the channel pore located in the plasma membrane has direct consequences on the channel gating, stability or turn-over rates has been discussed in some studies but requires more attention to resolve the molecular mechanism [Citation78,Citation99,Citation100]. Besides gating or stability effects on the channel, changes in channel positioning or in the multi-subunit complex organization could also be possible.
To summarize, the VGCC pore-forming subunit as well as the auxiliary β and α2δ subunits play an essential role in voltage-induced calcium release, gene regulation, neuronal synaptogenesis, and transsynaptic signaling, which do not necessarily involve the Ca2+ transients of the channel complexes. The modular expression as multi-subunit complexes provides a first basis for the diversity and functional range of these molecules. We aim to further develop the idea that the functional diversity of VGCCs could not only arise from different basic channel kinetic and ion-conducting properties but rather from variations in their interaction partners or molecular environment. Some interesting findings pointing in this direction come from studies of alternative splice variants of VGCCs, which will be in the focus of the last section of this review.
Alternative splicing: How VGCC transcript variants shape neuronal phenotypes or vice versa?
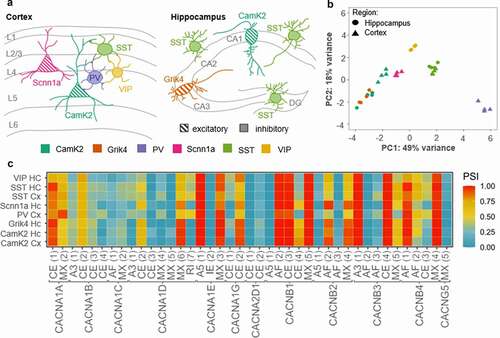
Alternative splicing is crucial for increasing the proteomic diversity of the finite number of genes, which allows for a massive expansion of the coding power of the metazoan genome [Citation101,Citation102]. To explain the high functional diversity of neuronal phenotypes observed in distinct brain areas and over development, many studies have focused on the regulatory role of alternative splicing for molecules involved in neurotransmission, including large multi exon ion channels and postsynaptic receptors [Citation103–108]. Nowadays, genome-wide analysis of splicing events strongly supports the idea of specialized splicing patterns in distinct neuronal phenotypes [Citation109–111]. In the case of VGCCs, nine of the ten genes that encode the pore-forming α1 calcium channel subunit are expressed in the mammalian brain, and each α1 subunit contains multiple sites that are hotspots for splicing events [Citation103,Citation112]. However, whether VGCCs are indeed spliced in a cell type-specific manner has not been comprehensively assessed, yet. For a first estimation, we have analyzed raw sequencing data generated by Furlanis and colleagues (accession code: GSE133291 [Citation110]; ) from ribosome-associated transcript isoforms in genetically defined neuron types of the mouse forebrain. The data indicate that some classes of neurons indeed stand out for the splicing events of VGCCs, annotated to date. Especially the class of parvalbumin (PV)-positive cells, which have been described as fast-spiking GABAergic interneurons [Citation113], show a distinct VGCC splicing pattern (). Hence, there might be a link between neuronal cell identity and VGCC splicing. The structural changes induced by alternative splicing might affect channel gating and conductance, which then leads to different functional properties of neuronal phenotypes. There is an extensive literature accumulated over the last decades that clearly demonstrates major effects of VGCCs’ splicing on the channel’s pharmacology, gating, surface expression, but also on its molecular interactions [Citation103,Citation114,Citation115]. Here, we want to present some examples, showing that alternative splicing has no direct consequence on the channel gating but rather affects their coupling to downstream interaction partners triggering different signaling pathways.
Figure 4. VGCC genes are spliced differently across neuronal cell types
One interesting VGCC splice event is the usage of two mutually exclusive exons 37a and 37b found in the C-terminus of the neuronal CaV2 calcium channels (CaV2.1 [Citation123], CaV2.2 [Citation124] and CaV2.3 [Citation116]). Independent studies have shown that the splicing of exons 37a and 37b generates two variants, EFa (or 37a) or EFb (or 37b), of the EF-hand-like Ca2+-binding motif that acts as a molecular switch for calcium-dependent facilitation and might modulate different neuronal phenotypes in a spatiotemporal manner [Citation105,Citation123–127]. The recent analysis of RNA-seq data obtained from specialized neuronal cell types mapped the differential usage of exon37 for CaV2.1 channels across excitatory and inhibitory neurons [Citation111]. Huntley et al. confirmed that the mutually exclusive usage of exon37 correlates with opposite short-term plasticity behavior observed for principal excitatory neurons versus inhibitory cells (like PV interneurons). These results are in line with the electrophysiological characterization of rat hippocampal neurons where the expression of CaV2.1[EFa] was shown to promote synaptic depression, while CaV2.1[EFb] favored synaptic facilitation [Citation105]. Thalhammer and colleagues have further demonstrated that CaV2.1[EFa] is more tightly coupled to presynaptic scaffold proteins and the neurotransmitter release machinery when compared to CaV2.1[EFb], which is characterized by a rather loose coupling [Citation105]. Although a contribution of differing biophysical properties for CaV2.1[EFa] and CaV2.1[EFb] cannot be excluded, the authors pointed out that the variation of the synaptic efficacy between these splice variants is likely due to a differential organization and molecular coupling at the presynaptic site [Citation105]. This suggests that the tighter coupling of CaV2.1[EFa], preferentially expressed in PV neurons, might be necessary to define the PV phenotype, whereas excitatory neurons use the CaV2.1[EFb] variant. As previously mentioned, the mutually exclusive usage of exon37 is conserved across neuronal CaV2 channels [Citation116,Citation123,Citation124]. For CaV2.2 channels, it has been shown that alternative splicing of the exon37 plays an important role in voltage-independent inhibition via G proteins. The inclusion of exon37a results in the expression of a tyrosine residue (Y1747), which is absent in exon37b, that triggers a voltage-independent inhibitory pathway that increases the sensitivity of CaV2.2 channels to opiates and the inhibitory neurotransmitter GABA [Citation128]. In a following study, the Lipscombe laboratory has further demonstrated the importance of activity-independent inhibition of CaV2.2 channels expressing exon37a for its function in nociceptors and morphine analgesia sensitivity in vivo, and thus the relevance for the pain pathway [Citation129].
As already indicated above, the C-terminus of neuronal VGCCs has been described for its central role for the channel’s synaptic targeting and organization [Citation108,Citation130], interaction with scaffolding proteins [Citation105,Citation131–135], G protein signaling [Citation128,Citation129], gating [Citation112,Citation125,Citation136–139], and consequently, for synaptic transmission and short-term plasticity[Citation105,Citation108]. Therefore, we now want to pay attention to another splice event in the distal part of the VGCC’s C-terminus that critically affects the length and binding sites expressed by this important structure. The expression of truncated VGCCs arising from alternative splicing events has been reported across neuronal calcium channel isoforms [Citation114,Citation140]. In the case of CaV2.1 channels, a premature stop codon results in the expression of a channel variant lacking exon47, termed ∆47, that exhibits a 150–250 amino acids (depending on the species) shorter C-terminus compared to the fully expressed exon 47 (+47) [Citation112,Citation141]. This truncation was shown to affect the channel’s trafficking, its molecular arrangement, as well as synaptic transmission properties including short-term plasticity [Citation108,Citation142]. However, only mild effects on the channel kinetics have been reported so far [Citation108,Citation142]. Moreover, a study by Aikawa et al. using a knockin mouse line, which exclusively expresses CaV2.1Δ47 (CACNA1ACtmKO/CtmKO), has shown that in cerebellar Purkinje cells, exon47 was not required to maintain such basic channel electrophysiological properties as current density-voltage relationships and channel inactivation. Instead, in the cerebellum, exon47 has a more important role in establishing channel interactions with scaffold proteins such as RIM-binding protein 2 and the auxiliary subunit β4. Surprisingly, the absence of exon47 did not only reduce interactions to scaffold proteins but also promoted the binding to GABAB2, a principal subunit of the G protein-coupled receptor for GABA, in the cerebellum of CACNA1ACtmKO/CtmKO mice. This enhancement of CaV2.1–GABAB2 interaction might contribute to the pathogenesis of absence seizures and motor incoordination observed in these animals [Citation143–147].
These examples demonstrate that changes in the structure of VGCCs induced by splicing events also affect their interaction to partner molecules and suggest that significant functional implications cannot be reduced to their ionic activity.
Outlook and perspectives
Although this review cannot fully capture the functional range of VGCCs, we aimed to draw attention to some side functions of VGCCs that go beyond their classical role as ion channels to induce local Ca2+ nanodomains necessary for neurotransmission or downstream Ca2+ signaling pathways. When associated with ryanodine receptors, VGCCs contribute to a functional complex to trigger voltage-induced Ca2+ release from intracellular stores, a process that is independent of their ionic activity. Further, VGCC auxiliary subunits as well as C-terminal domains of the α1 channel pore participate in gene regulation. Their activity- and Ca2+-dependent subcellular shuttling implicate a pathway how VGCCs communicate their own activity status to the nucleus and integrate transient Ca2+ signaling into longer-lasting transcriptional processes. These transcriptomic alterations should be considered especially when evaluating the phenotype of channelopathies, where most of the studies have primarily focused on explaining the pathological consequences based on the loss or gain of calcium channel function so far. Moreover, we have shown that α2δ auxiliary subunits are directly involved into neuronal network development and maintenance by fostering excitatory and inhibitory synaptogenesis and transsynaptic signaling. We have introduced some examples where structural changes in critical VGCC domains result in distinct synaptic plasticity behaviors and contribute, at least to some extent, to the specification of neuronal phenotypes. Notably, these changes in the channel structure do not necessarily involve differences in basic channel gating but might rather involve different protein environments or downstream pathways. Considering the many interaction partners of VGCCs, and from a molecular dynamics point of view, we want to point out the possibility that these large molecules could also serve as seeding points for molecular interactions. The question remains open at this point to what extent VGCCs can shape their environment by acting as a basic element bringing together important signaling molecules or how specific interaction partners that are present, e.g. in distinct neuronal phenotypes, regulate VGCCs’ functional diversity.
Disclosure of interest
The authors declare that there are no competing interests associated with this manuscript.
Abbreviations
| α1ACT | = | C-terminal fragment of the neuronal CaV2.1 VGCC |
| A3 | = | alternative 3ʹ splice site |
| A5 | = | alternative 5ʹ splice site |
| AF | = | alternative 3ʹ splice site |
| BK | = | big potassium |
| Ca2+ | = | calcium ions |
| CamK2 | = | Ca2+/calmodulin-dependent protein kinase II |
| CCAT | = | calcium channel associated transcriptional regulator |
| CE | = | cassette exon |
| CHCB2/HP1γ | = | chromobox protein 2/heterochromatin protein 1γ |
| CREB | = | cAMP response element-binding protein |
| DREAM | = | downstream regulator element antagonist modulator |
| ER | = | endoplasmic reticulum |
| GABA | = | gamma-aminobutyric acid |
| GAD65/GAD67 | = | glutamic acid decarboxylase 65/67 |
| Grik4 | = | glutamate receptor, ionotropic, kainate 4 |
| HVA | = | high voltage-activated |
| IRES | = | internal ribosomal entry site |
| Kv | = | voltage-gated potassium channels |
| KCa | = | callcium-activated potassium channels |
| KIKO | = | knockin-knockout compound heterozygote mice |
| LRRM | = | leucine -rich repeat transmembrane proteins |
| mEPSCs | = | miniature excitatory postsynaptic currents |
| mIPSCs | = | miniature inhibitory postsynaptic currents |
| MX | = | mutually exclusive exon |
| NFAT | = | nuclear factor of activated T-cells |
| NMDA | = | N-methyl-D-aspartate |
| ORAI | = | calcium release-activated calcium channel protein |
| PCA | = | principal component analysis |
| PP2A | = | protein phosphatases 2A |
| PSI | = | percentage spliced in |
| PV | = | parvalbumin |
| RI | = | retained intron |
| RNA-seq | = | high-throughput RNA sequencing |
| RyR | = | ryanodine receptor |
| SCA6 | = | spinocerebellar ataxia type 6 |
| Scnn1a | = | α subunit of the epithelial sodium channel ENaC |
| SST | = | somatostatin |
| Tet | = | tetracycline |
| TRP | = | transient receptor potential channels |
| VGCC | = | voltage-gated calcium channel |
| VIP | = | vasoactive intestinal peptide |
Acknowledgements
We thank the Scheiffele Lab from the University of Basel for sharing the extensive dataset of ribosome-associated mRNAs from major neuron classes published in Furlanis et al. 2019 and for interesting discussions about neuronal cell class-specific alternative splicing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Clapham D. Calcium signaling. Cell. 1995;80(2):259–268.
- Brini M, Calì T, Ottolini D, et al. Intracellular calcium homeostasis and signaling. 2013.
- Campbell A Intracellular calcium. 2015.Intracellular Ca2+ – Principles and Terminology. Intracellular Calcium, 2014:39-80.
- Catterall WA. Voltage-Gated Calcium Channels. Cold Spring Harbor Perspectives in Biology. 2011; 3:a003947
- Tsien RW, Lipscombe D, Madison DV, et al. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11(10):431–438.
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18(2):89–98.
- Carafoli E, Krebs J. Why calcium? How calcium became the best communicator. J Biol Chem. 2016;291(40):20849–20857.Carafoli E, Krebs J. Why Calcium? How Calcium Became the Best Communicator. J Biol Chem 2016; 291:20849-57.
- Zamponi GW, Striessnig J, Koschak A, et al. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67:821–870.
- Dolphin AC. Voltage‐gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594(19):5369–5390.Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 2016; 594:5369-90.
- Striessnig J. Voltage-gated calcium channels - from basic mechanisms to disease. J Physiol. 2016;594(20):5817–5821.
- Lacinova L. Voltage-dependent calcium channels. Gen Physiol Biophys. 2005;24 Suppl 1:24.
- Campiglio M, Flucher BE. The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J Cell Physiol. 2015;230(9):2019–2031.
- Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13(3):298–307.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol 2003; 13:298-307.
- Dolphin AC. Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13(8):542.
- Melzer W, Herrmann-Frank A, Lüttgau H. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. 1995;1241:59–116. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes.
- Leong P, MacLennan DH. Complex interactions between skeletal muscle ryanodine receptor and dihydropyridine receptor proteins. Biochem Cell Biol. 1998;76(5):681–694.
- Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–729.Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev 1997; 77:699-729.
- Lu X, Xu L, Meer G. Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J Biol Chem. 1994;269(9):6511–6516.
- Marty I, Robert M, Villaz M, et al. Biochemical evidence for a complex involving dihydropyridine receptor and ryanodine receptor in triad junctions of skeletal muscle. Proc Natl Acad Sci U S A. 1994;91(6):2270–2274.
- Ouardouz M, Nikolaeva MA, Coderre E, et al. Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron. 2003;40(1):53–63.
- Chavis P, Fagni L, Lansman JB, et al. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382(6593):719–722.
- Mouton J, Marty I, Villaz M, et al. Molecular interaction of dihydropyridine receptors with type-1 ryanodine receptors in rat brain. Biochem J. 2001;354(3):597–603.
- Kim S, Yun H-M, Baik J-H, et al. Functional interaction of neuronal Cav1.3 L-type calcium channel with ryanodine receptor Type 2 in the rat hippocampus *. J Biol Chem. 2007;282(45):32877–32889.
- Furuichi T, Furutama D, Hakamata Y, et al. Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J Neurosci. 1994;14(8):4794.
- Kuwajima G, Futatsugi A, Niinobe M, et al. Two types of ryanodine receptors in mouse brain: skeletal muscle type exclusively in Purkinje cells and cardiac muscle type in various neurons. Neuron. 1992;9(6):1133–1142.
- Walton PD, Airey JA, Sutko JL, et al. Ryanodine and inositol trisphosphate receptors coexist in avian cerebellar purkinje neurons. J Cell Biol. 1991;113(5):1145–1157.
- Giannini G, Conti A, Mammarella S, et al. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J Cell Biol. 1995;128(5):893–904.
- Striessnig J, Pinggera A, Kaur G, et al. L-type Ca2+ channels in heart and brain. Wiley Interdisciplinary Reviews: Membrane Transport and Signaling. 2014;3(2):15–38. DOI:https://doi.org/10.1002/wmts.102
- Sahu G, Wazen R-M, Colarusso P, et al. Junctophilin proteins tether a Cav1-RyR2-KCa3.1 tripartite complex to regulate neuronal excitability. Cell Rep. 2019;28(9):2427–2442.e6.
- Vierra NC, Kirmiz M, Van Der List D, et al. Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. eLife. 2019;8:8:e49953.
- Stys PK. White matter injury mechanisms. Curr Mol Med. 2004;4(2):113–130.
- Barbado M, Fablet K, Ronjat M, et al. Gene regulation by voltage-dependent calcium channels. 2009;1793:1096–1104. Biochim Biophys Acta, Mol Cell Res.
- Greenberg M, Ziff E, Greene L. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science. 1986;234(4772):80.
- Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322(6079):552–555.
- Dolmetsch RE, Pajvani U, Fife K, et al. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294(5541):333–339.
- Zhao R, Liu L, Rittenhouse AR. Ca2+ influx through both L- and N-type Ca2+ channels increases c-fos expression by electrical stimulation of sympathetic neurons. Eur J Neurosci. 2007;25(4):1127–1135.
- Hernández-Ochoa EO, Contreras M, Cseresnyés Z, et al. Ca2+ signal summation and NFATc1 nuclear translocation in sympathetic ganglion neurons during repetitive action potentials. Cell Calcium. 2007;41(6):559–571.
- Leclerc GM, Boockfor FR. Calcium influx and DREAM protein are required for GnRH gene expression pulse activity. Mol Cell Endocrinol. 2007;267(1–2):70–79.
- Gomez-Ospina N, Tsuruta F, Barreto-Chang O, et al. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127(3):591–606.
- Kordasiewicz HB, Thompson RM, Clark HB, et al. C-termini of P/Q-type Ca 2+ channel α1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum Mol Genet. 2006;15(10):1587–1599.
- Gomez-Ospina N, Panagiotakos G, Portmann T, et al. A promoter in the coding region of the calcium channel gene CACNA1C generates the transcription factor CCAT. PLoS One. 2013;8(4):e60526–e60526.
- Ge J, Ju Y, Xue Z, et al. Distal C terminus of CaV1.2 channels plays a crucial role in the neural differentiation of dental pulp stem cells. PLoS One. 2013;8(11):e81332–e81332.
- Du X, Wang J, Zhu H, et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell. 2013;154(1):118–133.
- Du X, Wei C, Hejazi Pastor DP, et al. α1ACT is essential for survival and early cerebellar programming in a critical neonatal window. Neuron. 2019;102(770–785):e7. DOI:https://doi.org/10.1016/j.neuron.2019.02.036
- Jun K, Piedras-Rentería ES, Smith SM, et al. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proc Natl Acad Sci USA. 1999;96(26):15245.
- Lu L, Sirish P, Zhang Z, et al. Regulation of gene transcription by voltage-gated L-type calcium channel, Cav1.3. J Biol Chem. 2015;290(8):4663–4676.
- Dolphin AC. β subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35(6):599–620.
- Altier C, Garcia-Caballero A, Simms B, et al. The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci. 2010;14(2):173.Altier C, Garcia-Caballero A, Simms B, You H, Chen L, Walcher J, et al. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci 2011; 14:173-80.
- Waithe D, Ferron L, Page KM, et al. β-Subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradation. The. J Biol Chem. 2011(286):9598–9611.Waithe D, Ferron L, Page KM, Chaggar K, Dolphin AC. Beta-subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradation. J Biol Chem 2011; 286:9598-611.
- Meir A, Bell DC, Stephens GJ, et al. Calcium channel beta subunit promotes voltage-dependent modulation of alpha 1 B by G beta gamma. Biophys J. 2000;79(2):731–746.Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel beta subunit promotes voltage-dependent modulation of alpha 1 B by G beta gamma. Biophys J 2000; 79:731-46.
- Neely A, Garcia-Olivares J, Voswinkel S, et al. Folding of active calcium channel β1b -subunit by size-exclusion chromatography and its role on channel function. J Biol Chem. 2004;279(21):21689–21694.Neely A, Garcia-Olivares J, Voswinkel S, Horstkott H, Hidalgo P. Folding of active calcium channel beta(1b) -subunit by size-exclusion chromatography and its role on channel function. J Biol Chem 2004; 279:21689-94.
- Hibino H, Pironkova R, Onwumere O, et al. Direct interaction with a nuclear protein and regulation of gene silencing by a variant of the Ca2+-channel beta 4 subunit. Proc Natl Acad Sci U S A. 2003;100(1):307–312.
- Colecraft HM, Alseikhan B, Takahashi SX, et al. Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541(2):435–452.
- Subramanyam P, Obermair GJ, Baumgartner S, et al. Activity and calcium regulate nuclear targeting of the calcium channel beta4b subunit in nerve and muscle cells. Channels (Austin, Tex). 2009;3(5):343–355.
- Etemad S, Obermair GJ, Bindreither D, et al. Differential neuronal targeting of a new and two known calcium channel β4 subunit splice variants correlates with their regulation of gene expression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34(4):1446–1461. DOI:https://doi.org/10.1523/JNEUROSCI.3935-13.2014
- Xu X, Lee YJ, Holm JB, et al. The Ca2+ Channel β4c subunit interacts with heterochromatin protein 1 via a PXVXL binding motif. J Biol Chem. 2011;286(11):9677–9687.
- Tadmouri A, Kiyonaka S, Barbado M, et al. Cacnb4 directly couples electrical activity to gene expression, a process defective in juvenile epilepsy. Embo J. 2012;31(18):3730–3744.
- Ronjat M, Kiyonaka S, Barbado M, et al. Nuclear life of the voltage-gated Cacnb4 subunit and its role in gene transcription regulation. null. 2013;7:119–125.
- De Jongh KS, Warner C, Catterall WA. Subunits of purified calcium channels. Alpha 2 and delta are encoded by the same gene. J Biol Chem. 1990;265:14738–14741.
- Jay SD, Sharp AH, Kahl SD, et al. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J Biol Chem. 1991;266(5):3287–3293.
- Wu J, Yan Z, Li Z, et al. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 Å resolution. Nature. 2016;537(7619):191.
- Tong X-J, López-Soto EJ, Li L, et al. Retrograde synaptic inhibition is mediated by α-neurexin binding to the α2δ subunits of N-type calcium channels. Neuron. 2017;95(326–340):e5.
- Brockhaus J, Schreitmüller M, Repetto D, et al., α-neurexins together with α2δ-1 auxiliary subunits regulate Ca 2+ Influx through Ca v 2.1 channels. J Neurosci. Internet. 2018;38(38):8277–8294. Available from http://www.jneurosci.org/content/early/2018/08/13/JNEUROSCI.0511-18.2018.abstract
- Bikbaev A, Ciuraszkiewicz-Wojciech A, Heck J, et al. Auxiliary α2δ1 and α2δ3 subunits of calcium channels drive excitatory and inhibitory neuronal network development. J Neurosci. 2020;40(25):4824. DOI:https://doi.org/10.1523/JNEUROSCI.1707-19.2020
- Heinrich L, Ryglewski S. Different functions of two putative drosophila α2δ subunits in the same identified motoneurons. Sci Rep. 2020;10(1):13670.
- Klugbauer N, Lacinová L, Marais E, et al. Molecular diversity of the calcium channel α 2 δ subunit. J Neurosci. 1999;19(2):684–691.
- Gong HC, Hang J, Kohler W, et al. Tissue-specific expression and gabapentin-binding properties of calcium channel α2δ subunit subtypes. J Membr Biol. 2001;184(1):35–43.
- Klugbauer N, Marais E, Hofmann F. Calcium channel α2δ subunits: differential expression, function, and drug binding. J Bioenerg Biomembr. 2003;35(6):639–647.
- Cole RL, Lechner SM, Williams ME, et al. Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol. 2005;491(3):246–269.
- Davies A, Hendrich J, Van Minh AT, et al. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007;28(5):220–228.
- Schlick B, Flucher BE, Obermair GJ. Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience. 2010;167(3):786–798.
- Edvardson S, Oz S, Abulhijaa FA, et al. Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet. 2013;50(2):118.
- Vergult S, Dheedene A, Meurs A, et al. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet. 2015;23(5):628–632.
- Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–299. DOI:https://doi.org/10.1016/j.neuron.2012.04.009
- Eroglu Ç, Allen NJ, Susman MW, et al. The gabapentin receptor α2δ-1 is the neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139(2):380–392. DOI:https://doi.org/10.1016/j.cell.2009.09.025
- Senatore A, Colleoni S, Verderio C, et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α(2)δ-1 subunit. Neuron. 2012;74(2):300–313. DOI:https://doi.org/10.1016/j.neuron.2012.02.027
- Kadurin I, Rothwell SW, Lana B, et al. LRP1 influences trafficking of N-type calcium channels via interaction with the auxiliary α(2)δ-1 subunit. Sci Rep. 2017;7(1):43802.
- Zhang F-X, Gadotti VM, Souza IA, et al. Suppress Cavα2δ subunit function to reduce inflammatory and neuropathic pain. Cell Rep. 2018;22(8):1956–1964.
- Chen J, Li L, Chen S-R, et al. The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 2018;22(9):2307–2321. DOI:https://doi.org/10.1016/j.celrep.2018.02.021
- Kurshan PT, Oztan A, Schwarz TL. Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat Neurosci. 2009;12(11):1415–1423.
- Held RG, Liu C, Ma K, et al. Synapse and active zone assembly in the absence of presynaptic Ca2+ channels and Ca2+ Entry. Neuron. 2020;107(667–683):e9. DOI:https://doi.org/10.1016/j.neuron.2020.05.032
- Schöpf CL, Geisler S, Stanika RI, et al. Presynaptic α2δ subunits are key organizers of glutamatergic synapses. bioRxiv. 2019;826016.
- Faria LC, Gu F, Parada I, et al. Epileptiform activity and behavioral arrests in mice overexpressing the calcium channel subunit α2δ-1. Neurobiol Dis. 2017;102:70–80.
- Risher WC, Kim N, Koh S, et al. Thrombospondin receptor α2δ-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol. 2018;217(10):3747–3765. DOI:https://doi.org/10.1083/jcb.201802057
- Tang A-H, Chen H, Li TP, et al. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature. 2016;536(7615):210.
- Biederer T, Kaeser PS, Blanpied TA. Transcellular nanoalignment of synaptic function. Neuron. 2017;96:680–696.
- Ichtchenko K, Hata Y, Nguyen T, et al. Neuroligin 1: a splice site-specific ligand for β-neurexins. Cell. 1995;81(3):435–443.
- Boucard AA, Chubykin AA, Comoletti D, et al. Code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to α- and β-neurexins. Neuron. 2005;48(2):229–236.
- Missler M, Hammer RE, Südhof TC. Neurexophilin binding to α-Neurexins: a single LNS domain functions as an independently folding ligand-binding unit. J Biol Chem. 1998;273(52):34716–34723.
- Sugita S, Saito F, Tang J, et al. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154(2):435–446.
- Ko J, Fuccillo MV, Malenka RC, et al. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64(6):791–798.
- De Wit J, Sylwestrak E, O’Sullivan ML, et al. LRRTM2 interacts with neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64(6):799–806.
- Uemura T, Lee S-J, Yasumura M, et al. Trans-synaptic interaction of GluRδ2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141(6):1068–1079.
- Reissner C, Runkel F, Missler M. Neurexins. Genome Biol. 2013;14(9):213.
- Gee NS, Brown JP, Dissanayake VUK, et al. The novel anticonvulsant drug, gabapentin (neurontin), binds to the subunit of a calcium channel. J Biol Chem. 1996;271(10):5768–5776.
- Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent ca2+ channel α2δ subunit in current stimulation and subunit interaction. Neuron. 1996;16(2):431–440.
- Müller CS, Haupt A, Bildl W, et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A. 2010;107(34):14950–14957. DOI:https://doi.org/10.1073/pnas.1005940107
- Voigt A, Freund R, Heck J, et al. Dynamic association of calcium channel subunits at the cellular membrane. Neurophotonics. 2016;3(4):041809.
- Kadurin I, Alvarez-Laviada A, Ng SFJ, et al. Calcium currents are enhanced by α2δ-1 lacking its membrane anchor. J Biol Chem. 2012;287(40):33554–33566.
- Davies A, Kadurin I, Alvarez-Laviada A, et al. The α(2)δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A. 2010;107(4):1654–1659.
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–463.
- Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12(10):715–729.
- Lipscombe D, Andrade A, Allen SE. Alternative splicing: functional diversity among voltage-gated calcium channels and behavioral consequences. Biochim Biophys Acta. 2013;1828(7):1522–1529.
- Zandany N, Marciano S, Magidovich E, et al. Alternative splicing modulates Kv channel clustering through a molecular ball and chain mechanism. Nat Commun. 2015;6(1):6488.
- Thalhammer A, Contestabile A, Ermolyuk YS, et al. Alternative splicing of P/Q-type Ca2+ channels shapes presynaptic plasticity. Cell Rep. 2017;20(2):333–343.
- Regan MC, Grant T, McDaniel MJ, et al. Structural mechanism of functional modulation by gene splicing in NMDA receptors. Neuron. 2018;98(521–529):e3.
- Bunda A, LaCarubba B, Bertolino M, et al. Cacna1b alternative splicing impacts excitatory neurotransmission and is linked to behavioral responses to aversive stimuli. Mol Brain. 2019;12(1):81.
- Heck J, Parutto P, Ciuraszkiewicz A, et al. Transient confinement of CaV2.1 Ca2+-channel splice variants shapes synaptic short-term plasticity. Neuron. 2019;103(1):66–79.e12.
- Zhang X, Chen MH, Wu X, et al. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell. 2016;166(1147–1162):e15. DOI:https://doi.org/10.1016/j.cell.2016.07.025
- Furlanis E, Traunmüller L, Fucile G, et al. Landscape of ribosome-engaged transcript isoforms reveals extensive neuronal-cell-class-specific alternative splicing programs. Nat Neurosci. 2019;22(10):1709–1717.
- Huntley MA, Srinivasan K, Friedman BA, et al. Genome-wide analysis of differential gene expression and splicing in excitatory neurons and interneuron subtypes. J Neurosci. 2020;40(5):958.
- Soong TW, DeMaria CD, Alvania RS, et al. Systematic identification of splice variants in human P/Q-type channel α12.1 subunits: implications for current density and Ca2+-dependent inactivation. J Neurosci. 2002;22(23):10142.
- Kawaguchi Y, Kubota Y. Neurochemical features and synaptic connections of large physiologically-identified GABAergic cells in the rat frontal cortex. Neuroscience. 1998;85(3):677–701.
- Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflügers Archiv - European Journal of Physiology. 2009;458(3):481–487. DOI:https://doi.org/10.1007/s00424-009-0635-5
- Lipscombe D, Andrade A. Calcium channel CaVα1 splice isoforms - tissue specificity and drug action. Curr Mol Pharmacol. 2015;8(1):22–31.
- Gray AC, Raingo J, Lipscombe D. Neuronal calcium channels: splicing for optimal performance. Cell Calcium. 2007;42(4–5):409–417.
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
- Team RC. R: a language and environment for statistical computing. Austria: Vienna; 2013.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Wickham H. Programming with ggplot2. ggplot2. Springer: 2016. page 241–253.
- Patro R, Duggal G, Love MI, et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–419.
- Trincado JL, Entizne JC, Hysenaj G, et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018;19(1):40.
- Bourinet E, Soong TW, Sutton K, et al. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2(5):407.
- Bell TJ, Thaler C, Castiglioni AJ, et al. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41(1):127–138.
- Chaudhuri D, Chang S-Y, DeMaria CD, et al. Alternative splicing as a molecular switch for Ca2+/Calmodulin-dependent facilitation of P/Q-Type Ca2+ Channels. J Neurosci. 2004;24(28):6334.
- Chaudhuri D, Alseikhan BA, Chang SY, et al. Developmental activation of calmodulin-dependent facilitation of cerebellar P-type Ca2+ current. J Neurosci. 2005;25(36):8282.
- Chang SY, Yong TF, Yu CY, et al. Age and gender-dependent alternative splicing of P/Q-type calcium channel EF-hand. Neuroscience. 2007;145(3):1026–1036.
- Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10(3):285–292.
- Andrade A, Denome S, Jiang Y-Q, et al. Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nat Neurosci. 2010;13(10):1249–1256.
- Maximov A, Bezprozvanny I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J Neurosci. 2002;22(16):6939.
- Maximov A, Südhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999;274(35):24453–24456.
- Hibino H, Pironkova R, Onwumere O, et al. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca2+ channels. Neuron. 2002;34(3):411–423.
- Kaeser PS, Deng L, Wang Y, et al. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144(2):282–295.
- Davydova D, Marini C, King C, et al. Bassoon specifically controls presynaptic P/Q-type Ca2+ Channels via RIM-binding protein. Neuron. 2014;82(1):181–194. DOI:https://doi.org/10.1016/j.neuron.2014.02.012
- Hirano M, Takada Y, Wong CF, et al. C-terminal splice variants of P/Q-type Ca(2+) channel Ca(V)2.1 α(1) subunits are differentially regulated by Rab3-interacting molecule proteins. The. J Biol Chem. 2017(292):9365–9381.
- Lee A, Wong ST, Gallagher D, et al. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399(6732):155.
- Lee A, Scheuer T, Catterall WA. Ca2+/Calmodulin-dependent facilitation and inactivation of P/Q-Type Ca2+. J Neurosci. 2000;20(18):6830.
- DeMaria CD, Soong TW, Alseikhan BA, et al. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411(6836):484.
- Moreno CM, Dixon RE, Tajada S, et al. Ca2+ entry into neurons is facilitated by cooperative gating of clustered CaV1.3 channels. eLife. 2016;5:e15744.
- Lipscombe D, Castiglioni AJ. Alternative Splicing in Voltage Gated Calcium Channels. Internet]. In: McDonough SI, editor. Calcium Channel Pharmacology. Boston, MA: Springer US; 2004. page 369–409. Available from: https://doi.org/https://doi.org/10.1007/978-1-4419-9254-3_11
- Krovetz HS, Helton TD, Crews AL, et al. C-terminal alternative splicing changes the gating properties of a human spinal cord calcium channel α1A Subunit. J Neurosci. 2000;20(20):7564.
- Adams PJ, Garcia E, David LS, et al. CaV2.1 P/Q-type calcium channel alternative splicing affects the functional impact of familial hemiplegic migraine mutations: implications for calcium channelopathies. Channels. 2009;3:(2):110–121.
- Aikawa T, Watanabe T, Miyazaki T, et al. Alternative splicing in the C-terminal tail of Cav2.1 is essential for preventing a neurological disease in mice. Hum Mol Genet. 2017;26(16):3094–3104. DOI:https://doi.org/10.1093/hmg/ddx193
- Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat Genet. 1997;15(1):62.
- Ishiguro T, Ishikawa K, Takahashi M, et al. The carboxy-terminal fragment of α1A calcium channel preferentially aggregates in the cytoplasm of human spinocerebellar ataxia type 6 Purkinje cells. Acta Neuropathol. 2010;119(4):447–464. DOI:https://doi.org/10.1007/s00401-009-0630-0
- Heyes S, Pratt WS, Rees E, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. 2015;134:36–54.
- Clark MB, Wrzesinski T, Garcia AB, et al. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol Psychiatry. 2020;25(1):37–47.