
ABSTRACT
The mitochondrial BKCa channel (mitoBKCa) is a splice variant of plasma membrane BKCa (Maxi-K, BKCa, Slo1, KCa1.1). While a high-resolution structure of mitoBKCa is not available yet, functional and structural studies of the plasma membrane BKCa have provided important clues on the gating of the channel by voltage and Ca2+, as well as the interaction with auxiliary subunits. To date, we know that the control of expression of mitoBKCa, targeting and voltage-sensitivity strongly depends on its association with its regulatory β1-subunit, which overall participate in the control of mitochondrial Ca2+-overload in cardiac myocytes. Moreover, novel regulatory mechanisms of mitoBKCa such as β-subunits and amyloid-β have recently been proposed. However, major basic questions including how the regulatory BKCa-β1-subunit reaches mitochondria and the mechanism through which amyloid-β impairs mitoBKCa channel function remain to be addressed.
Introduction
Mitochondrial ion channels play key roles in maintaining and regulating mitochondrial volume, biogenesis, membrane potential, Ca2+ homeostasis, metabolism and a broad spectrum of physiological processes derived from mitochondrial physiology including cell death. Amongst other mitochondrial ion channels so far described, mitoBKCa channel received special attention since its pharmacological activation exerts cardioprotection, as early noted by the O’Rourke laboratory in 2002 [Citation1]. The genetic ablation of BKCa in cardiac myocytes expanded these early observations and revealed that mitoBKCa is a splice variant of plasma membrane BKCa, and that its targeting to the inner mitochondrial membrane (IMM) strongly depends on a short stretch of amino acids located at the C-terminal (the DEC sequence) [Citation2]. The molecular recognition of the mitoBKCa-DEC segment by members of the mitochondrial outer membrane import system (TOM) assures proper target of the channel into the IMM [Citation3]. Moreover, recent findings have revealed that the association of mitoBKCa with auxiliary BK-β1 subunit regulates its expression, targeting, and shifts the channel’s voltage sensitivity, which has important physiological consequences on mitochondrial Ca2+ handling [Citation4]. Novel mechanisms of regulation of mitoBKCa channels such as amyloid-β (Aβ) are also emerging. Aβ is a peptide that accumulates in brain in Alzheimer´s Disease and inhibits the activity of mitoBKCa channels [Citation5]. Confirmation of this effect by other laboratories might have important implications in our understanding of AD and possibly on the development of new treatments for neurodegenerative diseases. In this review, we will expand our discussion of the basic properties of mitoBKCa contained in the most recent papers. We will start discussing the evolution and conservation of the mitochondrial targeting sequence (DEC) amongst vertebrates and the most recently discovered mechanisms that control the expression, targeting and activation of this channel in mitochondria. In the last part of this review, we discuss the novel modes of regulation of mitoBKCa channels and its fundamental roles in mitochondrial and cell physiology.
The molecular nature of mitoBKCa
In rodent cardiomyocytes, mitoBKCa is a splice variant of the plasma membrane BKCa, KCNMA1 [Citation2]. Unlike the canonical mitochondrial targeting relying on signal peptides at the N-terminal, the mitoBKCa possesses a 50 amino acid sequence at the C-terminal denominated as “DEC” after the last three amino acids () that promotes the import of the channel to the IMM [Citation2].
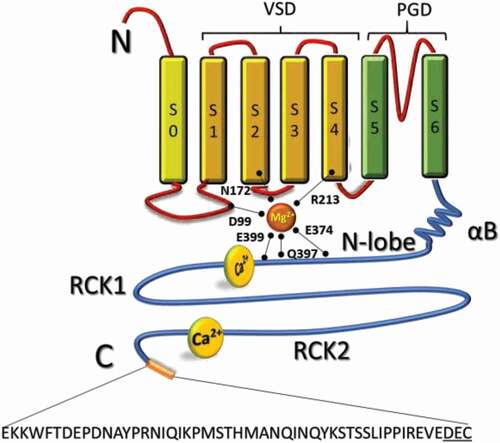
Figure 1. Structural components of the mitoBKCa channel α subunit. Schematic topology of mitoBKCa α subunit. The α-subunit is composed by 7 transmembrane domains (S0-S6) and N-terminal (cytoplasmic) and C-terminal (matrix) opposites. The S1-S4 domains constitute the voltage sensing domain (VSD) and the S5-S6 domain conform the pore gating domain (PGD). The C-terminal domain (CTD) is connected to the transmembrane domain through an alpha helix/beta-sheet linker (αB), each connecting S6 to the rest of the N-lobe of the Regulator of Potassium Conductance (RCK) 1 domain (residues 344–613). The “gating-ring” contains residues D99; N172; R213; E374; Q397 and E399 important for activation of the channel by Mg2+; and a second RCK domain (residues 718–1056). High affinity Ca2+-binding sites located at RCK 1 and 2 conform the “Ca2+-bowl”. At the end of the CTD a 50 amino acid insert contains the DEC sequence specific to target mitoBKCa channel. Four α subunits form a functional channel
The genetic origin of mitoBKCa-DEC sequence
To understand the evolutionary relationship between the BKCa and the DEC sequence we performed a BLASTp and tBLASTx search for both sequences in the NCBI database (https://www.ncbi.nlm.nih.gov). Interestingly, the hits for the DEC sequence were solely present in proteins annotated as BKCa or SLO-like channels, suggesting a novel and probably unique mechanism for mitochondrial import of these proteins. To determine if the hits obtained for the BKCa belonged to bona fide BKCa channels we searched for the Ca2+ bowl sequence and the highly conserved GYG (or GxGD) pore motif. For our surprise, while bona fide BKCa channels are widely conserved throughout the animal kingdom (), the DEC sequence is only present in vertebrates showing a high degree of conservation among this group (), suggesting a conserved mechanism for mitochondrial targeting.
Figure 2. Conservation of BKCa-DEC sequence. (A) A simplified metazoan phylogeny is represented describing the presence or absence of BKCa and DEC sequences in the examined taxa. Taxonomic groups belonging to vertebrates and invertebrates are depicted in black and gray font, respectively. The tree topology was done by phyloT based on the NCBI taxonomy and visualized in the interactive Tree of Life tool (https://itol.embl.de/itol.cgi). Solid color boxes indicate that the correspondent sequence was identified in all the organisms searched for that taxonomic group. Half-filled boxes indicate that at least one, but not all, of the organisms belonging to that taxonomic group possess the sequence. A white box represents the cases where the sequence was not found; in the case of dipnoi we cannot assure complete absence since only genomic traces and transcriptomic data were available. The search of BKCa (KCNMA1) and its DEC sequence were done in NCBI database, using their respective Homo sapiens sequence as initial query for BLASTp and tBLASTx. Additional rounds of BLAST searches were performed using hit sequences from the first round to identify potential distantly related homologs that might not be detected by using the initial query sequences. To corroborate that the sequences obtained belonged to bona fide BKCa channels a search for the Ca2+ bowl sequence [Citation6] and the GYG (or GxGD) pore motif [Citation71] was performed posteriori. (B) Alignment of DEC sequences found in vertebrates. The colored bar at left indicates the taxonomic group, pink for mammals, yellow for birds, green for reptiles, turquoise for amphibians, light blue for bony fishes (actinopterygians plus coelacanths), dark blue for Chondrichthyes (cartilaginous fishes) and purple for cyclostomes (jawless fish)
![Figure 2. Conservation of BKCa-DEC sequence. (A) A simplified metazoan phylogeny is represented describing the presence or absence of BKCa and DEC sequences in the examined taxa. Taxonomic groups belonging to vertebrates and invertebrates are depicted in black and gray font, respectively. The tree topology was done by phyloT based on the NCBI taxonomy and visualized in the interactive Tree of Life tool (https://itol.embl.de/itol.cgi). Solid color boxes indicate that the correspondent sequence was identified in all the organisms searched for that taxonomic group. Half-filled boxes indicate that at least one, but not all, of the organisms belonging to that taxonomic group possess the sequence. A white box represents the cases where the sequence was not found; in the case of dipnoi we cannot assure complete absence since only genomic traces and transcriptomic data were available. The search of BKCa (KCNMA1) and its DEC sequence were done in NCBI database, using their respective Homo sapiens sequence as initial query for BLASTp and tBLASTx. Additional rounds of BLAST searches were performed using hit sequences from the first round to identify potential distantly related homologs that might not be detected by using the initial query sequences. To corroborate that the sequences obtained belonged to bona fide BKCa channels a search for the Ca2+ bowl sequence [Citation6] and the GYG (or GxGD) pore motif [Citation71] was performed posteriori. (B) Alignment of DEC sequences found in vertebrates. The colored bar at left indicates the taxonomic group, pink for mammals, yellow for birds, green for reptiles, turquoise for amphibians, light blue for bony fishes (actinopterygians plus coelacanths), dark blue for Chondrichthyes (cartilaginous fishes) and purple for cyclostomes (jawless fish)](/cms/asset/1c1b3251-9bf4-48d4-abd5-5c4bc40b940a/kchl_a_1919463_f0002_oc.jpg)
Despite these results, the activity of mitoBKCa channels has also been observed in invertebrates such as Caenorhabditis elegans [Citation7] and Drosophila melanogaster [Citation8], both lacking the DEC sequence, suggesting the existence of additional mitochondrial targeting mechanisms in those taxonomic groups. In addition, BKCa-like currents have been observed in planar lipid bilayers reconstituted of mitochondrial membrane fractions from the potato Solanum tuberosum [Citation9], and the protist Dictyostelium discoideum [Citation10]. While the molecular identity of BKCa was assigned based on western blots with the use of an anti-KCa1.1 antibody [Citation9,Citation10], our blast search shows that neither S. tuberosum nor D. discoideum possess a bona fide BKCa channel encoded in their genome, neither the epitope sequence for the anti-KCa1.1 is present in their sequences. Hence, more experiments including loss of function and knockout mutants need to be performed to elucidate the molecular identity responsible for these currents in these evolutionary distant organisms.
The presence of the DEC sequence in all the groups of vertebrates might indicate that a channel with characteristics such as large conductance for K+ and exquisitely regulated by Ca2+, plays an important role in mitochondrial physiology, that once selected during evolution has suffered minor changes in the subsequent younger taxa. Yet, our understanding of the precise role(s) that mitoBKCa plays in such evolutionary distant organisms and in multiple organs and cell types in the same organism has just begun.
Structure of BKCa and mitoBKCa
As a splice variant of the plasma membrane BKCa, the overall structure of mitoBKCa might be conserved. The basic architecture of the pore forming α-subunit can be divided in a transmembrane domain consisting of seven transmembrane segments (S0-S6) and a cytoplasmic domain (), while three major structural domains can be recognized: i) a voltage-sensor domain formed by charged residues located at the S2, S3 and S4 segments [Citation11,Citation12,Citation13,Citation14]; ii) a pore-gate domain (S5-S6) through which K+ ions are conducted; and iii) a cytoplasmic domain that contains Mg2+ binding sites and the regulators for conductance of K+ or RCK domains that bind Ca2+. Four α-subunits encoded by the KCNMA1 gene form a functional mitoBKCa channel () and four pairs of RCKs (RCK1-RCK2) form the Ca2+-sensing apparatus, the so-called “gating-ring” occupying two-thirds of the whole BKCa structure.
Biophysical properties of mitoBKCa
Over the past 20 years, a large collection of papers has described the most fundamental biophysical properties of mitoBKCa channel (). Most of the work has been performed using the patch-clamp technique applied to mitoplasts (inner mitochondrial membrane devoid of outer mitochondrial membrane). With this technique, Siemen and coworkers were the first to establish that mitoBKCa channel from a glioma cell-line LN229 has large conductance for K+ as well as a voltage and Ca2+ sensitivity [Citation15], like its counterpart at the plasma membrane [Citation16,Citation17,Citation18]. A pioneer assessment of a physiological role of mitoBKCa in cardioprotection, was done by the O’Rourke laboratory revealing that cardiac mitoBKCa was sensitive to changes in matrix Ca2+ and could be blocked by charybdotoxin (ChTx) applied to the external face of the channel [Citation1]. Moreover, pharmacological activation of mitoBKCa with NS1619 proved to be cardioprotective reducing the infarct size of hearts treated with this mitoBKCa opener [Citation1]. These early studies did not only shape the pharmacological profile of mitoBKCa channel but also represented a milestone in the understanding of the physiological role of BKCa in mitochondrial and cell physiology. The studies that followed these seminal works revealed that the conductance slope of mitoBKCa varies among different tissues and cell types [Citation19,Citation20]. Moreover, the unitary conductance of cardiac mitoBKCa has proven to be diverse (), ranging between 190 and 300 pS [Citation1,Citation21,Citation22]; in a recent report [Citation23], a cardiac mitoBKCa channel from mice displayed a conductance of 145 pS in 150 mM symmetric KCl and 100 µM Ca2+. This variability suggests the existence of heterogeneous conductances for K+ in cardiac mitochondria, as recently demonstrated by Citation22, where a conductance of 190 pS was assigned to mitoBKCa, among other conductances observed in the same preparation of mitoplasts, those ranging between 60 and 370 pS [Citation22]. We had recently found that cardiac mitoBKCa channel has a conductance ranging between 290 and 320 pS under symmetric 150 mM KGluconate and 10 µM matrix Ca2+ [Citation4]. It is possible that different ionic conditions employed in other studies caused such variability in the reported conductances for mitoBKCa.
Table 1. Biophysical and pharmacological properties of mitoBKCa channels
Activation of BKCa channel
Intracellular Ca2+ concentration ([Ca2+]i) and membrane depolarization can allosterically activate plasma membrane BKCa channel [Citation17,Citation24,Citation25,Citation26]. Likewise, Ca2+ and membrane potential exquisitely activate mitoBKCa. Since mitoBKCa is encoded by the same gene, KCNMA1, we can expect structural and functional conservation. In this regard, BKCa channel open probability increases as a function of Ca2+ concentration and, as has been described for mammalian Slo1 channels, the Ca2+-activation curve is a function of membrane voltage [Citation24].
Voltage activation of BKCa channel and Ca2+ binding to the gating ring
It is well established that BKCa channel gating is also regulated by voltage, involving charged amino acids located in S2-S4 transmembrane segments [Citation11,Citation12,Citation13,Citation14,Citation27,Citation28,Citation29,Citation30,Citation31,Citation32,Citation33,]. Allosteric interactions between Ca2+ or voltage sensors can open the channel independently as well as synergistically, enabling the channel to functionally couple intracellular Ca2+ signals with the electrical activity of the cell [Citation34]. The intracellular gating ring of eukaryotic BKCa channel comprises eight RCK domains (). Each BKCa channel subunit contains two nonidentical RCK domains (RCK1 and RCK2) linked in tandem [Citation35], thus forming an intracellular gating ring of four RCK1–RCK2 tandems.
Arrangements on the RCK domain transduce in the opening of BKCa
The structural changes that occur after binding of Ca2+ to the RCK domains have provided important clues to understanding the channel opening. Experimental studies pioneered by the Olcese laboratory [Citation36,Citation37], revealed conformational changes of the RCK1 and RCK2 domains induced by Ca2+, as well as an elegant optical demonstration that Ca2+ binding to the intracellular BK regions allosterically facilitates the activation of the voltage sensing apparatus of the channel [Citation38]. Moreover, the recently determined crystal structure of Slo1 from Aplysia californica revealed that binding to the Ca2+ bowl and RCK1 sites in the C-terminal domain (CTD) leads to a near rigid-body lateral tilting (away from pore) of the N-lobes formed by the upper part of each RCK1 domain () [Citation39,Citation40,Citation41]. This lateral tilting moves the RCK1 attachment point for each S6-RCK1 C-linker laterally and downward, pulling on S6 to potentially open the pore gate. Simultaneously, the lateral tilting of the N-lobe moves the αB helix located at the top of each N-lobe () both upward and laterally to push on the bottom of the S4–S5 linker/VSD to potentially open the channel. In addition, it has recently been demonstrated that in human BKCa, the αB helix links the binding of Ca2+ at the RCK domains to the VGD of the channel, confirming that interaction between the αB helices at the top of the N-lobes of the CTD and the cytoplasmic surfaces of the S4-S5 linkers/VSD is required to open the channel [Citation42]. In agreement with this new model for channel opening, early observations have shown that both RCK1 and 2 domains can move independently from each other upon binding of Ca2+, indicating a high degree of flexibility for this domain [Citation43]. Moreover, Giraldez and Rothberg expanded these observations establishing that ligand binding to the RCK domains stabilized the active conformation of the BKCa channel [Citation44]. Despite these important advances, questions regarding the activation of mitoBKCa by voltage and Ca2+ remain open. In excitable cells, large non-physiological amounts of Ca2+ are required to activate the BKCa channel. When expressed alone the BKCa α subunit shows a voltage of half activation (V1/2) of 18 mV at 10 µM [Ca2+]i, which is shifted toward negative and relatively physiological values (V1/2 = −77 mV) when co-expressed with its regulatory β1 subunit [Citation45]. Strikingly, in cardiac mitochondria one population of mitoBKCa channels showed a V1/2 = −55 mV at 12 µM [Ca2+]i, indicative of functional association with auxiliary β1 subunits [Citation4]. Shifting the voltage sensitivity of mitoBKCa by association with regulatory subunits and its allosteric activation by elevating mitochondrial matrix Ca2+, helps to define the physiological window where opening the channel might occur. In this context, largely hyperpolarized mitochondrial membrane potential (ΔΨ ~-200 mV) might keep the channel mostly closed, maintaining the large driving force for Ca2+ and the physiological processes that depend on it. On the other hand, under stress conditions such as ischemia, opening of this large conductance for K+ has proven to be cardioprotective when treated with NS1619, a BKCa opener. Moreover, opening of mitoBKCa correlates with a higher capacity of mitochondria to handle Ca2+ [Citation1,Citation2,Citation4]. Although proximity with the sarcoplasmic reticulum (SR) ensures mitochondrial Ca2+ uptake, rises in matrix Ca2+ must be tightly controlled particularly under high stress conditions such as ischemia and/or metabolic dysfunction. Thus, opening of mitoBKCa might depolarize mitochondria reducing the driving force for Ca2+, thus preventing the initiation of apoptosis and cell death. We will discuss the experimental evidence that support this hypothesis in the following sections.
Expression of mitoBKCa channels in adult cardiomyocytes
Adult rodent cardiomyocytes express a splice variant of plasma membrane BKCa. This splice variant localizes exclusively at the IMM and chemical activation of cardiac mitoBKCa channel reduces the infarct size after ischemic insult [Citation1]. Moreover, hearts from the BK-KO (KCNMA1−/-) treated with the BKCa opener NS1619 did not show this protection against ischemic insult [Citation2]. The study by Singh and coworkers elegantly demonstrates the importance of mitoBKCa channel in cardiac function; however, the mechanism(s) through which mitoBKCa prevents cardiac damage remains to be fully elucidated.
The physiological role of mitoBKCa channel in controlling Ca2+ overload
To understand the role that mitoBKCa channel might play in protecting cardiac tissue after an ischemic insult, it is necessary to review the function of plasma membrane BKCa channel in other cell systems. The rhythm of vital physiological processes depends on the dynamics of Ca2+ entry and membrane potential, both triggers of BKCa activity. Action potentials (AP) in neurons and smooth muscle cells depend on the activation of voltage-dependent calcium channels (VDCC) which in turn increase cytosolic Ca2+ [Citation46] and activate neighboring BKCa channels [Citation47,Citation83,Citation84]. This functional coupling causes a massive K+ efflux through BKCa channels that rapidly repolarizes (<1 ms) the membrane potential by shutting down the VDCCs, shaping and ensuring propagation of the AP. In smooth muscle cells, activation of BKCa channels has a negative-feedback effect in contractility by reducing entry of Ca2+ via the VDCCs [Citation48]. A similar feedback mechanism might occur in cardiac myocytes, where Ca2+ and K+ play major roles in contraction-relaxation processes. During cardiac AP, a depolarization of the plasma membrane activates VDCC. Influx of Ca2+ induces the release of more Ca2+ from the SR through the activation of the ryanodine receptors (RyR). This rapid elevation of cytosolic Ca2+ ensures activation of the myofilaments contracting the myocytes. The delicate balance between contraction and relaxation largely depends on the rapid extrusion/removal of Ca2+ from the cytoplasm, which occurs mainly through the active recapture of Ca2+ into the SR via the Ca2+-ATPase (SERCA) and through extrusion of Ca2+ via the plasmalemmal Na+/Ca2+ exchanger (NCX). Detachment of Ca2+ from its binding sites on the troponins lead to the relaxation of myocytes. This dynamic and perfectly coordinated mechanism commonly known as cardiac excitation-contraction coupling (ECC) accounts for the proper pumping of 6000 l of blood per day in the adult human heart. Adult cardiomyocytes express a large battery of K+ channels responsible to restore the membrane potential that terminates the cardiac AP [Citation49]. Intriguingly, the large conductance for K+, voltage-dependent and Ca2+-activated BKCa channels do not take part in this process mostly due to their exclusive expression in mitochondria [Citation2]. It has been previously hypothesized, and excellently reviewed by [Citation50], that the activation of a large conductance for K+ might help to modulate mitochondrial Ca2+ overload, a critical step preventing mitochondrial permeability transition pore (mPTP) opening and cell death. In metazoans, mitochondrial Ca2+ uptake occurs mainly through the mitochondrial calcium uniporter (MCU) [Citation51], that uses the large driving force for Ca2+ established by the activity of the electron transport chain [Citation52]. Rise in the mitochondrial matrix Ca2+ would activate the mitoBKCa channel [Citation15], which in turn would depolarize the mitochondrial membrane potential reducing the mitochondrial Ca2+ driving force [Citation53], preventing the mitochondrial Ca2+ overload, and thus the formation and opening of the mPTP [Citation21,Citation54,Citation55,Citation56]. In agreement with this hypothesis, we observed that blocking the mitoBKCa channel with paxilline impaired the ability of mitochondria to control Ca2+ overload. Our observations also indicate that mitoBKCa is functionally associated with auxiliary subunits of the β1-type [Citation4]. To understand the physiological role of this association, we must briefly review the well-documented modes of regulation exerted by BK-auxiliary subunits on plasma membrane BKCa.
Association of plasma membrane BKCa with auxiliary subunits
Expression of plasma membrane BKCa pore forming α-subunit is commonly accompanied by the expression of auxiliary β (1–4), γ (1–4), or both types of subunits. Auxiliary BKCa-subunits β and γ (mostly tissue-specific) modify the kinetics of the channel, Ca2+ and voltage sensitivities, and toxin sensitivity [see Citation57,for a detailed review on this topic]. Moreover, auxiliary β subunits can also act as modulators of channel density at the plasma membrane and mitochondria via endocytic processes [Citation4,Citation19].
Auxiliary BKCa-β-subunits (KCNMB1–4)
BKCa-β subunits possess two transmembrane domains (T1 and T2) connected by an extracellular loop, with both N-terminal and C-terminal domains located cytosolically. As mentioned earlier, association of BKCa-α subunit with auxiliary β1 subunits at high [Ca2+]i (10 µM), shifts the V1/2 of activation from 18 mV to −77 mV relative to the expression of the α-subunit alone [Citation45]. This functional association also prevents inactivation and rectification of the channel [Citation58]. In addition, association of BKCa-α subunit with auxiliary β2, and β3 subunits (comprising four splicing variants, a-d, in humans and primates) mediate fast inactivation and instantaneous current rectification [Citation59,Citation60]. Association with BKCa-β2 affects the movements and equilibrium of the S3-S4 region, promoting opening of the channel by favoring the activated state of the voltage-sensor [Citation61]. Overall, the auxiliary β2 subunits shifts BKCa V1/2 toward more negative membrane potentials, ranging from 27 mV [Citation61] to 75 mV shift at 3–4 µM [Ca2+]i [Citation62]. However, the most distinctive characteristic of β2-containing BKCa channels is their fast inactivation [Citation58,Citation63].
Regulatory BKCa-γ-subunits
Auxiliary γ subunits belong to the Leucine-Rich Repeat (LRR) superfamily. The four γ subunits have an N-terminal signal peptide, an extracellular LRRC domain with an N-terminal cysteine-rich segment (LRR-NT), six LRRs, and a C-terminal cysteine-rich segment (LRR-CT), a single transmembrane domain, and a short cytosolic C-terminal tail [Citation64]. Each γ subunit has a unique tissue-specific expression pattern and modulates the BKCa voltage dependence in heterologous expression systems [Citation65]. Of the identified γ subunits, only γ1 has been established as a definitive BKCa channel regulator in native cells. This subunit shifts the V1/2 of BKCa from 168 to 10 mV in the absence of Ca2+ [Citation66], and from 31 to −85 mV at 10 µM free [Ca2+]i [Citation67]. Auxiliary γ1 subunit also induces resistance to mallotoxin [Citation68]. Homotetramers of BKCa channels can accommodate up to four γ1 subunits, one γ1 per α-subunit; however, a single γ1 is sufficient to produce the full gating shift of the channel [Citation67,Citation69,Citation70]. As explained before, functional association of BKCa channel with its regulatory subunits modulates their biophysical properties, and ensures proper targeting and activation of the channel, crucial steps for cellular excitability, maintenance of Ca2+ homeostasis, triggering of signaling cascades, neurotransmitter release, among other physiological processes [Citation29,Citation71,Citation77,Citation78]. It is known that in the cardiovascular system, the expression and association of plasma membrane BKCa channels with auxiliary β1 subunit regulates vascular tone and blood pressure [Citation79,Citation80]. Moreover, this association is also preserved in mitochondria from adult cardiomyocytes [Citation4]. Nevertheless, to date the association of mitoBKCa channel with auxiliary γ-subunits remains unknown.
The expression, targeting and activity of mitoBKCa depends on its association with regulatory β1 subunits
We recently observed that the capacity of cardiac mitochondria to handle Ca2+ is linked to the expression of the BKCa-β1 subunit. Mitochondria from β1-KO mice show a reduced capacity to retain Ca2+ and early opening of the mPTP, which correlates with lower expression and low Po of mitoBKCa channel [Citation4]. These results indicate that regulatory β1-subunit controls the activity of mitoBKCa and consequently mitochondrial Ca2+ handling. Despite the evidence, it is hard to reconcile the activation of mitoBKCa channel in the context of mitochondrial physiology (ΔΨ ~-200 mV, [Ca2+]mit < 200 nM), where mitoBKCa channel must remain closed. Despite this, it is worth noticing that mitoBKCa displays a remarkable high Po at negative membrane potentials in different cell types [Citation1,Citation15,Citation81]. As noted, mitoBKCa displays a hyperpolarized V1/2 of activation relative to that of the BKCa-α subunit when it is expressed alone [Citation45,Citation85,Citation86,Citation87,Citation88,Citation89]. Opening of the channel at hyperpolarized membrane potentials is enhanced upon its association with its regulatory β1 subunit [Citation4,Citation21,Citation90]. Yet, the mechanisms by which the auxiliary BKCa-β-subunits are targeted to the mitochondria and the roles that these subunits might play in health and disease remain to be determined. A new line of evidence suggests that regulatory β1 subunits might participate in the translation of mechanical stimuli into gating of BKCa channels [Citation91]; however, as we will discuss in the next section, the mechanosensitivity of BKCa channels has yet to be fully determined.
Additional regulatory mechanisms of mitoBKCa channels
It has been recently published that a subpopulation of mitoBKCa channels is modulated by mechanical stimulation. The authors found a slight increment in the Po of mitoBKCa channels from human astrocytoma cells in response to mechanical stimulation (from 0.016 at 0 mmHg to 0.3 at −40 mmHg at +20 mV) [Citation92]. Inherent mechanosensitive ion channels have evolved to detect and transduce mechanical forces into electrical signals, evoking substantial changes in their Po in response to a mechanical stimulus. Amongst them, bacterial channels MscL and MscS [Citation93,Citation94], and eukaryotic channels PIEZO1, TRAAK and TREK1, as well as the most recently described members of the OSCA family [Citation95,Citation96,Citation97]. Voltage-dependent and Ca2+-activated BKCa channels sense and gate the pore in response to changes in both membrane potential and elevation in cytosolic [Ca2+], increasing their Po [Citation11,Citation12,Citation13,Citation14,Citation17,Citation24,Citation25,Citation26,Citation29,Citation30,Citation31,Citation27,Citation28,Citation32,Citation33,Citation98]. Those extensively studied mechanisms of activation strongly contrast with a rather negligible change in the Po displayed by BKCa in response to large mechanical stimulus. Even more, when compared to changes in Po observed in well-characterized mechanosensitive ion channels such as PIEZO1 and 2 [Citation99, Citation100], the assessment that BKCa channels are capable to respond to a mechanical stimulus must be carefully revised. Despite the efforts to identify a structural domain on BKCa channel that acts as a “mechanosensor,” this remains as an open question. A study conducted by Zhao and Sokabe suggested the presence of a mechanosensitive domain in BKCa, which is the linker that connects the transmembrane segment S6 with the RCK1 domain [Citation101]. Moreover, shortening the linker results in an increased membrane-stretch sensitivity, whereas the opposite effect was observed by lengthening the linker, suggesting this site as the sensor of membrane tension. Paradoxically, the mutant with the longer linker also showed a reduced voltage and calcium sensitivity, indicating that the mutation of this region (S6) might affect the overall function of the channel. In addition, the stress-regulated exon (STREX), which is a cysteine-rich domain (CRD) located between RCK1 and RCK2 domains in the STREX-BKCa splice variant, has been proposed to be an essential element for the stretch sensitivity of BKCa channel [Citation102]. STREX anchors the C-terminal of the BKCa to the plasma membrane [Citation102,Citation103] by a palmitoylation modification throughout the cysteine residues C12 and C13 within the CRD [Citation103,Citation104]. It is worth noting that the C-terminal from ZERO-BKCa (BKCa channel without the STREX insert) remains in the cytoplasmic side [Citation103] and ZERO-BKCa alone does not respond to mechanical stimulus as shown by Citation102. Furthermore, single amino acid substitution from Ala674 to Thr674 within the STREX (ERA sequence) on BKCa channels completely abolished the stretch sensitivity [Citation102]. An interaction of STREX with the cytoskeleton that ultimately may translate into a slight opening of the channel could not be ruled out. Further experiments are required for BKCa channels to be considered as mechanosensitive, together with a mechanism of activation and a physiological meaning for this property, thus far this phenomenon should not be considered as an inherent property of BKCa channels.
In the study by Citation92,the mRNA containing the STREX exon was detected in human astrocytoma cells. However, the splice variants of mitoBKCa containing the DEC sequence alone or together with STREX were not detected. Thus, it is not clear if mitoBKCa channels from human astrocytoma cells contain the STREX exon and whether the expression of this splice variant is sensitive to mechanical stimuli. Complementary research would help to assess mitoBKCa as an inherent mechanosensitive channel, including but not limited to channel reconstitution in proteoliposomes and stretch application through the patch-clamp pipette, together with loss or gain of function assays that might depend on the expression of different splice variants such as STREX.
Modulation of mitoBKCa by amyloid-β (Aβ)
Alzheimer’s disease (AD) is the most common neurodegenerative disease characterized by neuronal loss, progressive cognitive deterioration associated with the reduction of daily activities and behavioral changes in elder people. The aggregation of Aβ peptides in the human brain has a neurotoxic effect and plays a key role in the development of AD [reviewed at Citation105]. Aβ is a self-aggregating peptide produced by the cleavage of a transmembrane glycoprotein, the amyloid precursor protein. In addition, mitochondrial dysfunction [see Citation106,for a review in this topic] and Ca2+ unbalance are among the most prominent hallmarks of AD [reviewed at Citation107]. In neurons, Aβ peptides promote Ca2+ release from endoplasmic reticulum (ER) increasing intracellular Ca2+ levels [Citation108]. Neighbor mitochondria take up this Ca2+ inducing loss of mitochondrial membrane potential, generating ROS, and leading to apoptosis and cell death [Citation108]. In addition, Aβ peptides can affect directly mitochondrial physiology since they accumulate in mitochondria [Citation106,Citation109]. Studies in vitro have shown that Aβ peptides are imported through the TOM complex and predominantly localized at the IMM [Citation109]. Interestingly, a recent study on mitoplasts from human astrocytoma cells has reported that different forms of Aβ, including monomers, oligomers and fibrils inhibit the activity of mitoBKCa channels in a concentration dependent manner. The highest concentration of Aβ fibrils tested (5 µM) produced an 80% inhibition, whereas Aβ monomers and oligomers inhibited 50% and 70% of mitoBKCa channel activity, respectively [Citation5]. All forms of Aβ inhibited mitoBKCa channel activity when applied at either side of the membrane [Citation5], indicative of an indirect effect on the channel. As it has been reported that Aβ oligomers modify the tension of the plasma membrane and disrupts the cytoskeleton [Citation110], Kravenska and coworkers proposed that Aβ forms induce a mechanical change that transduces into closure of mitoBKCa channel [Citation5]. As we stated in a previous section of this review, the opening of mitoBKCa correlates with a higher capacity of mitochondria to handle Ca2+ in cardiomyocytes [Citation1,Citation2,Citation4]. If this is the case in neurons, we can hypothesize that the presence of Aβ in the IMM might contribute to the development of AD through the inhibition of mitoBKCa, which ultimately could lead to mitochondrial damage and cell death. Thus, it becomes relevant to confirm these findings in proper AD models to determine the molecular mechanisms and signaling pathways through which Aβ affects the biophysical properties of mitoBKCa channel and how this affects mitochondrial and neuronal physiology. This will improve our understanding of AD development and perhaps would help us to design specific strategies to prevent or treat AD.
Concluding remarks
Evidence about the constitution, regulation, origin, and evolution of the mitochondrial BKCa channel is emerging. More importantly, it has been shown that for a successful targeting of mitoBKCa to the mitochondria needs to bear the DEC sequence and, in some tissues, interact with the auxiliary subunit β1. Here we showed that the DEC sequence is solely associated with BKCa channels, and it is highly conserved and exclusively present in vertebrates. Although these findings might contribute to our understanding of the physiological role of mitoBKCa in the organisms that bear the DEC sequence, their full significance remains to be clarified. Moreover, new questions arise about the mechanisms of mitochondrial targeting; especially considering that large conductances for K+ have been described as mitoBKCa channels in organisms that do not contain the DEC sequence. Regulation of mitoBKCa channels by auxiliary β1-subunit and amyloid-β peptides was recently proposed. Although most of the questions remain open, the modulation of mitoBKCa through these mechanisms could be of high relevance in the development of pathophysiological conditions such as ischemia and neurological diseases where mitochondria have a crucial role. Thus, the study of the signaling pathways and the molecules implied in regulation of mitoBKCa might contribute to the understanding and future treatment or prevention of these conditions.
Acknowledgments
The authors thank the enormous support of Professor Riccardo Olcese, PhD, for all the important contributions to this paper. This work was supported by the doctoral fellowship from Fundação para a Ciência e a Tecnologia (FCT; SFRH/BD/146484/2019) (CSC), the CONACyT postdoctoral fellowship EPE-2016291121 and EPE-2017291231 (ALGC), and by UC MEXUS-CONACyT Postdoctoral Fellowship FE-13-248 (EB) and AHA-WSA 15POST22490015 (EB).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Xu W, Liu Y, Wang S, et al. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298:1029–1033.
- Singh H, Lu R, Bopassa JC, et al. mitoBKCa is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc Natl Acad Sci U S A. 2013;110:10836–10841.
- Zhang J, Li M, Zhang Z, et al. The mitochondrial BKCa channel cardiac interactome reveals BKCa association with the mitochondrial import receptor subunit Tom22, and the adenine nucleotide translocator. Mitochondrion. 2017;33:84–101.
- Balderas E, Torres NS, Rosa-Garrido M, et al. MitoBKCa channel is functionally associated with its regulatory beta1 subunit in cardiac mitochondria. J Physiol. 2019;597:3817–3832.
- Kravenska Y, Nieznanska H, Nieznanski K, et al. The monomers, oligomers, and fibrils of amyloid-beta inhibit the activity of mitoBKCa channels by a membrane-mediated mechanism. Biochim Biophys Acta Biomembr. 2020;1862:183337.
- Schreiber M, Salkoff L. A novel calcium-sensing domain in the BK channel. Biophys J. 1997;73:1355–1363
- Wojtovich AP, Sherman TA, Nadtochiy SM, et al. SLO-2 is cytoprotective and contributes to mitochondrial potassium transport. PLoS One. 2011;6:e28287.
- Gururaja Rao S, Bednarczyk P, Towheed A, et al. BKCa (Slo) Channel Regulates Mitochondrial Function and Lifespan in Drosophila melanogaster. Cells. 2019;8. DOI:https://doi.org/10.3390/cells8090945
- Koszela-Piotrowska I, Matkovic K, Szewczyk A, et al. A large-conductance calcium-activated potassium channel in potato (Solanum tuberosum) tuber mitochondria. Biochem J. 2009;424:307–316.
- Laskowski M, Kicinska A, Szewczyk A, et al. Mitochondrial large-conductance potassium channel from Dictyostelium discoideum. Int J Biochem Cell Biol. 2015;60:167–175.
- Díaz L, Meera P, Amigo J, et al. Role of the S4 segment in a voltage-dependent calcium-sensitive potassium (hSlo) channel. J Biol Chem. 1998;273:32430–32436.
- Horrigan FT, Aldrich RW. Allosteric voltage gating of potassium channels II. Mslo channel gating charge movement in the absence of Ca(2+). J Gen Physiol. 1999;114:305–336.
- Horrigan FT, Cui J, Aldrich RW. Allosteric voltage gating of potassium channels I. Mslo ionic currents in the absence of Ca(2+). J Gen Physiol. 1999;114:277–304.
- Stefani E, Ottolia M, Noceti F, et al. Voltage-controlled gating in a large conductance Ca2+-sensitive K+channel (hslo). Proc Natl Acad Sci U S A. 1997;94:5427–5431.
- Siemen D, Loupatatzis C, Borecky J, et al. Ca2+-Activated K Channel of the BK-Type in the Inner Mitochondrial Membrane of a Human Glioma Cell Line. Biochim Biophys Res Comm. 1999;257:549–554.
- Barrett JN, Barrett EF, Dribin LB. Calcium-dependent slow potassium conductance in rat skeletal myotubes. In Press. 1981; 82.
- Latorre R, Oberhauser A, Labarca P, et al. Varieties of calcium-activated potassium channels. Ann Rev Physiol. 1989;51:385–399.
- Latorre R, Vergara C, Hidalgo C. Reconstitution in planar lipid bilayers of a Ca2+-dependent K+ channel from transverse tubule membranes isolated from rabbit skeletal muscle. Proc Natl Acad Sci USA. 1982;79:805–809.
- Balderas E, Zhang J, Stefani E, et al. Mitochondrial BKCa channel. Front Physiol. 2015;6:104.
- Singh H, Stefani E, Toro L. Intracellular BK(Ca) (iBK(Ca)) channels. J Physiol. 2012;590:5937–5947.
- Ohya S, Kuwata Y, Sakamoto K, et al. Cardioprotective effects of estradiol include the activation of large-conductance Ca(2+)-activated K(+) channels in cardiac mitochondria. Am J Physiol Heart Circ Physiol. 2005;289:H1635–H1642.
- Soltysinska E, Bentzen BH, Barthmes M, et al. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS One. 2014;9:e103402.
- Frankenreiter S, Bednarczyk P, Kniess A, et al. cGMP-Elevating Compounds and Ischemic Conditioning Provide Cardioprotection Against Ischemia and Reperfusion Injury via Cardiomyocyte-Specific BK Channels. Circulation. 2017;136:2337–2355.
- Barrett JN, Magleby KL, Pallotta BS. Properties of single calcium-activated potassium channels in cultured rat muscle. J Physiol. 1982;331:211–230.
- Blatz AL, Magleby KL. Ion conductance and selectivity of single calcium-activated potassium channels in cultured rat muscle. J Gen Physiol. 1984;84:1–23.
- Meech RW, Standen NB. Potassium activation in helix aspersa neurons under voltage clamp: a component mediated by calcium influx. J Physiol. 1975;249:211–239.
- Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110:257–281.
- Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305.
- Lee US, Cui J. BK channel activation: structural and functional insights. Trends Neurosci. 2010;33:415–423.
- Ma Z, Lou XJ, Horrigan FT. Role of charged residues in the S1-S4 voltage sensor of BK channels. J Gen Physiol. 2006;127:309–328.
- Pantazis A, Gudzenko V, Savalli N, et al. Operation of the voltage sensor of a human voltage- and Ca2+-activated K+ channel. Proc Natl Acad Sci U S A. 2010;107:4459–4464.
- Rothberg BS, Magleby KL. Kinetic structure of large-conductance Ca2+-activated K+ channels suggests that the gating includes transitions through intermediate or secondary states. A mechanism for flickers. J Gen Physiol. 1998;111:751–780.
- Rothberg BS, Magleby KL. Gating kinetics of single large-conductance Ca2+-activated K+ channels in high Ca2+ suggest a two-tiered allosteric gating mechanism. J Gen Physiol. 1999;114:93–124.
- Hoshi T, Pantazis A, Olcese R. Transduction of voltage and Ca2+ signals by Slo1 BK channels. Physiology (Bethesda). 2013;28:172–189.
- Yuan P, Leonetti MD, Pico AR, et al. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science. 2010;329:182–186.
- Yusifov T, Javaherian AD, Pantazis A, et al. The RCK1 domain of the human BKCa channel transduces Ca2+ binding into structural rearrangements. J Gen Physiol. 2010;136:189–202.
- Yusifov T, Savalli N, Gandhi CS, et al. The RCK2 domain of the human BKCa channel is a calcium sensor. Proc Natl Acad Sci U S A. 2008;105:376–381.
- Savalli N, Pantazis A, Yusifov T, et al. The contribution of RCK domains to human BK channel allosteric activation. J Biol Chem. 2012;287:21741–21750.
- Hite RK, Tao X, MacKinnon R. Structural basis for gating the high-conductance Ca(2+)-activated K(+) channel. Nature. 2017;541:52–57.
- Tao X, Hite RK, MacKinnon R. Cryo-EM structure of the open high-conductance Ca(2+)-activated K(+) channel. Nature. 2017;541:46–51.
- Yuan P, Leonetti MD, Hsiung Y, et al. Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature. 2012;481:94–97.
- Geng Y, Deng Z, Zhang G, et al. Coupling of Ca(2+) and voltage activation in BK channels through the alphaB helix/voltage sensor interface. Proc Natl Acad Sci U S A. 2020;117:14512–14521.
- Miranda P, Giraldez T, Holmgren M. Interactions of divalent cations with calcium binding sites of BK channels reveal independent motions within the gating ring. Proc Natl Acad Sci U S A. 2016;113:14055–14060.
- Giraldez T, Rothberg BS. Understanding the conformational motions of RCK gating rings. J Gen Physiol. 2017;149:431–441.
- Meera P, Wallner M, Jiang Z, et al. A calcium switch for the functional coupling between a (hslo) and b subunits (KV,Cab) of maxi K channels. FEBS Lett. 1996;382:84–88.
- Schiller J, Schiller Y, Stuart G, et al. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505(Pt 3):605–616.
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905.
- Jaggar JH, Porter VA, Lederer WJ, et al. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256.
- Jeevaratnam K, Chadda KR, Huang CL, et al. Cardiac Potassium Channels: physiological Insights for Targeted Therapy. J Cardiovasc Pharmacol Ther. 2018;23:119–129.
- Bauer TM, Murphy E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ Res. 2020;126:280–293.
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364.
- Bernardi P. Mitochondria in muscle cell death. Ital J Neurol Sci. 1999;20:395–400.
- Testai L, Martelli A, Marino A, et al. The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/reperfusion injury. Biochem Pharmacol. 2013;85:1634–1643.
- Aon MA, Cortassa S, Wei AC, et al. Energetic performance is improved by specific activation of K+ fluxes through K(Ca) channels in heart mitochondria. Biochim Biophys Acta. 2010;1797:71–80.
- Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. 2015;78:129–141.
- Wang X, Yin C, Xi L, et al. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am J Physiol Heart Circ Physiol. 2004;287:H2070–H2077.
- Gonzalez-Perez V, Lingle CJ. Regulation of BK Channels by Beta and Gamma Subunits. Annu Rev Physiol. 2019;81:113–137.
- Zeng XH, Xia XM, Lingle CJ. Redox-sensitive extracellular gates formed by auxiliary beta subunits of calcium-activated potassium channels. Nat Struct Biol. 2003;10:448–454.
- Uebele VN, Lagrutta A, Wade T, et al. Cloning and functional expression of 2 families of {beta}-subunits of the large conductance calcium-activated K+ channel. J Biol Chem. 2000;275:23211–23218.
- Xia XM, Ding JP, Zeng XH, et al. Rectification and rapid activation at low Ca2+ of Ca2+-activated, voltage-dependent BK currents: consequences of rapid inactivation by a novel beta subunit. J Neurosci. 2000;20:4890–4903.
- Savalli N, Kondratiev A, De Quintana SB, et al. Modes of operation of the BKCa channel beta2 subunit. J Gen Physiol. 2007;130:117–131.
- Orio P, Latorre R. Differential effects of beta 1 and beta 2 subunits on BK channel activity. J Gen Physiol. 2005;125:395–411.
- Xia XM, Ding JP, Lingle CJ. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J Neurosci. 1999;19:5255–5264.
- Dolan J, Walshe K, Alsbury S, et al. The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genomics. 2007;8:320.
- Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci U S A. 2012;109:7917–7922.
- Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature. 2010;466:513–516.
- Gonzalez-Perez V, Xia XM, Lingle CJ. Functional regulation of BK potassium channels by gamma1 auxiliary subunits. Proc Natl Acad Sci U S A. 2014;111:4868–4873.
- Almassy J, Begenisich T. The LRRC26 protein selectively alters the efficacy of BK channel activators. Mol Pharmacol. 2012;81:21–30.
- Carrasquel-Ursulaez W, Alvarez O, Bezanilla F, et al. Determination of the Stoichiometry between alpha- and gamma1 Subunits of the BK Channel Using LRET. Biophys J. 2018;114:2493–2497.
- Gonzalez-Perez V, Ben Johny M, Xia XM, et al. Regulatory gamma1 subunits defy symmetry in functional modulation of BK channels. Proc Natl Acad Sci U S A. 2018;115:9923–9928.
- Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66:852–875.
- Kulawiak B, Bednarczyk P. Reconstitution of brain mitochondria inner membrane into planar lipid bilayer. Acta Neurobiol Exp. 2005;65:271–276
- Gu XQ, Siemen D, Parvez S et al. Hypoxia increases BK channel activity in the inner mitochondrial membrane. Biochem Biophys Res Commun. 2007;358:311–316
- Cheng Y, Gu XQ, Bednarczyk P et al. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell Physiol Biochemistry. 2008;22:127–136
- Skalska J, Piwonska M, Wyroba E et al. A novel potassium channel in skeletal muscle mitochondria. Biochimica et Biophysica Acta. 2008; 1777:651–659
- Skalska J, Bednarczyk P, Piwonska M et al. Calcium ions regulate K+ uptake into brain mitochondria: The evidence for a novel potassium channel. Int J Mol Sci. 2009; 10:1104–1120
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurons. J Physiol. 1987;389:187–203.
- Meredith AL, Wiler SW, Miller BH, et al. BK calcium-activated potassium channels regulate circadian behavioral rhythms and pacemaker output. Nat Neurosci. 2006;9:1041–1049.
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535.
- Ledoux J, Werner ME, Brayden JE, et al. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda). 2006;21:69–78.
- Fahanik-Babaei J, Eliassi A, Saghiri R. How many types of large conductance Ca(+)(2)-activated potassium channels exist in brain mitochondrial inner membrane: evidence for a new mitochondrial large conductance Ca(2)(+)-activated potassium channel in brain mitochondria. Neuroscience. 2011;199:125–132.
- Bednarczyk P, Koziel A, Jarmuszklewicz W, et al. Large-conductance Ca2+-activated potassium channel in mitochondria of endothelial EA.hy926 cells. Am J Physiol Heart Circ Physiol. 2013;304:H1415–H1427
- Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron. 2001;31:1027–1034
- Berkefeld H, Sailer CA, Bildl W, et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620
- Bao L, Cox DH. Gating and ionic currents reveal how the BKCa channel’s Ca2+ sensitivity is enhanced by its beta1 subunit. J Gen Physiol. 2005;126:393–412.
- Brenner R, Perez GJ, Bonev AD, et al. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876.
- Nimigean CM, Magleby KL. The beta subunit increases the Ca2+ sensitivity of large conductance Ca2+-activated potassium channels by retaining the gating in the bursting states [In Process Citation]. J Gen Physiol. 1999;113:425–440.
- Sweet TB, Cox DH. Measuring the influence of the BKCa {beta}1 subunit on Ca2+ binding to the BKCa channel. J Gen Physiol. 2009;133:139–150.
- Tanaka Y, Meera P, Song M, et al. Molecular constituents of maxi KCa channels in human coronary smooth muscle. Predominant a + b subunit complexes. J Physiol. 1997;502:545–557.
- Bautista L, Castro MJ, Lopez-Barneo J, et al. Hypoxia inducible factor-2alpha stabilization and maxi-K+ channel beta1-subunit gene repression by hypoxia in cardiac myocytes: role in preconditioning. Circ Res. 2009;104:1364–1372.
- Xin F, Cheng Y, Ren J, et al. The extracellular loop of the auxiliary beta1-subunit is involved in the regulation of BKCa channel mechanosensitivity. Am J Physiol Cell Physiol. 2018;315:C485–C493.
- Walewska A, Kulawiak B, Szewczyk A, et al. Mechanosensitivity of mitochondrial large-conductance calcium-activated potassium channels. Biochim Biophys Acta Bioenerg. 2018;1859:797–805.
- Sukharev SI, Blount P, Martinac B, et al. A large-conductance mechanosensitive channel in E. coli encoded by mscL alone. Nature. 1994;368:265–268.
- Levina N, Totemeyer S, Stokes NR, et al. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. Embo J. 1999;18:1730–1737.
- Brohawn SG, Su Z, MacKinnon R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci U S A. 2014;111:3614–3619.
- Murthy SE, Dubin AE, Whitwam T, et al. OSCA/TMEM63 are an Evolutionarily Conserved Family of Mechanically Activated Ion Channels. Elife. 2018;7.
- Syeda R, Florendo MN, Cox CD, et al. Piezo1 Channels Are Inherently Mechanosensitive. Cell Rep. 2016;17:1739–1746.
- Latorre R, Castillo K, Carrasquel-Ursulaez W, et al. Molecular Determinants of BK Channel Functional Diversity and Functioning. Physiol Rev. 2017;97:39–87.
- Coste B, Mathur J, Schmidt M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60.
- Shin KC, Park HJ, Kim JG, et al. The Piezo2 ion channel is mechanically activated by low-threshold positive pressure. Sci Rep. 2019; 9: 6446
- Zhao H, Sokabe M. Tuning the mechanosensitivity of a BK channel by changing the linker length. Cell Res. 2008;18:871–878.
- Naruse K, Tang QY, Sokabe M. Stress-Axis Regulated Exon (STREX) in the C terminus of BK(Ca) channels is responsible for the stretch sensitivity. Biochem Biophys Res Commun. 2009;385:634–639.
- Tian L, Jeffries O, McClafferty H, et al. Palmitoylation gates phosphorylation-dependent regulation of BK potassium channels. Proc Natl Acad Sci U S A. 2008;105:21006–21011.
- Jeffries O, Geiger N, Rowe IC, et al. Palmitoylation of the S0-S1 linker regulates cell surface expression of voltage- and calcium-activated potassium (BK) channels. J Biol Chem. 2010;285:33307–33314.
- Sanabria-Castro A, Alvarado-Echeverria I, Monge-Bonilla C. Molecular Pathogenesis of Alzheimer’s Disease: an Update. Ann Neurosci. 2017;24:46–54.
- Wang W, Zhao F, Ma X, et al. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegeneration. 2020;15:30.
- Reiss AB, Arain HA, Stecker MM, et al. Amyloid toxicity in Alzheimer’s disease. Rev Neurosci. 2018;29:613–627.
- Ferreiro E, Oliveira CR, Pereira CMF. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008;30:331–342.
- Hanson-Petersen CA, Alikhani N, Behbahani H, et al. The amyloid b-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. PNAS. 2008;105(35):13145–13150.
- Gao Q, Fang Y, Zhang S, et al. Dynamic effect of beta-amyloid 42 on cell mechanics. J Biomech. 2019;86:79–88.