
ABSTRACT
KCNMA1-linked channelopathy is an emerging neurological disorder characterized by heterogeneous and overlapping combinations of movement disorder, seizure, developmental delay, and intellectual disability. KCNMA1 encodes the BK K+ channel, which contributes to both excitatory and inhibitory neuronal and muscle activity. Understanding the basis of the disorder is an important area of active investigation; however, the rare prevalence has hampered the development of large patient cohorts necessary to establish genotype-phenotype correlations. In this review, we summarize 37 KCNMA1 alleles from 69 patients currently defining the channelopathy and assess key diagnostic and clinical hallmarks. At present, 3 variants are classified as gain-of-function with respect to BK channel activity, 14 loss-of-function, 15 variants of uncertain significance, and putative benign/VUS. Symptoms associated with these variants were curated from patient-provided information and prior publications to define the spectrum of clinical phenotypes. In this newly expanded cohort, seizures showed no differential distribution between patients harboring GOF and LOF variants, while movement disorders segregated by mutation type. Paroxysmal non-kinesigenic dyskinesia was predominantly observed among patients with GOF alleles of the BK channel, although not exclusively so, while additional movement disorders were observed in patients with LOF variants. Neurodevelopmental and structural brain abnormalities were prevalent in patients with LOF mutations. In contrast to mutations, disease-associated KCNMA1 single nucleotide polymorphisms were not predominantly related to neurological phenotypes but covered a wider set of peripheral physiological functions. Together, this review provides additional evidence exploring the genetic and biochemical basis for KCNMA1-linked channelopathy and summarizes the clinical repository of patient symptoms across multiple types of KCNMA1 gene variants.
Introduction
KCNMA1-linked channelopathy is a recently characterized neuromuscular disorder essentially defined by the presence of a mutation in the KCNMA1 gene, associated with various combinations of movement disorders, seizures, developmental delay, and intellectual disability. The disorder does not yet have a standardized clinical correlation, diagnostic criteria, or therapeutic approach. This review serves to identify the key factors contributing to barriers in these areas and move discussions toward a better mechanistic understanding of the disorder. Since the prior review in 2019 [Citation1], the cohort of patients identified with KCNMA1-linked channelopathy has nearly doubled, and the number of studies addressing the functional effects of KCNMA1 variants on BK channel activity has expanded. This larger dataset offers the opportunity to update the phenotypic range and variability of KCNMA1-linked channelopathy, and to begin to evaluate potential associations between KCNMA1 variants, BK channel activity, and patient symptoms.
Functional effects of KCNMA1 variants on BK current
The KCNMA1 gene is located on human chromosome 10q22 and encodes the α subunit of the BK (“Big K+”) large conductance voltage and Ca2+-dependent K+ channel (KCa1.1) [Citation2]. The BK channel conducts outward K+ currents in response to changes in intracellular Ca2+ and membrane depolarization and is formed as a homotetramer of the KCNMA1 gene product [Citation3]. Each α subunit is composed of seven transmembrane domains (S0-S6) and an intracellular C-terminal gating ring (). S1-S4 contain positively charged residues mediating voltage sensing, while S5-S6 house a K+ selective pore. The gating ring contains two Ca2+ binding domains, regulator of conductance of K+ (RCK) RCK1 and RCK2, that mediate allosteric opening in response to local Ca2+ increases [Citation4]. BK channels are distributed through a variety of both excitable and non-excitable tissues, with alternative transcriptional splicing and post-translational modifications contributing to the specificity of cellular responses [Citation3].
Figure 1. Graphic of the α subunit of the BK channel, annotated with all known patient mutations. The S0-S6 transmembrane domains represent the pore-forming portion of the subunit, while S7-S8 and S9-S10 comprise the Ca2+-sensitive RCK1 and RCK2 domains respectively. The + symbols in domains S2-S4 mark the location of residues conferring voltage-sensitivity. Mutation types are denoted by color. C413Y/N449fs is a double mutation in a single patient
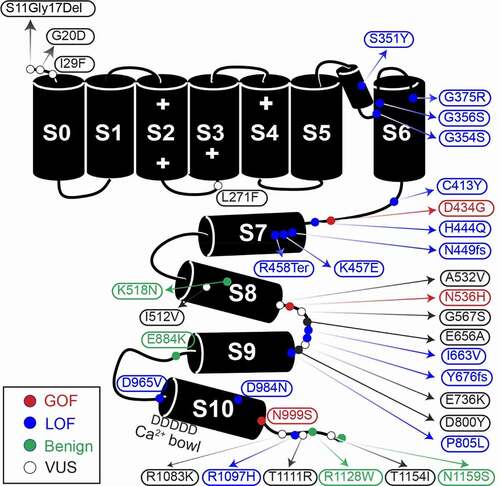
In this review, we summarize the functional classifications for 37 variants at varying stages of channel-level investigation. Over half of KCNMA1-linked channelopathy patients harbor variants that have been shown to change BK current levels and properties, and in this section, we review the evidence supporting the gain of function (GOF), loss of function (LOF), and putative benign designations with respect to BK channel activity. These studies have been conducted in heterologous cells with homotetramers of mutant BK channels. Thus, there exists a relatively large gap in knowledge concerning how KCNMA1 variants affect neuronal activity [Citation1]. It is also important to note that KCNMA1-linked channelopathy is at an early stage of definition with respect to causation. Most KCNMA1 variants have not yet been definitively shown to cause or correlate with neurological dysfunction in patients. Caveats related to this are further discussed in the sections that follow.
At the BK channel level, GOF mutations corroborated across multiple labs include D434G and N999S (also called N995S, N1036S, and N1053S), located in the intracellular gating ring (). D434G is located in the AC region of RCK1, an area known to regulate Ca2+-dep gating [Citation4], while N999S is located at the distal C-terminus of the RCK2 domain within a structurally unresolved region. Both GOF mutations increase BK current [Citation5–10]. However, N999S was recently shown to generate a larger increase in BK current when directly compared to D434G, suggesting a stronger GOF mutation effect [Citation5]. This relative difference may stem from distinct mechanistic underpinnings. N999S appears to exert this effect through a voltage-dependent but Ca2+-independent manner [Citation6–10], producing larger hyperpolarizing shifts in the V1/2 (40–60 mV) that are unaffected by mutation of the RCK1 or RCK2 Ca2+ binding sites [Citation6]. In contrast, D434G increases the Ca2+ sensitivity via an increased Hill coefficient, but results in smaller V1/2 shifts (<40 mV) that require the Ca2+ site in RCK1 [Citation8]. Both mutations additionally speed activation and slow deactivation of BK channels [Citation5,Citation7,Citation8]. More recently, N536H demonstrated a voltage-dependent GOF effect as well [Citation11]. This mutation also exhibits a strong hyperpolarizing effect on BK channel activation (30–90 mV V1/2 shift) and speeds activation, and like N999S, exceeds the D434G GOF phenotype in direct comparison [Citation11]. N536H is located at the C-terminus of the αE helix RCK1 and increases voltage-dependent activation without significant effect on Ca2+-sensitivity. Altogether, these three mutations have the most data supporting their functional GOF classification with respect to BK channel activity under several conditions tested.
Up to 14 confirmed and putative LOF alleles have been identified ( and ), almost all of which arise de novo, and are associated with a range of detrimental effects on the BK channel [Citation1]. Four mutations are associated with complete loss of channel current in heterologous cells (S351Y, G356R, G375R, and N449fs) and one with reduced current amplitude (D984N) [Citation12]. S351Y and G356R are located in the pore, G375R in S6, and N449 in the AC domain of RCK1. The mechanisms underlying the loss of current are not yet known, and the expression and trafficking of these mutants has not been thoroughly studied. Other LOF mutations are associated with varying levels of reduced BK current through multiple mechanisms including a shift in the voltage-dependence of activation to more depolarized potentials (C413Y, H444Q, K457E, P805L, D965V, and R1097H), decreased channel expression (P805L), slower channel activation (G354S, H444Q, D965V), and decreased single-channel conductance with altered selectivity (G354S) [Citation12–15]. Befitting its effects, G354S is located in the selectivity filter within the pore [Citation15]. K457E is located in the αB helix of RCK1 [Citation14]. The K457 residue was previously shown to interact with Phosphatidylinositol 4,5-bisphosphate (PIP2) to promote BK channel activation, and a charge-neutralizing mutation decreased PIP2 sensitivity and altered β subunit modulation [Citation16–18]. D965V mutation is located in RCK2, in a series of aspartate residues that comprise the calcium bowl [Citation4,Citation19]. Little is known about the potential mechanisms for other LOF mutations. C413Y and H444Q are both located within the AC region of RCK1, P805L in the loop between S9 and S10 of RCK2, and R1097H is in the unstructured distal C-terminal of the RCK2. Two additional variants are mutations predicted to produce truncated channel proteins, but have not yet been verified in functional studies (R458Ter and Y676Lfs) [Citation20,Citation21]. These mutations would produce channels missing most of the intracellular gating ring or a truncation that excludes RCK2, respectively. Lastly, four variants introduced in BK channels had no effect on either BK current levels or properties and have been initially categorized as benign (K518N, E884K, R1128W, and N1159S) [Citation5,Citation6,Citation22,Citation23]. In total, with respect to data on BK channel activity from the currently available in vitro studies, 11% of known alleles can be classified as GOF, 37% as LOF or putative LOF, 11% as putative benign, and 41% remain uncategorized.
Table 1. Summary of KCNMA1 variants and major clinical presentations
Clinical evaluation
KCNMA1-linked channelopathy typically presents with either epileptic seizures or movement dysfunction, or both, often without remarkable brain imaging or EEG [Citation1]. Following the identification of a mutation in the KCNMA1 gene, it remains challenging to sort through the etiology of the various symptoms associated with the channelopathy, which may be either episodic or present persistently (). Patients commonly experienced both epileptic seizure and movement disorder in this study and in previous studies [Citation1,Citation9,Citation20,Citation24], which have been difficult to disentangle. Episodic symptoms can be particularly difficult to delineate, as they can resemble paroxysmal dyskinesia, tonic or atonic seizures, and cataplexy. Neurologists prioritized capturing these events on EEG to determine whether they were caused by epileptic activity. Prior interictal EEG findings across the patient cohort included generalized and focal spike and wave complexes, Lennox-Gastaut syndrome, and background slowing, and clinical seizures with EEG correlate have been described as tonic-clonic, absence, myoclonic, atonic, and tonic [Citation1,Citation9,Citation20,Citation21]. The presence of paroxysmal events without concurrent EEG abnormalities has typically led to a workup for a movement disorder. However, many patients with abnormal episodic movements are initially diagnosed with epilepsy due to interictal epileptiform abnormalities, despite non-epileptic EEG findings during the clinical events.
Figure 2. Schematic illustrating the observed spectrum of clinical courses and diagnostic outcomes following molecular diagnosis of KCNMA1-linked channelopathy. If there is more than one clinical phenotype for episodic or paroxysmal symptoms, each symptom or episode should be evaluated independently on EEG, given the co-occurrence of epilepsy and other non-epileptic paroxysms. Long-term EEG is often helpful. PNKD3 denotes PNKD associated with a confirmed KCNMA1 mutation. GTC-Generalized Tonic-Clonic, EEG-electroencephalogram, PNKD- paroxysmal nonkinesigenic dyskinesia, PED- paroxysmal exertion-induced dyskinesia, PKD- paroxysmal kinesigenic dyskinesia
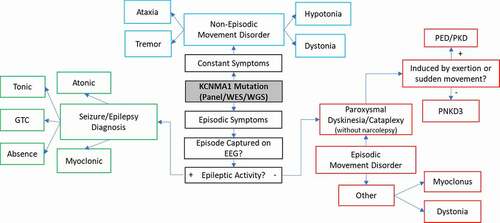
Once a movement disorder has been delineated from seizure activity by the history, clinical phenotype, and EEG, the type and extent are evaluated (). Paroxysmal non-kinesigenic dyskinesia (PNKD) is typically used to describe sudden-onset movement abnormalities which are not otherwise initiated themselves by other movements (i.e. paroxysmal kinesigenic dyskinesia or PKD). The term dyskinesia is nonspecific, and the movement abnormalities seen in PNKD are characterized by one or more involuntary, hyper- or hypokinetic findings, including dystonia, atonia, weakness, chorea, athetosis, rhythmic repetitive movements, or a combination thereof [Citation25], reviewed in [Citation26]. PNKD was originally linked to a specific KCNMA1 variant, D434G, in a familial pedigree [Citation9]. Following the later association with a separate N995S mutation [Citation23], PNKD3 emerged as a more specific designation for “PNKD associated with KCNMA1 mutations” (OMIM 609,446). Previously reviewed movement attacks typified PNKD3, described as paroxysmal “drop attacks” involving behavioral arrest, dystonic postures, and often perioral dyskinesias [Citation1,Citation9,Citation24]. Furthermore, alcohol, fatigue, emotion, and stress have been reported as triggers [Citation9,Citation24], consistent with a diagnosis of PNKD [Citation25]. In general, patients with PNKD experience a few attacks daily, which typically last from 10 minutes to several hours, and are characteristically triggered not by exertion or movement as in other paroxysmal dyskinesias, but by alcohol, caffeine, emotion, and sleep deprivation [Citation27]. Notably, KCNMA1 patients had PNKD episodes that were generally shorter and more frequent than those described in the traditional diagnostic criteria [Citation9,Citation11,Citation24–26], sometimes occurring up to hundreds per day [Citation28–30], supporting the distinction for PNKD3.
PNKD3 events have features which overlap considerably with the cataplexy seen in patients with type 1 narcolepsy [Citation31]. Both PNKD3 and cataplectic attacks can be triggered by emotion or excitement, are usually brief (seconds), and result in loss of muscle control. However, the classic description of cataplexy involves a sudden and near-complete loss of tone with absent deep tendon reflexes, while the spells seen in KCNMA1-linked channelopathy are often accompanied by preserved tone and the presence of deep tendon reflexes [Citation24,Citation32]. This is consistent with our (author SK) direct examination of tone and reflexes in two patients during a typical PNKD episode, as well as video examination of multiple cases where events were characterized by semi-rigid posturing consistent with preserved or increased tone and inconsistent with atonia). However, cataplectic events, especially in children, can have a broad phenotype that overlaps with the paroxysmal dyskinesias seen in KCNMA1-linked channelopathy [Citation31]. Although the somewhat broader PNKD term is preferred to describe the episodic movement abnormality seen in KCNMA1-linked channelopathy, there is disagreement regarding the nomenclature for patient episodes [Citation33] and thus we have included the term “cataplexy without narcolepsy” in [Citation33]. Other episodic and non-episodic movement disorders observed among KCNMA1 patients include dystonia, hypotonia, myoclonus, ataxia, and tremor () [Citation1,Citation12,Citation15,Citation21].
Overall, description and categorization of patient symptoms following identification of a KCNMA1 mutation was often a complicated and highly involved process. The lack of initial neurological findings, the phenotypic overlap between PNKD3 and other diagnoses, and the subtle differences between symptoms observed among channelopathy patients and traditional diagnostic criteria have all contributed to this difficulty. Summarizing the symptomatic range identified among patients with KCNMA1-linked channelopathy therefore provides a basic set of descriptors for the spectrum of diagnoses following identification of a KCNMA1 mutation ().
Core clinical presentations of KCNMA1-linked channelopathy
KCNMA1 pathogenicity is currently supported by several familial pedigrees showing transmission of the KCNMA1 variants D434G and Y676Lfs [Citation9,Citation21], as well R1097H and T1154I (). In addition, the most common de novo mutation, N999S, is now found in 12 patients with similar symptoms [Citation6,Citation23,Citation24,Citation34–36]. These variants support the conclusion that KCNMA1 plays a pathogenic role in KCNMA1-linked channelopathy, although it may not represent the entire molecular picture of the disorder in all cases. In this review, we focus on the core symptoms of seizure, movement abnormalities, neurodevelopmental abnormalities, and a fourth category encompassing brain magnetic resonance imaging (MRI) abnormalities (). The rationale for these core features is first based on a combination of epilepsy and/or movement disorder exhibited by essentially all patients. In the prior cohort, brain abnormalities were only observed in six patients presenting to a single clinic [Citation12]. Similar abnormalities have now been documented in the newly expanded cohort. Given the relevance of structural central nervous system (CNS) abnormalities to other core phenotypes, this phenotype was also assessed from patient reports and prior publications. The relevance of other non-CNS structural and skeletal anomalies to core phenotypes is currently less clear and detailed elsewhere [Citation12].
Emerging genotype–phenotype relationships
KCNMA1-linked channelopathy is a rare disorder, with the available allele frequencies estimated to be 1:100,000 for KCNMA1 mutations [Citation37]. Together with the present limitation that the majority of KCNMA1 mutations have arisen de novo, there is limited ability to construct adequate patient cohorts to link patient symptoms with mutations and establish genetic causality. Initially, pedigree analysis from a single family describing the first pathogenic KCNMA1 mutation suggested that PNKD was qualitatively associated with D434G, a GOF mutation [Citation9]. PNKD was then found among patients harboring a second type of GOF mutation, N995S [Citation5,Citation6,Citation23,Citation24,Citation35], suggestive of a correlation. In contrast, previous studies suggested that ataxia, axial hypotonia, and tremor occurred among patients harboring a reduced BK channel function [Citation1,Citation12,Citation15,Citation21]. However, apart from qualitative movement disorder findings, prior studies uncovered few distinctions between GOF and LOF variants with respect to core KCNMA1-linked channelopathy patient phenotypes. Previous conclusions regarding seizure phenotypes were limited to noting the wide variety of epilepsy diagnoses across mutation types and a qualitative enrichment of absence seizures among GOF patients [Citation1]. Documentation of neurodevelopmental phenotypes was likewise limited to the identification of intellectual disability and developmental delay across mutation types, while autism spectrum disorder (ASD) was limited to two LOF patients [Citation1].
In this review, we present a summary of the available genotype and phenotype data to date. Individual patients () were grouped according to the type of mutation they carried (GOF, LOF, or VUS genotype) and the presence or absence of symptoms for each symptom category (phenotype; ). GOF and LOF designations were applied if any channel-level study supported the conclusion of increased or decreased activity. VUS designation was made at the time of genetic diagnosis, identifying variants with insufficient or conflicting evidence to support the conclusion of an independent disease-causing mutation. Based on prevalence, movement disorder was stratified into PNKD3 versus all other movement disorders. Patients with seizure were divided into those experiencing absence seizures alone and those experiencing at least one epilepsy diagnosis other than absence. The 32 patients identified in this and other recent studies were aggregated with the 37 previously reviewed patients [Citation1]. The putative truncation variants R458Ter and Y676fs were considered LOF mutations for the purposes of this summary. A group of patients with putative benign variants were combined with VUS, as only four such patients were identified, with limited phenotypic data available (). Furthermore, studies probing functional alterations are limited, and negative data under a single set of conditions is not considered definitive. Any symptom for which a patient’s description was not specified was not included in the genotype-phenotype summary or in the total patient number in .
Table 2. Number of patients with core KCNMA1-linked channelopathy symptoms by mutation type
Movement disorder
We found that movement disorders are a consistent symptom of KCNMA1-linked channelopathy across mutation types, present in 51/69 total patients (). Consistent with prior review [Citation1], GOF patients with movement disorders almost exclusively exhibit PNKD3, while LOF patients harbor a heterogeneous array of movement disorders. This relationship was upheld in the expanded cohort, with 23/27 GOF patients exhibiting PNKD versus 5/17 LOF patients. A single GOF patient who was not diagnosed with PNKD exhibited a movement disorder that was similar to PNKD, though did not have a formal diagnosis. By comparison, 12/17 LOF patients had non-PNKD movement disorders. Among the 10 new GOF patients identified since prior review, 6 were diagnosed with PNKD3, including 4 N995S patients, or were PNKD-like (N536H) [Citation6,Citation11,Citation23,Citation24,Citation35,Citation36].
Five new LOF patients were identified in this review: H444Q, D965V, and three patients with R1097H mutations [Citation13]. While patients with LOF mutations disproportionately experience movement disorders other than PNKD3, these new patients complicate a simple GOF versus LOF paradigm with respect to movement disorder. Only the D965V patient presented with purportedly “LOF” symptoms- a non-classic ataxia rather than a paroxysmal dyskinesia (). Yet interestingly, the H444Q patient and two R1097H patients exhibited PNKD3 of varying severity (). It is worth noting that the H444Q patient also exhibited multiple ectopic mutations of unknown significance that may be producing phenotypic overlap with the KCNMA1 mutation. Similarly, the previously identified R458Ter patient had a second CACNA1H mutation, which has been previously linked with childhood generalized and absence epilepsy [Citation20,Citation38–40]. The classification of E884K as a putative benign mutation is also interesting, as this patient reportedly experienced PNKD3 [Citation23] (). Eighteen additional patients with apparently benign or untested (VUS) mutations have been identified since prior review, 13 of which experience movement symptoms. Four patients, A532V, T1111R and the two T1154I patients experience symptoms that appear similar to PNKD3 episodes (). Five other VUS/no effect patients experience LOF-like symptoms including myoclonus, ataxia, axial hypotonia, and dystonia, while the remaining patients experience symptoms associated with neither GOF nor LOF mutations.
In summary, while PNKD3 is present in a higher percentage of GOF than LOF patients, this relationship was not exclusive, as 5 LOF and one patient with a putative benign variant received the same diagnosis. Two of these LOF patients, H444Q and R458Ter, exhibited at least one other significant genome mutation, raising the possibility for multifactorial etiology of PNKD3 [Citation20].
Epilepsy
Although there is preliminary evidence that movement disorder phenotypes may segregate based on functional mutation type, seizure heterogeneity, and prevalence remained similar to that observed in prior reviews. Here, 35/69 patients had some form of seizure or epilepsy, compared to 18/37 previously [Citation1], but no difference was observed between the proportions of GOF and LOF patients experiencing seizure symptoms (). Four of the 10 recently identified GOF patients reported an epilepsy diagnosis, all reporting multiple, including absence, atonic, myoclonic, and generalized tonic-clonic (GTC), as well as one report of an epileptic encephalopathy (). The relative predominance of absence seizures among GOF patients observed previously [Citation1] was less robustly supported in the expanded cohort (). While two recently identified GOF patients did demonstrate absence seizures, both patients experienced other seizure types as well ().
New evidence has broadened the range of LOF epilepsy diagnoses, though only two of the five new LOF patients exhibited seizure activity. One R1097H patient experienced seizure characterized as atonic and GTC, while the D965V patient expanded the phenotypic spectrum to include focal temporal lobe epilepsy (). Twelve patients with newly described putative benign or VUS mutations also exhibited some form of seizure activity (). Six patients reported specific epilepsy diagnoses, including absence, myoclonic, tonic, GTC, and atonic. The remaining patients either did not have, or did not specify, a particular seizure type (). In summary, although epilepsy is diagnosed in about half of the patient population, no additional, more precise diagnostic correlations are supported by the existing cohort.
Neurodevelopmental abnormalities
With the expansion of the KCNMA1 patient population, a greater proportion of LOF patients exhibited atypical neurocognitive and developmental phenotypes than did GOF patients. Sixteen of 17 LOF patients exhibited such phenotypes mutations compared to 17/30 GOF patients (). However, the distinction between GOF and LOF patient symptoms appears to mostly depend on the contrast between D434G and LOF, as there are no differences in neurocognitive phenotypes between N999S and LOF patients. Diagnoses fell primarily within the previously described spectrum of intellectual disability, developmental delay, and ASD and were present across mutation types (). There were also three reports of behavioral or conduct disorder and other diagnoses, including nonspecific motor delay and learning disability (). In this review, attribution of neurodevelopmental abnormalities to LOF mutations is not considered definitive, as all eleven GOF patients identified over the past year exhibited these phenotypes (). Additionally, it cannot be ascertained at this time whether these phenotypes are primary effects of KCNMA1 mutations or secondary to channelopathy-provoked seizures.
Brain MRI Abnormalities
Patients of all mutation types exhibited MRI abnormalities, though structural changes were present in a greater proportion among patients with LOF mutations than among patients with GOF mutations (). As was the case with neurocognitive phenotypes, this observation appears to be mediated primarily by the difference between D434G and LOF patients. Among all patients, LOF individuals were also the most abundantly represented with eight patients, followed by six VUS patients and three GOF patients [Citation6,Citation12,Citation15,Citation20,Citation21,Citation24] (). Given the commonality of these symptoms among VUS patients, the enrichment of structural abnormalities among LOF patients versus GOF patients is likely to require reexamination. Cerebellar atrophy was the most common abnormal finding on MRI. In multiple previously identified cases, this atrophy was progressive and accompanied by more widespread cortical atrophy [Citation6,Citation12,Citation15,Citation20,Citation21], though these findings were more mild in recently identified patients with N536H [Citation11] and intron 17 mutations (). Cerebellar atrophy was not, however, the sole finding. The patient with the VUS I29F showed evidence of perinatal hypoxic ischemic encephalopathy, while G20D and G567S presented with the more general findings of progressive white matter disease and cortical atrophy, respectively ().
Current limitations for genotype-phenotype analysis
At present, the disorder “KCNMA1-linked channelopathy” is so named to reflect that it is solely defined by a neurological diagnosis in conjunction with a mutation in the KCNMA1 gene. While the broad goal of this review summary is to highlight emerging patterns, a primary limitation to developing robust correlations is imposed by the size and composition of the small cohort for this rare disorder. Only four variants have any degree of familial pedigree associated with patient symptomology, while independent de novo variants provide the remainder of the associational data. The number of patients harboring specific variants varies greatly in the current cohort, with N999S and D434G dominating the GOF group and a more heterogeneous set of individual variants populating the LOF group. The preliminary observations regarding genotype–phenotype relationships, as well as the functional and symptomatic classifications, presented in this review will continue to emerge as the patient cohort is expanded.
There are several important additional caveats to translating KCNMA1 mutation effects assessed from in vitro patch-clamp experiments into even a basic understanding of the molecular role of patient variants in KCNMA1-linked channelopathy. First, although KCNMA1 causality is commonly assumed to be established, the level of evidence varies greatly between the different variants at both the genetic and channel levels. In epilepsy, it has been proposed that pathogenicity cannot be ascribed to a variant without considering the presence and functional classification of variants in other channels and modulatory subunits [Citation41]. With respect to patient genetics, the variety of genes included across different epilepsy panels [Citation42] suggests that additional ion channel variants may have gone undetected in KCNMA1 patients, depending on the commercially available panel. WES is also limited by genome coverage and the reporting of additional significant variants [Citation42–44]. Complete genetic data were not available for some patients reported in prior publications. Thus, it is an open question whether in some cases, KCNMA1 could actually represent a putative risk variant in linkage disequilibrium with the disease allele [Citation45].
Following this, the functional categorizations for patient variants are not definitive. Given the complex gating and expression mechanisms for the BK channel [Citation3], the absence of a detectable channel effect under one set of conditions may not be entirely predictive of the full potential for changes in cellular BK currents and a binary designation of pathogenicity. Investigation of variants presently categorized as GOF and LOF could differ under additional Ca2+ conditions and under endogenous cellular conditions where BK channels undergo regulated trafficking and assembly into multi-subunit complexes. Very few variants have been studied with respect to expression, and for those that have, the data are limited to heterologous cells. Most notably though, these KCNMA1 variants have been studied in the equivalent of a homozygous configuration in vitro, in which all four α subunits of the BK channel tetramer carry the mutation. However, all but four patients harbor heterozygous alleles. The stoichiometry of wildtype (WT) to mutant subunits in heterozygous patient tissues in vivo is currently unknown. These WT/mutant interactions could potentially mitigate, or exacerbate, phenotypes at the channel or patient levels via mechanisms not assessed in existing datasets.
There are further limitations imposed by incomplete or inaccurate patient data. Standardized survey and direct submission data provided by patients were subjective, self-reported and dependent on participant communication. Patients also had non-identical medical trajectories through different clinics. Some patients may therefore have been more likely to receive a more specific epilepsy or movement disorder diagnosis, depending on the specialty clinic at which they were evaluated. Nevertheless, these patients were effectively blinded with respect to the functional categorization of their channel variants (GOF vs LOF), suggesting that selection bias may have little effect on genotype-phenotype correlation emerging from this small sample size.
Lastly, it remains unclear whether the neurodevelopmental problems are primary effects of KCNMA1 mutations, or secondarily resulting from epilepsy. Patient data were relatively unhelpful in addressing this issue, as among the 44 patients with these findings, half experienced seizures, while half did not (). Comorbid intellectual disability and epilepsy are often concurrently associated with an underlying genetic syndrome in humans [Citation46]. However, previous evidence shows that untreated seizures in childhood are associated with developmental delay later in life [Citation47]. Likewise, cerebellar atropy may represent a consequence of seizures rather than a direct effect of channel dysfunction [Citation48]. BK channel function has been implicated in a range of atypical cognitive phenotypes in animal models that could inform observations from patients [Citation49–56]. Furthermore, BK α expression is upregulated in the late embryonic period and early prenatal period [Citation53,Citation57,Citation58], and this is thought to be linked to functional maturation of affected neuronal populations [Citation59]. However, it is difficult to speculate further about the etiology and correlation of these patient phenotypes until more longitudinal developmental data are available.
The limitations of genotype and phenotype assessment also hamper the understanding of KCNMA1 channelopathy disease mechanisms. For example, LOF KCNMA1 mutations could theoretically produce a paradoxical GOF effect at the cellular level through upregulated expression of other ion channels. Epileptogenesis in Dravet Syndrome has been hypothesized to result from the upregulation of sodium channels in response to SCN1A haploinsufficiency [Citation60,Citation61]. In individual sodium channel gene deletions, expression of other sodium channels in various excitable cells is increased [Citation62–64]. Likewise, the lack of a clear distinction in seizure phenotypes may complicate the GOF versus LOF paradigm. Given that BK channels permeate outward K+ current following activation, LOF mutations could be broadly predicted to produce widespread hyperexcitability, as seen in seizure. However, the absence of a strong correlation between LOF mutations and seizure suggests additional factors are at work. Moreover, it has been hypothesized that both GOF and LOF mutations could promote global hyperexcitability through disruption of excitation and inhibition in the cortex [Citation1,Citation59,Citation65]. Evidence from in vitro studies supports this possibility, as BK channels are expressed in both excitatory and inhibitory rodent neurons with context-dependent BK current effects [Citation59]. Another possibility stems from the ability of BK channel activation to exert bidirectional effects on neuronal firing activity [Citation66]. The ability of BK channel activation to increase excitability via reduction of action potential repolarization time [Citation59,Citation66–68] could predict a strong correlation between GOF variants and seizure. Yet epilepsy is not diagnosed in many GOF patients (), even among those harboring the strong GOF D434G and N999S mutant channels [Citation5,Citation6,Citation8,Citation9,Citation36]. Lastly, in rodents, pharmacologic BK channel inhibition, which could mimic a KCNMA1 LOF mutation, paradoxically produces anti-epileptogenic effects [Citation69]. Reconciliation of these gaps in the mechanistic underpinnings of KCNMA1-associated neuropathology will be required. Development of animal models of patient mutations for in vivo study will be useful both in connecting in vitro channel phenotypes to brain function, and for any type of allele-based therapeutic testing [Citation70]. Toward this, transgenic flies expressing SLO-E366G have been proposed as an animal model for the human D434G variant [Citation71].
Single nucleotide polymorphisms
While rare mutations underlie the described cases of KCNMA1-linked channelopathy, single nucleotide polymorphisms (SNPs) comprise the majority of the sequence variation between individuals [Citation72]. SNPs are single base pair changes ranging in number from 4.1 to 5 million per individual genome that may or may not be deleterious [Citation72]. Genome-wide association studies (GWAS) and linkage analysis are combined with WES to explore the phenotypic consequences for SNPs. This review includes SNPs to further probe the landscape of KCNMA1 variants linked to disease and to evaluate the overlap with potential KCNMA1-linked channelopathy pathogenesis.
KCNMA1 harbors 178,978 SNPs, including 175,864 intronic, 598 synonymous, and 823 missense variants [Citation73]. Unlike KCNMA1 mutations, no systematic themes have emerged with respect to disease-associated SNPs (). However, KCNMA1 SNPs have been linked to multiple neurological disorders including ASD [Citation74,Citation75], epileptic encephalopathy [Citation6], epilepsy [Citation76], and Alzheimer’s Disease [Citation77]. The ASD-linked A138V SNP was previously evaluated for functional channel effects in vitro and found to produce a reduced BK current under specific conditions [Citation74,Citation78]. Other neurological conditions linked to KCNMA1 include migraines [Citation79,Citation80] and noise-induced hearing loss [Citation81]. KCNMA1 has also been implicated in multiple substance use disorders [Citation82–84], and alcohol dependence has been of particular interest due to ethanol’s capacity to activate the BK channel in model organisms [Citation85]. Increased risk of both hypertension and myocardial infarction have been ascribed to variants in the BK channel’s α subunit [Citation86,Citation87], as well as increased all-cause mortality among patients with a previous heart failure event [Citation88]. Interestingly, one variant in the channel’s ß subunit (KCNMB1) has been repeatedly shown to exert a protective effect against cardiovascular disease [Citation89–94]. KCNMA1 has also been implicated in inflammatory pathology, including angioedema in response to specific antihypertensives [Citation95], chronic rhinosinusitis [Citation96], and primary biliary cholangitis [Citation97]. GWAS has further uncovered KCNMA1-linked variations in drug and nutrient metabolism, including selenium [Citation98], thiopurine chemotherapy [Citation99,Citation100], and ranibizumab for age-related macular degeneration [Citation101]. KCNMA1 variants were also associated with changes in macroscopic metabolism, as demonstrated by a genome-wide association with obesity [Citation102]. A more recent study identified an association between two KCNMA1 variants and osteoporosis [Citation103]. Lastly, multiple studies have closely linked KCNMA1 variants to myopia [Citation104,Citation105]. On the other hand, stringent statistical standards have eliminated numerous KCNMA1 SNP variants from consideration such as disease-causing in epilepsy, hyperlipidemia, and alcohol dependence [Citation41,Citation106,Citation107]. Taken together, SNP analysis suggests the potential involvement of KCNMA1 and the BK channel in a wide range of human diseases, and the variant associations are not delimited by the core features of KCNMA1-linked channelopathy. Nevertheless, the functional consequences of specific variants in SNP-linked disease mechanisms are difficult to hypothesize based on the limited data at present.
Table 3. Disease-linked KCNMA1 SNPs
Summary
This review expands the phenotypic spectrum for KCNMA1-linked channelopathy and presents an updated summary of the clinical picture observed among patients. New functional characterizations of BK channel activity from novel KCNMA1 variants suggest that PNKD could be more prevalent among GOF than LOF patients, while non-PNKD movement disorders are currently observed in higher proportion among LOF than GOF patients. Additionally, both neurodevelopmental and structural brain abnormalities were present in a higher percentage of LOF than GOF patients. Yet, these preliminary relationships are not exclusive and, so far, fail to implicate similar genetic distinctions by mutation type for epilepsy. While the small patient cohort and limitations in the associated diagnostic and BK channel data preclude constructing genotype-phenotype correlations, there is now a growing body of evidence connecting four core neuropathologies (movement disorder, epilepsy, neurodevelopmental, and structural brain abnormalities) to KCNMA1 patient mutations. Meanwhile, SNP linkage analysis reveals the potential for KCNMA1 function to modify an even wider variety of neurological and peripheral pathologies. Future studies expanding and refining the genotypic and phenotypic findings presented here, in tandem with a more comprehensive, stratified analysis of the biochemical basis of KCNMA1-linked channelopathy, may illuminate a path toward improved mechanistic understanding and therapeutics.
Acknowledgments
We gratefully acknowledge patients, families, and physicians for their participation in the KCNMA1 Channelopathy International Advocacy Foundation (KCIAF) registry questionnaire or providing direct submission information, and Alyssa Mendel and the Coordination of Rare Diseases at Sanford (CoRDS) program for providing patient registry data collation and access.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bailey CS, Moldenhauer HJ, Park SM, et al. KCNMA1-linked channelopathy. J Gen Physiol. 2019;151(10):1173–1189.
- Dworetzky SI, Trojnacki JT, Gribkoff VK. Cloning and expression of a human large-conductance calcium-activated potassium channel. Brain Res Mol Brain Res. 1994;27(1):189–193.
- Gonzalez-Perez V, Lingle CJ. Regulation of BK channels by beta and gamma subunits. Annu Rev Physiol. 2019;81:113–137.
- Yang H, Zhang G, Cui J. BK channels: multiple sensors, one activation gate. Front Physiol. 2015;6:29.
- Moldenhauer HJ, Matychak KK, Meredith AL. Comparative gain-of-function effects of the KCNMA1-N999S mutation on human BK channel properties. J Neurophysiol. 2020;123(2):560–570.
- Li X, Poschmann S, Chen Q, et al. De novo BK channel variant causes epilepsy by affecting voltage gating but not Ca2+ sensitivity. Eur J Hum Genet. 2018;26(2):220–229.
- Wang B, Rothberg BS, Brenner R. Mechanism of increased BK channel activation from a channel mutation that causes epilepsy. J Gen Physiol. 2009;133(3):283–294.
- Junqiu Yang GK, Saxena A, Zhang G, et al. An epilepsy/dyskinesia-associated mutation enhances BK channel activation by potentiating Ca2+ sensing. Neuron. 2010;66:871–883.
- Du W, Bautista JF, Yang H, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37(7):733–738.
- Diez-Sampedro A, Silverman WR, Bautista JF, et al. Mechanism of increased open probability by a mutation of the BK channel. J Neurophysiol. 2006;96(3):1507–1516.
- Zhang G, Gibson RA, McDonald M, et al. A gain-of-function mutation in KCNMA1 causes dystonia spells controlled with stimulant therapy. Mov Disord. 2020;35(10):1868–1873.
- Liang L, Li X, Moutton S, et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet. 2019;28(17): 2937–295.
- Moldenhauer H, Park SM, Meredith AL. Characterization of new human KCNMA1 loss-of-function mutations. Biophys J. 2020;118(3):114a.
- Buckley C, Williams J, Munteanu T, et al. Status dystonicus, oculogyric crisis and paroxysmal dyskinesia in a 25 year-old woman with a novel KCNMA1 variant, K457E. Tremor Other Hyperkinet Mov. 2020;10:49.
- Du X, Carvalho-de-Souza JL, Wei C, et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc Natl Acad Sci. 2020;117(11):6023–6034.
- Tang QY, Zhang Z, Meng XY, et al. Structural determinants of phosphatidylinositol 4,5-bisphosphate (PIP2) regulation of BK channel activity through the RCK1 Ca2+ coordination site. J Biol Chem. 2014;289(27):18860–18872.
- Liu HW, Hou PP, Guo XY, et al. Structural basis for calcium and magnesium regulation of a large conductance calcium-activated potassium channel with beta1 subunits. J Biol Chem. 2014;289(24):16914–16923.
- Hou P, Zeng W, Gan G, et al. Inter-alpha/beta subunits coupling mediating pre-inactivation and augmented activation of BKCa beta2. Sci Rep. 2013;3:1666.
- Schreiber M, Salkoff L. A novel calcium-sensing domain in the BK channel. Biophys J. 1997;73(3):1355–1363.
- Yesil G, Aralasmak A, Akyuz E, et al. Expanding the phenotype of homozygous KCNMA1 mutations; dyskinesia, epilepsy, intellectual disability, cerebellar and corticospinal tract atrophy. Balkan Med J. 2018;35(4):336–339.
- Tabarki B, AlMajhad N, AlHashem A, et al. Homozygous KCNMA1 mutation as a cause of cerebellar atrophy, developmental delay and seizures. Hum Genet. 2016;135(11):1295–1298.
- Reiss SM, Prenatal Diagnostic AL. Testing challenges with novel gene alterations in KCNMA1-linked channelopathy: a case report. Annual Clinical Genetics Meeting; 2021 April. https://acmg.planion.com/Web.User/AbstractDet?ACCOUNT=ACMG&ABSID=10843&CONF=AM21&ssoOverride=OFF&CKEY=
- Zhang ZB, Tian MQ, Gao K, et al. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov Disord. 2015;30(9):1290–1292.
- Heim J, Vemuri A, Lewis S, et al. Cataplexy in patients harboring the KCNMA1 p.N999S mutation. Mov Disord Clin Pract. 2020;7(7):861–862.
- Bruno MK, Lee HY, Auburger GW, et al. Genotype-phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology. 2007;68(21):1782–1789.
- McGuire S, Chanchani S, Khurana DS. Paroxysmal dyskinesias. Semin Pediatr Neurol. 2018;25:75–81.
- Bhatia KP. Paroxysmal dyskinesias. Mov Disord. 2011;26(6):1157–1165.
- Sanders L. DIAGNOSIS: KCNMA1: just 21 people are known to have this rare genetic condition. Can you help us find more? N Y Times. 2018 August;23:2018.
- Sanders L. DIAGNOSIS: the boy slumped to the floor. Could these be seizures? N Y Times. 2020 June;3:2020.
- Braverman A Epidose #4: looking for a village. Netflix; 2019. Scott Rudin EB, Garrett Basch, P.G. Morgan, Jonathan Chinn, Simon Chinn.
- Parmar A, Murray BJ, Narang I. Clinical characteristics of cataplectic attacks in type 1 narcolepsy. Curr Neurol Neurosci Rep. 2020;20(9):38.
- Gibson R, Galentine W, Gunduz MT, et al. Novel phenotype of paroxysmal spells due to KCNMA1 de novo gene mutation mimicking epilepsy and responding to stimulant therapy. Abst. 3.446. In: American Epilespy Society Meeting; 2018. Washington, DC.
- Lima FCB, Do Nascimento Junior EB, Teixeira SS, et al. Thinking outside the box: cataplexy without narcolepsy. Sleep Med. 2019;61:118–121.
- Ewida A, Waseem F, Fahimi G, et al. Yet another N995S case, but with a second mutation (Exon 1, c.34_36dupAGC (p.Ser12dup)). In: American Epilespy Society Meeting; 2019. Baltimore, MD.
- Wang J, Yu S, Zhang Q, et al. KCNMA1 mutation in children with paroxysmal dyskinesia and epilepsy: case report and literature review. Transl Sci Rare Dis. 2017;2:8.
- Ewida A, Ghonim HT, Waseem F, et al. An unusual case of paroxysmal kinesigenic dyskineSIA (PKD) with epilepsy associated with KCNMA1 gene mutation. In: American Epilepsy Society Meeting; 2019. Baltimore, MD.
- Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291.
- Chen Y, Lu J, Pan H, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54(2):239–243.
- Heron SE, Khosravani H, Varela D, et al. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Annals of Neurology. 2007;62(6):560–568.
- Khosravani H, Bladen C, Parker DB, et al. Effects of Cav3. 2 channel mutations linked to idiopathic generalized epilepsy. Annals of Neurology. 2005;57(5):745–749.
- Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145(7):1036–1048.
- Chambers C, Jansen LA, Dhamija R. Review of commercially available epilepsy genetic panels. J Genet Couns. 2016;25(2):213–217.
- Rimmer A, Phan H, Mathieson I, et al. Integrating mapping-, assembly-and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46(8):912–918.
- O’Rawe J, Jiang T, Sun G, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5(3):28.
- Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229.
- Devinsky O, Asato M, Camfield P, et al. Delivery of epilepsy care to adults with intellectual and developmental disabilities. Neurology. 2015;85(17):1512–1521.
- Berg AT, Loddenkemper T, Baca CB. Diagnostic delays in children with early onset epilepsy: impact, reasons, and opportunities to improve care. Epilepsia. 2014;55(1):123–132.
- Hagemann G, Lemieux L, Free SL, et al. Cerebellar volumes in newly diagnosed and chronic epilepsy. J Neurol. 2002;249(12):1651–1658.
- Mirkowski MN Cognitive function in BK channel knock-out mice [Thesis]. London, Ontario, Canada: Western University; 2013.
- Matthews EA, Weible AP, Shah S, et al. The BK-mediated fAHP is modulated by learning a hippocampus-dependent task. Proc Natl Acad Sci. 2008;105(39):15154–15159.
- Kshatri A, Cerrada A, Gimeno R, et al. Differential regulation of BK channels by fragile X mental retardation protein. J General Physiol. 2020;152:6.
- Choi T-Y, Lee S-H, Kim Y-J, et al. Cereblon maintains synaptic and cognitive function by regulating BK channel. J Neurosci. 2018;38(14):3571–3583.
- Higgins JJ, Tal AL, Sun X, et al. Temporal and spatial mouse brain expression of cereblon, an ionic channel regulator involved in human intelligence. J Neurogenet. 2010 Mar;24(1):18–26.
- Song T, Liang S, Liu J, et al. CRL4 antagonizes SCFFbxo7-mediated turnover of cereblon and BK channel to regulate learning and memory. PLoS Genet. 2018;14(1):e1007165.
- Choi TY, Lee SH, Kim SJ, et al. BK channel blocker paxilline attenuates thalidomide-caused synaptic and cognitive dysfunctions in mice. Sci Rep. 2018;8(1):17653.
- Hébert B, Pietropaolo S, Même S, et al. Rescue of fragile X syndrome phenotypes in Fmr1KO mice by a BKCa channel opener molecule. Orphanet J Rare Dis. 2014;9(1):124.
- Hafidi A, Beurg M, Dulon D. Localization and developmental expression of BK channels in mammalian cochlear hair cells. Neuroscience. 2005;130(2):475–484.
- MacDonald SH, Ruth P, Knaus HG, et al. Increased large conductance calcium-activated potassium (BK) channel expression accompanied by STREX variant downregulation in the developing mouse CNS. BMC Dev Biol. 2006;6:37.
- Contet C, Goulding SP, Kuljis DA, et al. BK channels in the central nervous system. Int Rev Neurobiol. 2016;128:281–342.
- Liu Y, Lopez‐Santiago LF, Yuan Y, et al. Dravet syndrome patient‐derived neurons suggest a novel epilepsy mechanism. Ann Neurol. 2013;74(1):128–139.
- Chopra R, Isom LL. Untangling the Dravet syndrome seizure network: the changing face of a rare genetic epilepsy: the paradox of Dravet syndrome. Epilepsy Curr. 2014;14(2):86–89.
- Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One. 2013;8(10):e77843.
- Chen C, Westenbroek RE, Xu X, et al. Mice lacking sodium channel β1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004;24(16):4030–4042.
- Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking nav1. 6 sodium channels. J Neurosci. 2006;26(27):7172–7180.
- N’Gouemo P. BKCa channel dysfunction in neurological diseases. Front Physiol. 2014;5:373.
- Montgomery JR, Meredith AL. Genetic activation of BK currents in vivo generates bidirectional effects on neuronal excitability. Proc Natl Acad Sci. 2012;109(46):18997–19002.
- Jaffe DB, Wang B, Brenner R. Shaping of action potentials by type I and type II large-conductance Ca2+-activated K+ channels. Neuroscience. 2011;192:205–218.
- Ly C, Melman T, Barth AL, et al. Phase-resetting curve determines how BK currents affect neuronal firing. J Comput Neurosci. 2011;30(2):211–223.
- Sheehan JJ, Benedetti BL, Barth AL. Anticonvulsant effects of the BK-channel antagonist paxilline. Epilepsia. 2009;50(4):711–720.
- Tanner MR, Beeton C. Differences in ion channel phenotype and function between humans and animal models. Front Biosci (Landmark Ed). 2018;23:43–64.
- Kratschmer P, Lowe SA, Buhl E, et al. Impaired pre-motor circuit activity and movement in a drosophila model of KCNMA1-linked dyskinesia. Mov Disord. 2021;36(5):1158–1169.
- Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
- Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311.
- Laumonnier F, Roger S, Guerin P, et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry. 2006;163(9):1622–1629.
- Levine JB, Morrow EM, Berdichevsky Y, et al. BKca channel in autism and mental retardation. Am J Psychiatry. 2007;164(6):977–978.
- Lee CG, Lee J, Lee M. Multi-gene panel testing in Korean patients with common genetic generalized epilepsy syndromes. PLoS One. 2018;13(6):e0199321.
- Burns L, Minster R, Demirci F, et al. Replication study of genome‐wide associated SNPs with late‐onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2011;156(4):507–512.
- Plante AE, Lai MH, Lu J, et al. Effects of single nucleotide polymorphisms in human KCNMA1 on BK current properties. Front Mol Neurosci. 2019;12:285.
- Anttila V, Nyholt DR, Kallela M, et al. Consistently replicating locus linked to migraine on 10q22-q23. Am J Hum Genet. 2008;82(5):1051–1063.
- Al-Karagholi MA, Ghanizada H, Nielsen CAW, et al. Opening of BKCa channels alters cerebral hemodynamic and causes headache in healthy volunteers. Cephalalgia. 2020;40(11):1145–1154.
- Zhang X, Ni Y, Liu Y, et al. Screening of noise-induced hearing loss (NIHL)-associated SNPs and the assessment of its genetic susceptibility. Environ Health. 2019;18(1):30.
- Kendler KS, Kalsi G, Holmans PA, et al. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol Clin Exp Res. 2011;35(5):963–975.
- Drgon T, Johnson CA, Nino M, et al. “Replicated” genome wide association for dependence on illegal substances: genomic regions identified by overlapping clusters of nominally positive SNPs. Am J Med Genet B Neuropsychiatr Genet. 2011;156(2):125–138.
- Obeidat M, Zhou G, Li X, et al. The genetics of smoking in individuals with chronic obstructive pulmonary disease. Respir Res. 2018;19(1):59.
- Dopico AM, Bukiya AN, Kuntamallappanavar G, et al. Modulation of BK channels by ethanol. Int Rev Neurobiol. 2016;128:239–279.
- Salvi E, Kutalik Z, Glorioso N, et al. Genome-wide association study using a high-density SNP-array and case-control design identifies a novel essential hypertension susceptibility locus in the promoter region of eNOS. Hypertension. 2012;59(2):248.
- Tomas M, Vazquez E, Fernandez-Fernandez JM, et al. Genetic variation in the KCNMA1 potassium channel α subunit as risk factor for severe essential hypertension and myocardial infarction. J Hypertens. 2008;26(11):2147–2153.
- Morrison AC, Felix JF, Cupples LA, et al. Genomic variation associated with mortality among adults of European and African ancestry with heart failure: the cohorts for heart and aging research in genomic epidemiology consortium. Circulation. 2010;3(3):248–255.
- Fernández-Fernández JM, Tomás M, Vázquez E, et al. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J Clin Investig. 2004;113(7):1032–1039.
- Kelley-Hedgepeth A, Peter I, Montefusco MC, et al. The KCNMB1 E65K variant is associated with reduced central pulse pressure in the community-based Framingham offspring cohort. J Hypertens. 2009;27(1):55.
- Kokubo Y, Iwai N, Tago N, et al. Association analysis between hypertension and CYBA, CLCNKB, and KCNMB1 functional polymorphisms in the Japanese population. Circ J. 2005;69(2):138–142.
- Nielsen T, Burgdorf KS, Grarup N, et al. The KCNMB1 Glu65Lys polymorphism associates with reduced systolic and diastolic blood pressure in the Inter99 study of 5729 Danes. J Hypertens. 2008;26(11):2142–2146.
- Kohler R. Single-nucleotide polymorphisms in vascular Ca2+-activated K+-channel genes and cardiovascular disease. Pflugers Arch. 2010 Jul;460(2):343–351.
- Sentí M, Fernández-Fernández JM, Tomás M, et al. Protective effect of the KCNMB1 E65K genetic polymorphism against diastolic hypertension in aging women and its relevance to cardiovascular risk. Circ Res. 2005;97(12):1360–1365.
- Rasmussen ER, Hallberg P, Baranova EV, et al. Genome-wide association study of angioedema induced by angiotensin-converting enzyme inhibitor and angiotensin receptor blocker treatment. Pharmacogenomics J. 2020;20:770–783.
- Purkey MT, Li J, Mentch F, et al. Genetic variation in genes encoding airway epithelial potassium channels is associated with chronic rhinosinusitis in a pediatric population. PLoS One. 2014;9(3):e89329.
- Ueno K, Aiba Y, Hitomi Y, et al. Integrated GWAS and mRNA microarray analysis identified IFNG and CD40L as the central upstream regulators in primary biliary cholangitis. Hepatol Commun. 2020;4(5):724–738.
- Savas S, Briollais L, Ibrahim-zada I, et al. A whole-genome SNP association study of NCI60 cell line panel indicates a role of Ca2+ signaling in selenium resistance. PLoS One. 2010;5(9):e12601.
- Choi R, Sohn I, Kim MJ, et al. Pathway genes and metabolites in thiopurine therapy in Korean children with acute lymphoblastic leukaemia. Br J Clin Pharmacol. 2019;85(7):1585–1597.
- Matimba A, Li F, Livshits A, et al. Thiopurine pharmacogenomics: association of SNPs with clinical response and functional validation of candidate genes. Pharmacogenomics. 2014;15(4):433–447.
- Akiyama M, Takahashi A, Momozawa Y, et al. Genome-wide association study suggests four variants influencing outcomes with ranibizumab therapy in exudative age-related macular degeneration. J Hum Genet. 2018;63(10):1083–1091.
- Jiao H, Arner P, Hoffstedt J, et al. Genome wide association study identifies KCNMA1contributing to human obesity. BMC Med Genomics. 2011;4(1):51.
- Park S, Daily JW, Song MY, et al. Gene-gene and gene-lifestyle interactions of AKAP11, KCNMA1, PUM1, SPTBN1, and EPDR1 on osteoporosis risk in middle-aged adults. Nutrition. 2020:110859. DOI: https://doi.org/10.1016/j.nut.2020.110859.
- Fan Q, Guo X, Tideman JWL, et al. Childhood gene-environment interactions and age-dependent effects of genetic variants associated with refractive error and myopia: the CREAM consortium. Sci Rep. 2016;6:25853.
- Wong Y-L, Hysi P, Cheung G, et al. Genetic variants linked to myopic macular degeneration in persons with high myopia: CREAM Consortium. PLoS One. 2019;14(8):e0220143.
- Kathiresan S, Manning AK, Demissie S, et al. A genome-wide association study for blood lipid phenotypes in the Framingham heart study. BMC Med Genet. 2007;8(1):1–10.
- Schuckit MA, Wilhelmsen K, Smith TL, et al. Autosomal linkage analysis for the level of response to alcohol. Alcohol Clin Exp Res. 2005;29(11):1976–1982.
- Tian WT, Huang XJ, Mao X, et al. Proline-rich transmembrane protein 2-negative paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 163 patients. Mov Disord. 2018;33(3):459–467.
- Wu H, Li H, Bai T, et al. Phenotype-to-genotype approach reveals head-circumference-associated genes in an autism spectrum disorder cohort. Clin Genet. 2020;97(2):338–346.
- Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067.
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443.