

Abstract
Incretin-based therapy such as GLP-1 receptor agonists and DPP-4 inhibitors for type 2 diabetes mellitus is characterized by glucose-dependent insulin secretion and glucose-inhibited glucagon secretion. Recently, autophagy deficiency in islet β cells has been shown to contribute to the pathogenesis of type 2 diabetes mellitus however, with the role of incretin has not been established. To evaluate the role of autophagy in incretin effects, 8-week-old male β cell-specific Atg7 knockout (Atg7Δβ cell) mice and wild-type mice were administered vildagliptin for 12 weeks. Vildagliptin treatment improved glucose intolerance and hypoinsulinemia; however, it failed to suppress serum glucagon levels after glucose loading in the Atg7Δβ cell mice. Ex vivo glucose-induced glucagon suppression was also blunted in the islets from vildagliptin-treated Atg7Δβ cell mice. The α cell mass was not affected by β cell autophagy deficiency or vildagliptin. However, glucagon mRNA expression was significantly increased by vildagliptin in the autophagy-deficient islets, and was significantly reduced by vildagliptin in wild-type islets. Pancreatic glucagon contents were not in agreement with the changes in mRNA expression, suggesting a dysregulation in glucagon translation and secretion. In vitro studies revealed that glucose-stimulated cAMP production was impaired in the autophagy-deficient islets exposed to exendin-4. Taken together, the results suggest that the constitutive autophagy in β cells could regulate incretin-induced glucagon expression and release in α cells, and that cAMP may play a role in this process.
Abbreviations
| ANOVA | = | analysis of variance |
| Atg7Δβ cell | = | beta cell-specific Atg7-deficient |
| AUC | = | area-under the curve |
| DPP-4 | = | dipeptidyl peptidase-4 |
| ELISA | = | enzyme-linked immunosorbent assay |
| ER | = | endoplasmic reticulum |
| GLP-1 | = | glucagon-like peptide-1 |
| GSIS | = | glucose-stimulated insulin secretion |
| KRB | = | Krebs-Ringer Bicarbonate |
| HG | = | high glucose |
| IHC | = | immunohistochemical |
| IPGTT | = | intraperitoneal glucose tolerance test |
| ITT | = | insulin tolerance test |
| LG | = | low glucose |
| NS | = | no significant difference |
| RT-PCR | = | real time polymerase chain reaction |
| T2DM | = | type 2 diabetes mellitus |
Introduction
Pancreatic islets are composed of several hormone-releasing cell types, including α cells and β cells. It is known that different types of islet cells interact with each other via a paracrine mechanism.Citation1 Glucagon-like peptide-1 (GLP-1), one of the incretin hormones, stimulates insulin secretion in β cells, inhibits glucagon release in α cells, and lowers blood glucose levels in a glucose-dependent manner.Citation2 Currently, incretin-based therapies such as GLP-1 receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors, which inhibit GLP-1 degradation and prolong its effects, are important treatment regimens for type 2 diabetes mellitus (T2DM).Citation3 In addition, incretin-based therapy has been reported to regenerate β cells and to inhibit β cell apoptosis in some animal models.Citation2 However, it is unclear whether incretin-based therapy affects the α cell mass.Citation4 A small human autopsy study described hyperplasia of pancreatic α cells in T2DM patients treated with incretin therapy.Citation5 However, extensive preclinical studies in rodents and nonhuman primates failed to detect α cell hyperplasia and even reported a decrease in α cell mass as a result of incretin therapy,Citation6-8 including vildagliptin,Citation9,10 a DPP-4 inhibitor.
Autophagy is a lysosomal degradation pathway that is essential for cell survival, differentiation, development, homeostasis, and the control of cellular metabolism, including in pancreatic β cells.Citation11 The β cell-specific autophagy-deficient mice by Atg7 deletion (Atg7Δβ cell mice) showed glucose intolerance and hypoinsulinemia.Citation12,13 Decreased β cell function and mass were observed in the mice, and were associated with a distended endoplasmic reticulum (ER), suggesting ER stress and mitochondrial dysfunction in the β cells.Citation12-15 Furthermore, autophagy deficiency was shown to contribute to β cell dysfunction and β cell loss in human T2DM.Citation16 Therefore, it is important to consider the effects of autophagy deficiency in the management of T2DM. There is one report on the use of incretin therapy in Atg7Δβ cell mice.Citation17 Exendin-4, a GLP-1 receptor agonist, decreased β cell death, increased insulin secretion, and improved glucose intolerance in Atg7Δβ cell mice. However, the influence on α cells, another target of GLP-1, was not described.
Therefore, we evaluated the long-term effects of a DPP-4 inhibitor on α cell mass and function in β cell-specific autophagy-deficient mice, to further examine the role of β cell autophagy in incretin effects.
Results
Vildagliptin enhanced insulin secretion and improved glucose intolerance in Atg7Δβ cell mice
In the Atg7Δβ cell mice, body weights were slightly lower and fed blood glucose levels were higher than those of wild-type mice, regardless of treatment (p < 0.005; ). In each mouse genotypes, the vildagliptin treatment did not affect these variables. However, according to the intraperitoneal glucose tolerance test (IPGTT), vildagliptin significantly reduced glucose intolerance in the Atg7Δβ cell mice (). There was no difference in glucose tolerance by the vildagliptin treatment in wild-type mice. As previously reported,Citation17 vildagliptin significantly increased fasting serum insulin levels in both genotypes (). Insulin tolerance test (ITT) results demonstrated no significant differences among the groups (), suggesting comparable insulin actions.
Figure 1. In vivo effects of vildagliptin on Atg7Δβ cell mice. During the 12-week experiments, (A) body weights and (B) fed blood glucose levels of the mice were monitored weekly. (C) After 12 weeks of vildagliptin treatment, IPGTT was performed, and (D) the area-under the curve (AUC) of IPGTT was calculated (n > 30 in each group). (E) Fasting serum insulin levels (n > 7 in each group) were measured. (F) ITT and (G) the AUC (n > 6 in each group) were examined. In (A)–(C) and (F), repeated measures ANOVA and post-hoc Tukey's HSD test were performed among the 4 groups: # indicates significant difference (p < 0.005) between wild-type and Atg7Δβ cell mice regardless of treatment; * indicates significant difference (p < 0.05) of the control-Atg7Δβ cell group from the other 3 groups. In (D), (E) and (G), Student's t tests were performed between control and vildagliptin groups in each mouse genotype; NS indicates no significant difference.
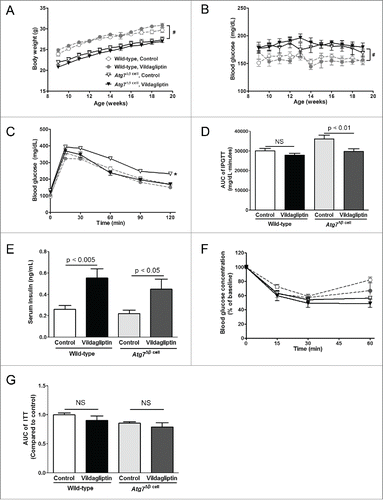
Vildagliptin blunted glucose-induced glucagon suppression in Atg7Δβ cell mice
Glucose loading significantly suppressed serum glucagon levels in both mouse genotypes (). However, the glucose-induced glucagon suppression disappeared in the vildagliptin-treated Atg7Δβ cell mice. Next, glucose stimulation of isolated islets was done ex vivo (). Vildagliptin induced significant glucagon suppression by high glucose in the wild-type islets, while it did not in the Atg7Δβ cell islets. The glucagon suppression index calculated from the ex vivo study demonstrated different responses to vildagliptin between the genotypes ().
Figure 2. Glucose-induced glucagon suppression by vildagliptin was impaired in Atg7Δβ cell islets. (A) After 12 weeks of vildagliptin treatment, serum was obtained before and 15 min after glucose loading, and glucagon levels were measured (n > 7 in each group). (B) Islets were isolated (n > 10 in each group), and glucose stimulation was performed. Supernatant glucagon levels were measured and adjusted by the cellular protein concentrations. (C) Suppression index of glucagon was calculated by the ratio of supernatant glucagon levels during HG for 1 h to those during LG for 1 h, and was compared between control and vildagliptin groups in each mouse genotype by the Student's t tests. HG, high glucose (15 mM); LG, low glucose (1.5 mM); NS, no significant difference * indicates significant difference (p < 0.05) by paired t-tests between before and after glucose stimulation.
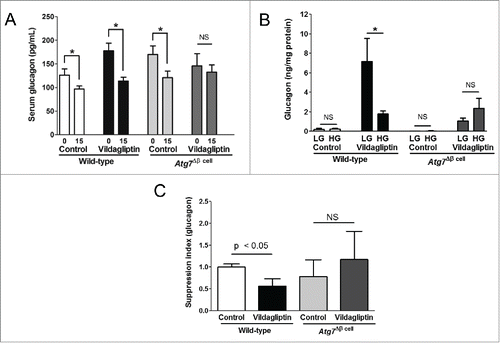
Vildagliptin affected glucagon synthesis but not α cell mass in Atg7Δβ cell mice
We assessed the α cell mass using point counting after immunohistochemical (IHC) staining for glucagon in pancreas sections (). Morphologies, distributions, and masses of α cells were comparable regardless of genotypes or vildagliptin treatment (). Quantitative real-time polymerase chain reaction (RT-PCR) with isolated islets demonstrated that mRNA expression levels of glucagon decreased in the wild-type islets while mRNA increased in the Atg7Δβ cell islets after vildagliptin treatment (). Changes in pancreatic glucagon contents by vildagliptin were opposite to the changes in glucagon transcripts in both mouse genotypes, although they were not significant ().
Figure 3. Effects of vildagliptin on the α cell mass and glucagon synthesis in Atg7Δβ cell mice. (A) Pancreas sections were stained with the anti-glucagon antibody. Representative images of islets from control (left) and vildagliptin-treated (right) Atg7Δβ cell mice are shown (scale bar, 100 μm). (B) The α cell mass was calculated by point counting (n = 4∼5 in each group). (C) The mRNA expression of glucagon compared to that of GAPDH in the islets was assessed by quantitative RT-PCR (n > 6 in each group). (D) Pancreatic glucagon contents were measured from pancreatic extracts (n > 5 in each group). Student's t tests were used between control and vildagliptin groups in each mouse genotype; NS indicates no significant difference.
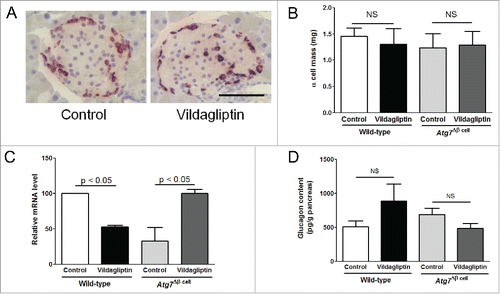
Glucose-stimulated cAMP responses were blunted in Atg7Δβ cell islets exposed to exendin-4 in vitro
Long-term administration of vildagliptin regulated α cell function to produce and secret glucagon in vivo. However, the cause of blunted glucose-induced glucagon suppression remains uncertain. Therefore, we examined GLP-1 signaling using in vitro exendin-4 treatments. Because cAMP is a key mediator of the GLP-1 pathway,Citation18 we determined if the cAMP response correlated with autophagy status. Although high glucose was challenged in the presence of exendin-4, cAMP did not increase in the Atg7Δβ cell islets, unlike in the wild-type islets (). However, an increase of ATP by high glucose levels in the presence of exendin-4 was unaffected regardless of β cell autophagy status (). Because cAMP is synthesized from ATP by adenylyl cyclase through the GLP-1 receptor,Citation18 these findings suggested that GLP-1 signaling was impaired in the autophagy-deficient islets.
Figure 4. Glucose-stimulated cAMP production by incretin was impaired in Atg7Δβ cell islets. The Atg7Δβ cell and wild-type islets were treated with or without 50 nM of exendin-4 in vitro for 2 h (n of experiments > 6), and then glucose stimulation was performed. Cellular cAMP (A), and ATP contents (B) were measured under low glucose (LG, 1.5 mM) or high glucose (HG, 15 mM), and adjusted by cellular protein. * indicates significant difference (p < 0.05) by paired t-tests between LG and HG. NS indicates no significant difference.
Discussion
In this study, and as previously reported, the effects of long-term vildagliptin treatment on glucose homeostasis of Atg7Δβ cell mice was favorable,Citation17 as shown by improved glucose tolerance from enhanced insulin secretion. However, the well-known incretin effect of glucose-induced glucagon suppression was abrogated (). Therefore, we can infer that β cell autophagy has a role in the incretin effects on α cells.
Constitutive autophagy deficiency in β cells did not affect α cell morphology and mass (), although the number of mice tested was too small to detect slight differences. However, autophagy deficiency in β cells did affect α cell function involving incretin. The expression of the glucagon transcript was increased by vildagliptin in the islets with autophagy-deficient β cells, contrary to in the wild-type islets (). In contrast, pancreatic glucagon content was not increased by vildagliptin in Atg7Δβ cell mice (). In the wild-type mice, in spite of the suppressed mRNA expression by vildagliptin, pancreatic contents did not decrease. These findings can be interpreted as follows. First, translational processes might be impaired in the autophagy-deficient islets, which could reduce the increase of glucagon peptides. Second, glucagon secretory processes might not be adequately inhibited by vildagliptin in the autophagy-deficient islets, which could also contribute to the lack of an increase of pancreatic contents, along with no-decrease of serum glucagon levels by glucose stimulation (). However, the discrepancy between glucagon mRNA levels and glucagon contents could have come from the experimental methods used. While glucagon mRNA levels were measured with isolated islets, glucagon contents were measured with whole pancreatic extracts. Comparable pancreatic weights (data not shown) and α cell masses () among the groups may have prevented erroneous results, but islet isolation procedures could have still affected the discrepancy. The effects of autophagy on incretin-stimulated glucagon production and secretion in the islets have not been previously reported, and the mechanisms require further verification.
Glucagon secretion is regulated by circulating insulin and glucose.Citation1,19 Therefore, our in vivo observations must have been affected by the hyperglycemia present in the Atg7Δβ cell mice, although the hyperglycemia was mild (). Therefore, we performed in vitro experiments to evaluate the role of autophagy in the GLP-1 pathway, to exclude the effects of hyperglycemia. It is well-known that GLP-1 potentiates glucose-stimulated cAMP increases in β cells, and that this contributes to insulin secretion in a glucose-dependent manner.Citation18 However, in the Atg7Δβ cell islets, we did not observe the cAMP response to glucose stimulation in the presence of exendin-4 (). Because we measured cAMP in whole islets, it was not possible to identify the cAMP response according to cell types. Although incretin induced cAMP in β cells and α cells via incretin receptors,Citation20 most of the mouse islets were composed of β cells, therefore the cAMP response in the mouse islets represents the response in the autophagy-deficient β cells. Generation of ATP, the precursor of cAMP, was not be affected by autophagy status in β cells (). This suggested that constitutive autophagy in β cells had a role in cAMP generation from ATP in response to GLP-1. The possible causes of impaired cAMP generation, including GLP-1 receptor desensitizationCitation21 and adenylyl cyclase down-regulation,Citation22 could have been affected by autophagy deficiency.
One concern with the use of autophagy-deficient islets in this study is that they are insulin deficient.Citation12 Glucagon secretion is closely regulated by insulin.Citation19 However, according to Abe et al,Citation17 exendin-4 increased glucose-dependent insulin secretion in the same mouse model, suggesting that insulin response to incretin was preserved even in autophagy-deficient β cells. Therefore, we suggest that β cell-specific autophagy deficiency directly affects α cell function, although indirect insulin effects cannot be excluded completely by the current data.
A limitation of this study design is that there could be confounding effects by genetic modulation. First, RIP-Cre mice can show Cre expression in the brain, or human growth hormone minigenes,Citation23,24 could affect the experimental results. The lower body weights in the Atg7Δβ cell mice () are consistent with this possibility, although we did not study the diet. Second, the mixed background of the Balb/c mice could have affected islet function, despite the use of littermates.
In summary, we found that long-term vildagliptin treatment failed to suppress glucagon secretion after glucose loading in the Atg7Δβ cell mice in vivo and ex vivo, and was associated with changes in α cell function of glucagon production. Impaired cAMP responses and resultant changes in insulin around α cells may have contributed to these results.Citation19 GLP-1 receptor agonists and DPP-4 inhibitors are known to inhibit glucagon secretion most likely via GLP-1 receptor–dependent stimulation of somatostatin secretion.Citation4 A recent study reported that both insulin and somatostatin were required in glucose-inhibited glucagon secretion.Citation25 Therefore, β cell autophagy may also have a role in somatostatin secretion. DPP-4 expression on the islet cell surface may also have influenced our results.Citation4
In conclusion, β cell autophagy is required for the incretin effects on cAMP generation and the glucagon suppression in response to glucose.
Material and Methods
Animals
Atg7F/F mice (C57Bl/6J background) were from Professor Masaaki Komatsu,Citation26 and were backcrossed with the Balb/c mouse strain. Mice with backcross numbers over 3 were used for the study, and were compared among the littermates. RIP-Cre mice (C57Bl/6J background) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Atg7F/F mice were crossed with RIP-Cre mice to generate Atg7Δβ cell mice (mixed background of C57Bl/6J and Balb/c). RIP-Cre littermates and Atg7F/F littermates were used as wild-type controls. Male mice were fed standard chow diets, and were randomized into control and vildagliptin groups at 8 weeks of age. In the vildagliptin group, vildagliptin was administered via drinking water (0.3 mg/mL) for 12 weeks.Citation9,27 The fed blood glucose levels and body weights were monitored weekly using a glucometer (One Touch, Johnson & Johnson, Milpitas, CA, USA) and standard scale, respectively. All animal experiments were conducted in accordance with the institutional guidelines of Seoul National University Hospital.
IPGTT and ITT
After completion of the 12 week vildagliptin or vehicle administration at 20 weeks of age, IPGTT and ITT were performed. The ITT was performed after 6 h of fasting, with a 0.75 unit/kg (body weight) intraperitoneal insulin injection, followed by serial monitoring of blood glucose for 60 min. The IPGTT was performed after overnight fasting, with a 1 g/kg (body weight) intraperitoneal glucose injection, followed by serial glucose monitoring. During the IPGTT, serum was also obtained from retro-orbital blood samples to measure insulin (enzyme-linked immunosorbent assay (ELISA) kit, ALPCO, Salem, MA, USA) and glucagon concentrations (ELISA kit, R&D System Inc., Minneapolis, MN, USA).
Ex vivo isolated islet experiments
Pancreatic islets were isolated from overnight fasted mice using the collagenase digestion technique.Citation12 For the glucose-regulated glucagon secretion assay, isolated pancreatic islets were starved in Krebs-Ringer bicarbonate (KRB) buffer containing 0.2% bovine serum albumin and 1.5 mM glucose for 1 h at 37°C. Then, the media was replaced with KRB containing 1.5 mM or 15 mM glucose. After a 1 h incubation, the supernatant was collected for the measurement of glucagon (ELISA kit, R&D System, Inc.), and the islets were harvested for measurement of cellular protein concentrations, ATP (Sigma-Aldrich, St Louis, MO, USA), and cAMP (Cell Biolabs, Inc., San Diego, CA, USA). The suppression index for glucagon was calculated using the supernatant glucagon levels obtained from islets incubated in 15 mM glucose divided by the glucagon level from supernatant incubated in 1.5 mM glucose. Exendin-4 (Sigma-Aldrich), a GLP-1 receptor agonist, was added for 2 h in some in vitro experiments.
Estimation of α cell mass
The pancreas was embedded in paraffin and sectioned to a thickness of 4 μm. IHC staining was performed with the anti-glucagon antibody (Sigma-Aldrich), and the α cell mass was then estimated by point counting.Citation12 In brief, the point numbers of glucagon-stained areas were counted at a magnification of 400× using a 48-point grid. The relative α cell area was calculated by dividing the point number of the glucagon-stained area by the point number over the entire pancreatic tissue sample on the same section. Two sections, separate from each other by 20 μm per mouse, were used for the mean relative area determination. The average number of islets was 20 ± 10 per section. The α cell mass was then estimated by multiplying α cell relative area by the pancreas weight.
Quantitative real-time polymerase chain reaction (RT-PCR) of glucagon mRNA
Total RNA was isolated from the islets, and the cDNA was synthesized and amplified using the SYBR Premix Ex Taq polymerase (TaKaRa, Otsu, Japan). The quantitative RT-PCR was performed, and analysis was performed using the LightCycler® 96 Real-Time PCR System (Roche, Mannheim, Germany). The forward primer sequence for glucagon was 5′- TGAAGACAAACGCCACTCAC-3′ and the reverse primer sequence was 5′- CAGCATGCCTCTCAAATTTCA-3′. Those of GAPDH used for standardization were 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAGTTGAGGTCA-3′, respectively.
Pancreatic glucagon contents
Pancreatic hormones were extracted using a previously published method.Citation28 ELISA was performed to measure the glucagon levels of the pancreatic extracts (R&D System, Inc.). The concentrations were normalized by pancreas weights.
Statistical analysis
All data are expressed as means ± standard error of the mean. A repeated measures analysis of variance (ANOVA) and subsequent post-hoc Tukey's Honestly Significant Difference (HSD) test were used for serial changes in body weight and blood glucose levels (). Student's t tests were employed to compare vildagliptin effects in the wild-type or in the knock-out groups. Paired t-tests were performed to evaluate glucagon, cAMP, and ATP responses to glucose stimulation. All analyses were performed using SPSS 14.0 for Windows (SPSS Inc., Chicago, IL, USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Professor Masaaki Komatsu for the kind donation of Atg7-floxed mice, and to Novartis for the supply of vildagliptin.
Funding
This study was supported by a grant from the Innovative Research Institute for Cell Therapy (A062260) by the Ministry of Health and Welfare, Republic of Korea, the Dong-A grant from the Korean Diabetes Association, and the research grant from the Seoul National University Hospital (03-2010-0300).
References
- Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obesity Metabol 2011; 13 Suppl 1:95-105; http://dx.doi.org/10.1111/j.1463-1326.2011.01450.x
- Lee YS, Jun HS. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic β-cells. Metab 2014; 63:9-19; http://dx.doi.org/10.1016/j.metabol.2013.09.010
- Cho YM, Wideman RD, Kieffer TJ. Clinical application of glucagon-like Peptide 1 receptor agonists for the treatment of type 2 diabetes mellitus. Endocrinol Metab 2013; 28:262-74; http://dx.doi.org/10.3803/EnM.2013.28.4.262
- Drucker DJ. Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes 2013; 62:3316-23; PMID:23818527; http://dx.doi.org/10.2337/db13-0822
- Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 2013; 62:2595-604; PMID:23524641; http://dx.doi.org/10.2337/db12-1686
- Parkes DG, Mace KF, Trautmann ME. Discovery and development of exenatide: the first antidiabetic agent to leverage the multiple benefits of the incretin hormone, GLP-1. Exp Opin Drug Discov 2013; 8:219-44; http://dx.doi.org/10.1517/17460441.2013.741580
- Nyborg NC, Molck AM, Madsen LW, Knudsen LB. The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes 2012; 61:1243-9; PMID:22338093; http://dx.doi.org/10.2337/db11-0936
- Engel SS, Williams-Herman DE, Golm GT, Clay RJ, Machotka SV, Kaufman KD, Goldstein BJ. Sitagliptin: review of preclinical and clinical data regarding incidence of pancreatitis. Int J Clin Practice 2010; 64:984-90; http://dx.doi.org/10.1111/j.1742-1241.2010.02382.x
- Sato K, Nakamura A, Shirakawa J, Muraoka T, Togashi Y, Shinoda K, Orime K, Kubota N, Kadowaki T, Terauchi Y. Impact of the dipeptidyl peptidase-4 inhibitor vildagliptin on glucose tolerance and β-cell function and mass in insulin receptor substrate-2-knockout mice fed a high-fat diet. Endocrinol 2012; 153:1093-102; http://dx.doi.org/10.1210/en.2011-1712
- Busch SJ, Hoffmann P, Sahota P, Johnson R, Kothny W, Meyer F, Foley JE. Studies in rodents with the dipeptidyl peptidase-4 inhibitor vildagliptin to evaluate possible drug-induced pancreatic histological changes that are predictive of pancreatitis and cancer development in man. Diabetes Obesity Metab 2013; 15:72-6; http://dx.doi.org/10.1111/j.1463-1326.2012.01678.x
- Quan W, Lee MS. Role of autophagy in the control of body metabolism. Endocrinol Metab 2013; 28:6-11; http://dx.doi.org/10.3803/EnM.2013.28.1.6
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab 2008; 8:318-24; PMID:18840362; http://dx.doi.org/10.1016/j.cmet.2008.08.013
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. Autophagy is important in islet homeostasis and compensatory increase of β cell mass in response to high-fat diet. Cell Metab 2008; 8:325-32; PMID:18840363; http://dx.doi.org/10.1016/j.cmet.2008.08.009
- Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging 2009; 1:425-37; PMID:20157526
- Kim MJ, Choi OK, Chae KS, Kim MK, Kim JH, Komatsu M, Tanaka K, Lee H, Chung SS, Kwak SH, et al. Mitochondrial Complexes I and II Are More Susceptible to Autophagy Deficiency in Mouse β-Cells. Endocrinol Metab 2015; 30:65-70; http://dx.doi.org/10.3803/EnM.2015.30.1.65
- Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, Marchetti P. Autophagy in human type 2 diabetes pancreatic β cells. Diabetologia 2009; 52:1083-6; PMID:19367387; http://dx.doi.org/10.1007/s00125-009-1347-2
- Abe H, Uchida T, Hara A, Mizukami H, Komiya K, Koike M, Shigihara N, Toyofuku Y, Ogihara T, Uchiyama Y, et al. Exendin-4 Improves β-Cell Function in Autophagy-Deficient β-Cells. Endocrinol 2013; 154:4512-24; http://dx.doi.org/10.1210/en.2013-1578
- Seino S. Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia 2012; 55:2096-108; PMID:22555472
- Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, Herrera PL, Polonsky KS, McGuinness OP, Kulkarni RN. Insulin signaling in α cells modulates glucagon secretion in vivo. Cell Metab 2009; 9:350-61; PMID:19356716; http://dx.doi.org/10.1016/j.cmet.2009.02.007
- Ding WG, Renstrom E, Rorsman P, Buschard K, Gromada J. Glucagon-like peptide I and glucose-dependent insulinotropic polypeptide stimulate Ca2+-induced secretion in rat α-cells by a protein kinase A-mediated mechanism. Diabetes 1997; 46:792-800; PMID:9133546; http://dx.doi.org/10.2337/diab.46.5.792
- Rajan S, Torres J, Thompson MS, Philipson LH. SUMO downregulates GLP-1-stimulated cAMP generation and insulin secretion. Am J Physiol Endocrinol Metab 2012; 302:E714-23; PMID:22234371; http://dx.doi.org/10.1152/ajpendo.00486.2011
- Delmeire D, Flamez D, Hinke SA, Cali JJ, Pipeleers D, Schuit F. Type VIII adenylyl cyclase in rat β cells: coincidence signal detector/generator for glucose and GLP-1. Diabetologia 2003; 46:1383-93; PMID:13680124; http://dx.doi.org/10.1007/s00125-003-1203-8
- Brouwers B, de Faudeur G, Osipovich AB, Goyvaerts L, Lemaire K, Boesmans L, Cauwelier EJ, Granvik M, Pruniau VP, Van Lommel L, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab 2014; 20:979-90; PMID:25470546; http://dx.doi.org/10.1016/j.cmet.2014.11.004
- Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, et al. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 2010; 59:3090-8; PMID:20802254; http://dx.doi.org/10.2337/db10-0624
- Elliott AD, Ustione A, Piston DW. Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic α-cell by lowering cAMP. Am J Physiol Endocrinol Metab 2015; 308:E130-43; PMID:25406263; http://dx.doi.org/10.1152/ajpendo.00344.2014
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Flock G, Baggio LL, Longuet C, Drucker DJ. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes 2007; 56:3006-13; PMID:17717280; http://dx.doi.org/10.2337/db07-0697
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, et al. Selective β-cell loss and α-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88:2300-8; PMID:12727989; http://dx.doi.org/10.1210/jc.2002-020735

