

ABSTRACT
Maintenance of pancreatic β-cell mass and function is fundamental to glucose homeostasis and to prevent diabetes. The PI3 K-Akt-mTORC1 pathway is critical for β-cells mass and function, while PDX1 has been implicated in β-cell development, maturation, and function. Here we tested whether Akt signaling requires PDX1 expression to regulate β-cell mass, proliferation, and glucose homeostasis. In order to address that, we crossed a mouse model overexpressing constitutively active Akt mutant in β-cells (β-caAkt) with mice lacking one allele of PDX1gene (β-caAkt/pdx1+/-). While the β-caAkt mice exhibit higher plasma insulin levels, greater β-cell mass and improved glucose tolerance compared to control mice, the β-caAkt/pdx1+/- mice are hyperglycemic and intolerant to glucose. The changes in glucose homeostasis in β-caAkt/pdx1+/- were associated with a 60% reduction in β-cell mass compared to β-caAkt mice. The impaired β-cell mass in the β-caAkt/pdx1+/- mice can be explained by a lesser β-cell proliferation measured by the number of Ki67 positive β-cells. We did not observe any differences in apoptosis between β-caAkt/pdx1+/- and β-caAkt mice. In conclusion, PDX1 contributes to β-cell mass expansion and glucose metabolism induced by activation of Akt signaling.
Introduction
Type 2 diabetes (T2D) is characterized by defective β-cell expansion and adaptation in conditions of increased insulin demand. In addition to alteration in mass, defects in insulin secretion also contribute to the development of hyperglycemia during the pathogenesis of T2D.Citation1,Citation2 The capacity of β-cells to increase its mass in response to insulin resistance is critical to maintain glucose homeostasis resulting from a dynamic balance of neogenesis, proliferation, cell size and apoptosis.Citation3 The contribution of these different components in regulation of β-cell mass under normal or regenerative conditions varies but replication of β-cells is a major determinant to maintain beta cell mass under basal conditions.Citation4,Citation5 How β-cells adapts to insulin resistance is not completely understood but published evidence suggest that Akt is involved.Citation1
Akt is a converging pathway for growth factors, insulin, incretins, and glucose in β-cells.Citation6–Citation11 Several lines of evidence suggest that activation of the IRS2/phosphoinositide 3-kinase (PI3 K)/Akt pathway plays a critical role in the regulation of β-cell mass and function.Citation8,Citation12-Citation20 Transgenic mice with gain and loss of function of Akt have provided further evidence for the role of this kinase in β-cells.Citation18,Citation20,Citation21 Overexpressing a constitutively active form of Akt1 specifically in islet β-cells (β-caAkt mice) induces striking increases in β-cell mass, proliferation, neogenesis and cell size as well as improved glucose tolerance and insulin secretion.Citation20,Citation21 The increase in β-cell proliferation was associated with increased cyclin D1, D2, p21 levels, and Cdk4.Citation5 On the other hand, overexpression of a kinase-dead Akt1 in β-cells provokes hypoinsulinemia and mild hyperglycemia accompanied by reduced glucose-stimulated insulin secretion and impaired glucose tolerance, increasing the susceptibility to diabetes.Citation18
Pancreatic duodenal homeobox-1 (PDX1), also known as insulin promoter factor 1 (IPF1) in humans, is a homeobox gene that plays critical roles in the developing pancreas.Citation22 PDX1 is required for the development of both pancreatic exocrine and endocrine cells since inactivating mutation in exon 1 of IPF1 in humans and targeted disruption of exon 2 in mice lead to pancreas agenesis.Citation22,Citation23 PDX1 heterozygous mice develop glucose intolerance later on in life due to alterations in β-cell survival.Citation24 In addition, disruption of PDX1 in developing β-cells using insulin-driven Cre recombinase results in hyperglycemia, compromised insulin-expressing cells, and increased glucagon-expressing cells.Citation25,Citation26 Interestingly, PDX1 expression becomes restricted to mature β-cells, suggesting that PDX1 plays a role in β-cells function. Indeed, conditional deletion of PDX1 in 80% of mature β-cells (after the first four weeks of age in mice) in a tamoxifen-inducible conditional knockout model induces diabetes by switching the identity of β-cells to a α-cell-like phenotype.Citation27 Importantly, PDX1 has a pro-survival role in β-cellCitation28 and it regulates the expression of a number of β-cell genes, e.g., insulin, islet amyloid, glucokinase, and the glucose transporter 2.Citation29–Citation34 Therefore, PDX1 is critical for pancreas development, β-cell maturation, and function. The published evidence underscores the importance of PDX1 in development, insulin secretion and β-cell survival, yet the role of PDX1 in proliferation is less known. For instance, it has been shown that PDX1 deficiency during β-cell adaptation to insulin resistance impairs β-cell proliferation and negatively impact β-cell expansion.Citation35
There are several lines of evidence linking insulin and/or Akt signaling pathway to PDX1: i) Foxo1 acts as a repressor of Foxa2 which activates PDX1 promoter; ii) Diabetes in the insulin receptor substrate knockout mice (IRS2KO) is rescued by genetically haploinsufficiency of Foxo1;Citation36 iii) Insulin protects islets from apoptosis via PDX1, partially mediated by AktCitation28 iv) GSK3β phosphorylates PDX1 and tags for proteasome degradationCitation37 and v) expression of a kinase dead mTOR decreases PDX1 protein content in islets.Citation38 However, the role played by PDX1 in Akt-induced β-cell expansion has not been addressed.
In the present study, we assessed the role of PDX1 in β-cell proliferation by crossing mice overexpressing a constitutively active Akt mutant in β-cell with the PDX1 haploinsufficient mice. Remarkably, deficiency in PDX1 blunted all the improvements in glucose tolerance, β-cell proliferation, and β-cell mass induced by increased Akt signaling. These studies show that β-cell expansion induced by Akt is mediated, at least in part, by PDX1.
Methods
Mice
All procedures were approved by University of Miami IACUC (#15-196). Mice overexpressing constitutively active Akt1 in β-cells (β-caAkt) have a C57BL/6 J background and have been described previously.Citation5,Citation20,Citation39 PDX1 haploinsufficient mice have been also described previously.Citation22,Citation24 We generated a new mouse model by crossing the β-caAkt mouse with the pdx1+/- mouse to study the role of PDX1 in the context of Akt-induced β-cell proliferation and expansion. Male mice were used in the experiments.
Metabolic studies
Fasting blood samples were obtained from the tail vein after fasting the mice overnight. Glucose was measured on whole blood using Accu-Chek II glucometer (Roche Diagnostics, Indianapolis, IN). Plasma insulin levels were determined using Rat insulin ELISA kit (Linco, Saint Louis, MO). Glucose tolerance tests were performed in 18-h–fasted animals by injecting glucose (2 g/kg) intraperitoneally as described previously.Citation40
Islet morphometry and proliferation analysis
Pancreata were fixed overnight in formalin solution, and embedded in paraffin using standard techniques as described previously.Citation20 Sections (5 μm) were placed on glass slides, deparaffinized with xylene, and stained for insulin by using guinea pig anti-human insulin AB (1:300; BioGenex Laboratories, San Ramon, California, USA) and the Histomouse-SP kit (Zymed Laboratories Inc., South San Francisco, California, USA) using 3-amino-9-ethyl carbazole (AEC) as chromogen according to the manufacturers’ instructions. Endogenous peroxidase was quenched with 0.6% H202/methanol for 30 minutes. The β-cell mass was calculated by point counting morphometry from five insulin-stained sections (5 µm) separated by 200 µm as previously described.Citation40 Proliferation was assessed in insulin- and rabbit anti-human Ki67 (1:200; # Ki67p/VPK451, Leicabiosystems)-stained sections. At least 2000 insulin-stained cells were counted for each animal.
Caspase 3 immunostaining
Sections were mounted on poly-L-lysine-coated slides, deparaffinized and microwaved. Guinea pig anti-insulin (DAKO, Carpinteria, CA) and rabbit anti-cleaved caspase 3 (1:50; Cell Signaling, Beverly, MA) were applied overnight at 4°C and washed in PBS. After washes, the slides were incubated simultaneously with anti-guinea pig-FITC and Anti Rabbit-Cy3 for 4 h at room temperature. Slides were washed and covered it with DAPI.
Imaging
Leica DM5500 immunofluorescence microscope with motorized stage and Image-Pro analysis software was used for imaging acquisition.
Statistical analysis
Quantitative data are presented as the mean ± s.e.m. One-way ANOVA (Analysis of Variance) followed by tukey posthoc test was used to compare all four groups. All statistical analysis was performed using Prism 7 and p < .05 was considered statistically significant.
Results
PDX1 is required for the β-caAkt mouse metabolic phenotype
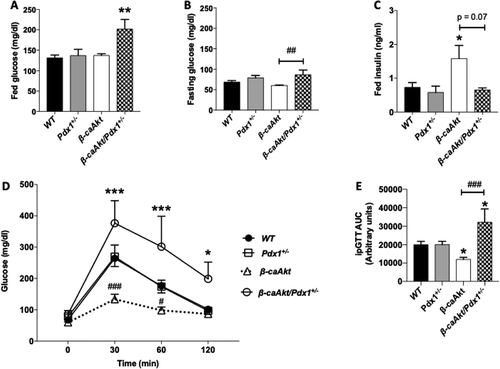
It is well documented that Akt regulates β-cell mass, survival, proliferation, and function.Citation5,Citation18,Citation20 Mice expressing a constitutively active Akt specifically in β-cells (β-caAkt) exhibit a remarkable increase in β-cell mass due to high proliferative rates, and greater insulin levels in the blood.Citation20,Citation21 Since PDX1 is critical for a proper function of mature β-cells by regulating important genes and insulin/Akt signaling modulates PDX1 expression, we tested the hypothesis that PDX1 mediates some of the biological functions induced by Akt. To address that, we generated β-caAkt mice deficient in one allele of pdx1 gene (pdx1+/-). The pdx1+/- and β-caAkt mice are normoglycemic in fed conditions (). However, glucose levels in the β-caAkt/pdx1+/- mice are higher compared to WT mice (). Fasting blood glucose levels are also increased in the β-caAkt/pdx1+/- mice compared to β-caAkt mice (). As expected, β-caAkt mice exhibit high plasma insulin levels compared to WT and pdx1+/- mice (). Interestingly, insulin levels in β-caAkt/pdx1+/- were restored to the levels observed in WT mice (). Examination of glucose tolerance in 8-week-old mice showed that while pdx1+/- mice have normal tolerance to glucose, β-caAkt mice exhibited improved glucose tolerance (). However, deletion of one allele of pdx1 induces glucose intolerance when compared to WT mice and pdx1+/- (,E).
Figure 1. Assessment of glucose homeostasis and glucose tolerance in β-caAkt and pdx1+/- intercross. (A) Fed glucose levels in blood; (B) Fasting glucose levels; (C) Fed plasma insulin levels; (D) Blood glucose levels after intraperitoneal glucose injection (2 g/kg) and (E) Area under the curve of values obtained in (C). * p < .05, ** p < .01 and *** p < .001 compared to WT. # p < .05, ## p < .01 and ### p < .001 compared to β-caAkt. N = 4–13.
Overexpression of caAkt induces increase in β-cell mass at least in part by a PDX1-dependent mechanism
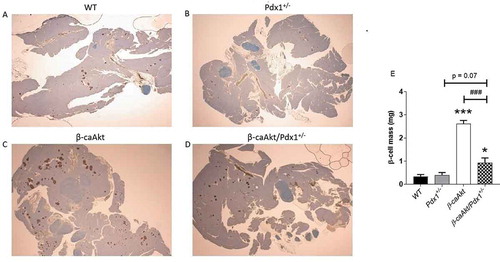
The data presented above suggest that the improved glucose tolerance and hyperinsulinemia found in β-caAkt mice were significantly affected by decreased PDX1 levels, suggesting that changes in β-cell mass could be responsible for these observations. β-cell mass in the β-caAkt/pdx1+/- intercross mice was then assessed by islet morphometry (). In adult pancreas from 12-14-week-old male mice, pdx1+/- mice exhibited comparable β-cell mass to WT mice (). Overexpression of the caAkt mutant was associated with six-fold induction in β-cell mass (). This striking increase in the β-caAkt mice was decreased by 75% in β-caAkt mice lacking one PDX1 allele (β-caAkt/pdx1+/- mice; ).
Figure 2. PDX1 deficiency limits β-cell mass expansion induced by Akt. (A-D) Representative pancreatic morphology in WT, pdx1+/-, β-caAkt and β-caAkt/pdx1+/- mice stained for insulin (red) and counterstained with hematoxylin (blue). (E) β-cell mass quantification performed at 10 weeks of age. * p < .05 and *** p < .001 compared to WT. ### p < .001 compared to β-caAkt. N = 4.
Normal levels of PDX1 are required for Akt-induced β-cell proliferation
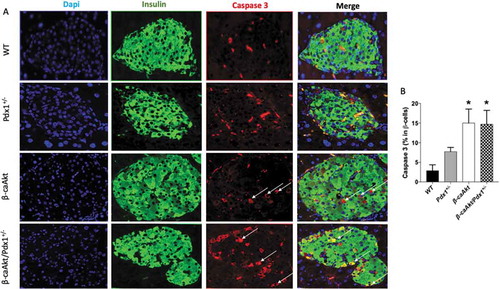
In order to understand the mechanism behind the blunted β-cell mass expansion in β-caAkt/pdx1+/- mice, we assessed proliferation and apoptosis in the four groups ( and , respectively). Analysis of proliferation by immunostaining for Ki67 in β-cell nuclei demonstrates that compared to WT mice, pdx1+/- mice exhibit a trend to decreased proliferation but this was not statistically significant (). Proliferation is remarkably increased in β-caAkt mice compared to WT mice. Interestingly, β-cell proliferation in the β-caAkt/pdx1+/- mice is not statistically different to WT mice (B). The β-caAkt mice exhibited increased apoptosis suggesting that these mice have high β-cell turnover due to concomitant elevation of proliferation and apoptosis ( and ). Apoptosis in β-caAkt/pdx1+/- mice was elevated to levels similar to those observed in β-caAkt mice (,B).
Figure 3. PDX1 expression is important for Akt-induced β-cell proliferation. (A) representative images of WT, pdx1+/-, β-caAkt and β-caAkt/pdx1+/- mice after staining paraffin pancreas slices for Dapi (blue), Insulin (green) and Ki67 (red). Arrows denote Ins+/Ki67+ cells. Images were taken with 40x objective (B) Quantification of Ins+/Ki67+ cells among all β-cells. *** p < .001 compared to WT. ## p < .01 compared to β-caAkt. N = 3–4.
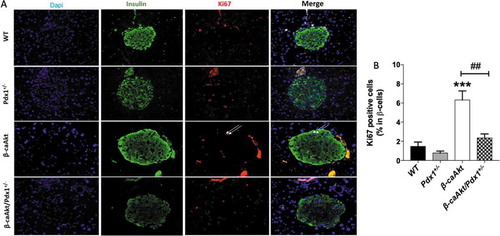
Figure 4. Quantification of β-cell death. (A) representative images of WT, pdx1+/-, β-caAkt, and β-caAkt/pdx1+/- mice after staining paraffin pancreas slices for Dapi (blue), Insulin (green) and Caspase 3 (red). Arrows denote Ins+/Caspase3+ cells. Images were taken with 40x objective (B) Quantification of Ins+/Caspase3+ cells among all β-cells. * p < .05 compared to WT. N = 3–4.
Discussion
The current studies were designed to assess the role of PDX1 in the phenotype observed in mice with activation of Akt signaling in pancreatic β-cells. We demonstrate that PDX1 haploinsufficiency is sufficient to limit β-cell expansion induced by activation of Akt signaling in vivo () and that PDX1 levels are essential for β-cell proliferative signals induced by activation of Akt (). Collectively, the current data place PDX1 as a central modulator of Akt-induced β-cell expansion.
Challenged by insulin resistance, β-cells are required to increase insulin secretion to maintain glucose at normal levels. Acutely, there is an increment in insulin secretion stimulated by increasing levels of glucose imposed by insulin resistance. The current evidence suggest that hyperinsulinemia is an important component to stimulate β-cell expansion in these conditions.Citation41,Citation42 Downstream of insulin receptor signaling, Akt/PKB pathway is critical for this response and its overactivation induces a remarkable increase in β-cell mass due to higher rates of β-cell proliferation ( and , andCitation20). Previous studies suggested that IRS2 and PDX1 are important for β-cells compensation to insulin resistance.Citation35,Citation43 Using two independent mouse models of insulin resistance – insulin receptor (IR)/IR substrate–1 (IRS-1) double heterozygous mice and liver-specific IR KO (LIRKO) mice – Kulkarni and coworkers showed that the compensatory islet-growth response to insulin resistance is impaired by PDX1 heterozygosity.Citation35 Our studies extend these observations by placing PDX1 downstream of Akt signaling and suggest that β-cell mass expansion in conditions of insulin resistance could be mediated at least in part by an IR/IRS2/Akt/PDX1 axis.
We confirmed in the present studies that β-caAkt and WT mice have similar blood glucose despite high plasma insulin levels as previously reported.Citation20 These results suggest peripheral insulin resistance. In contrast, Tuttle et al. have shown a 20-25% decrease in glucose levels concomitantly with an 4-5-fold increase in plasma insulin.Citation21 Although they did not show any difference in the insulin tolerance test between β-caAkt and WT mice, the fact that a 4-5-fold increase in plasma insulin only slightly impact glucose levels also suggest a degree of peripheral insulin resistance and/or insulin clearance.
The fed hyperglycemia and glucose intolerance in βcaAkt/pdx1+/- mice was associated with increased β-cell mass compared to WT and pdx1+/- mice, highlighting the important role of PDX1 in β-cell function and not only in β-cell mass. These findings suggest that PDX1 levels are essential in coordinating appropriate function in proliferative conditions induced by Akt and underline the role of this transcription factor in maintaining a differentiated phenotype in conditions of increase proliferation. In contrast to previous studies, our results showed that pdx1+/- mice exhibited normal glucose tolerance. This is most likely explained by the use of young male mice (2-month-old) in our studies.
Evidence that Akt-PDX1 axis plays a major role in β-cell proliferation is based on the following findings: i) PDX1 haploinsufficiency partially blocks the proliferation and β-cell mass observed in β-caAkt mice (); ii) Studies performed in islets from β-caAkt mice and caAkt MIN6 cells showed that Akt regulates protein levels of cyclin-dependent kinase 4 and cyclin Ds;Citation5,Citation39 The mechanisms whereby PDX1 regulates β-cell proliferation are unclear but it is possible that PDX1 could regulate transcription of critical cell cycle regulators by binding directly to cis regulatory elements or indirectly by regulating transcription of other molecules that will alter transcription of components of the cell cycle machinery;Citation5,Citation44 iii) decreased expression of β-catenin in β cells from PDX1 heterozygous islet provides additional potential mechanisms in coordinating β cell expansion by the Akt/PDX1 axis.Citation35 Future studies can be designed to assess additional mechanisms by performing gene expression in islets from β-caAkt/pdx1+/- mice.
PDX1 also regulates β-cell mass by regulating cell survival.Citation24,Citation28 The current studies showed a trend to increased apoptotic rates in PDX1 heterozygous mice () by staining β-cell (insulin+ cells) for caspase3, confirming previous findings.Citation24 Interestingly, higher apoptotic rates were also observed in β-caAkt mice (). However, we did not observe any difference between the β-caAkt and β-caAkt/pdx1+/- mice indicating that Akt-induced apoptosis in β-cells islets in the β-caAkt mice is not mediated by PDX1. It also indicates that the differences in β-cell mass between the β-caAkt and β-caAkt/pdx1+/- mice are not explained by changes in apoptosis. The unexpected increased apoptosis in β-caAkt mice was observed previously by Tuttle et al.Citation21 Given that Akt has been described as an oncogene, it is not unexpected to observe a concomitant increase in proliferation and apoptosis.Citation45 Indeed, high tissue turnover (increased proliferation and apoptosis) could provide an advantage to tissues with high growth rate after activation of oncogenic pathways.Citation45
In summary, the current work uncovers that Akt/PDX1 axis regulates β-cell proliferation and survival.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Meneur C, Bernal-Mizrachi E. Natural history of beta-cell adaptation and failure in type 2 diabetes. Mol Aspects Med. 2015;42:19–41. doi:10.1016/j.mam.2014.12.002.
- Arvan P, Bernal-Mizrachi E, Liu M, Pietropaolo M, Satin L, Schnell S, Soleimanpour SA. Molecular aspects of pancreatic beta cell failure and diabetes. Mol Aspects Med. 2015;42:1–2. doi:10.1016/j.mam.2015.01.002.
- Bonner-Weir S. Life and death of the pancreatic beta cells. Trends in Endocrinol Metab. 2000;11:375–378. doi:10.1016/S1043-2760(00)00305-2.
- Stewart AF, Hussain MA, Garcia-Ocana A, Vasavada RC, Bhushan A, Bernal-Mizrachi E, Kulkarni RN. Human beta-cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64:1872–1885. doi:10.2337/db14-1843.
- Fatrai S, Elghazi L, Balcazar N, Cras-Meneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55:318–325. doi:10.2337/diabetes.55.02.06.db05-0757.
- Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi:10.1016/S0092-8674(00)80546-2.
- Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 2000;105:199–205. doi:10.1172/JCI7917.
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–903. doi:10.1038/36116.
- Xuan S, Kitamura T, Nakae J, Politi K, Kido Y, Fisher PE, Morroni M, Cinti S, White MF, Herrera PL, et al. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J Clin Invest. 2002;110:1011–1019. doi:10.1172/JCI0215276.
- Kulkarni RN, Holzenberger M, Shih DQ, Ozcan U, Stoffel M, Magnuson MA, Kahn CR. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet. 2002;31:111–115. doi:10.1038/ng872.
- Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein Kinase C activation mediates glucagon-like peptide-1–induced pancreatic ß-cell proliferation. Diabetes. 2001;50:2237–2243. doi:10.2337/diabetes.50.10.2237.
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet. 1999;23:32–40. doi:10.1038/12631.
- Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, Schubert M, Fisher TL, Dow MA, Leshan R. Upregulation of insulin receptor substrate-2 in pancreatic β cells prevents diabetes. J Clin Invest. 2003;112(10):1521–1532. doi:10.1172/JCI18581.
- Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. 1996;122:1409–1416.
- Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ. Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-alpha or EGF, augments pancreatic beta-cell proliferation. Diabetes. 2002;51:966–976. doi:10.2337/diabetes.51.4.966.
- Lingohr MK, Dickson LM, Wrede CE, Briaud I, McCuaig JF, Myers MG Jr., Rhodes CJ. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol Cell Endocrinol. 2003;209:17–31. doi:10.1016/j.mce.2003.08.003.
- Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a Mammalian Target of Rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in {beta}-cells. J Biol Chem. 2005;280:2282–2293. doi:10.1074/jbc.M412179200.
- Bernal-Mizrachi E, Fatrai S, Johnson JD, Ohsugi M, Otani K, Han Z, Polonsky KS, Permutt MA. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114:928–936. doi:10.1172/JCI200420016.
- Srinivasan S, Bernal-Mizrachi E, Ohsugi M, Permutt MA. Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. Am J Physiol Endocrinol Metab. 2002;283:E784–93. doi:10.1152/ajpendo.00177.2002.
- Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. 2001;108:1631–1638. doi:10.1172/JCI200113785.
- Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, Birnbaum MJ. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7:1133–1137. doi:10.1038/nm1001-1133.
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi:10.1038/371606a0.
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995.
- Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, Misler S, Edlund H, Polonsky KS. Increased islet apoptosis in Pdx1+/– mice. J Clin Invest. 2003;111(8):1147–1160. doi:10.1172/JCI200316537.
- Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, Wright CVE. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol. 2008;314:406–417. doi:10.1016/j.ydbio.2007.10.038.
- Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–1768. doi:10.1101/gad.12.12.1763.
- Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, Yang C, Pannikar A, Doliba N, Zhang T. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014;19:259–271. doi:10.1016/j.cmet.2013.12.002.
- Johnson JD, Bernal-Mizrachi E, Alejandro EU, Han Z, Kalynyak TB, Li H, Beith JL, Gross J, Warnock GL, Townsend RR, et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci U S A. 2006;103(51):19575–19580. doi:10.1073/pnas.0604208103.
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. Embo J. 1993;12:4251–4259. doi:10.1002/j.1460-2075.1993.tb06109.x.
- Chakrabarti SK, James JC, Mirmira RG. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J Biol Chem. 2002;277:13286–13293. doi:10.1074/jbc.M111857200.
- Serup P, Jensen J, Andersen FG, Jorgensen MC, Blume N, Holst JJ, Madsen OD. Induction of insulin and islet amyloid polypeptide production in pancreatic islet glucagonoma cells by insulin promoter factor 1. Proc Natl Acad Sci U S A. 1996;93:9015–9020. doi:10.1073/pnas.93.17.9015.
- Macfarlane WM, Campbell SC, Elrick LJ, Oates V, Bermano G, Lindley KJ, Aynsley-Green A, Dunne MJ, James RFL, Docherty K. Glucose regulates islet amyloid polypeptide gene transcription in a PDX1- and calcium-dependent manner. J Biol Chem. 2000;275(20):15330–15335. doi:10.1074/jbc.M908045199.
- Watada H, Kajimoto Y, Umayahara Y, Matsuoka T, Kaneto H, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. The human glucokinase gene beta-cell-type promoter: an essential role of insulin promoter factor 1/PDX-1 in its activation in HIT-T15 cells. Diabetes. 1996;45:1478–1488. doi:10.2337/diab.45.11.1478.
- Waeber G, Thompson N, Nicod P, Bonny C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol. 1996;10(11):1327–1334. doi:10.1210/mend.10.11.8923459.
- Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest. 2004;114(6):828–836. doi:10.1172/JCI21845.
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J Clin Invest. 2002;110(12):1839–1847. doi:10.1172/JCI200216857.
- Boucher M-J, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem. 2006;281(10):6395–6403. doi:10.1074/jbc.M511597200.
- Alejandro EU, Bozadjieva N, Blandino-Rosano M, Wasan MA, Elghazi L, Vadrevu S, Satin L, Bernal-Mizrachi E. Overexpression of Kinase-Dead mTOR impairs glucose homeostasis by regulating insulin secretion and not beta-cell mass. Diabetes. 2017;66:2150–2162. doi:10.2337/db16-1349.
- Blandino-Rosano M, Alejandro EU, Sathyamurthy A, Scheys JO, Gregg B, Chen AY, Rachdi L, Weiss A, Barker DJ, Gould AP. Enhanced beta cell proliferation in mice overexpressing a constitutively active form of Akt and one allele of p21Cip. Diabetologia. 2012;55:1380–1389. doi:10.1007/s00125-012-2465-9.
- Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, Mirmira RG, Sonenberg N, Liu M, Rüegg MA, et al. Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat Commun. 2017;8:16014. doi:10.1038/ncomms16014.
- Takamoto I, Terauchi Y, Kubota N, Ohsugi M, Ueki K, Kadowaki T. Crucial role of insulin receptor substrate-2 in compensatory beta-cell hyperplasia in response to high fat diet-induced insulin resistance. Diabetes Obes Metab. 2008;10(Suppl 4):147–156. doi:10.1111/j.1463-1326.2008.00951.x.
- Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, et al. Glucokinase and IRS-2 are required for compensatory cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117(1):246–257. doi:10.1172/JCI17645.
- Brissova M, Blaha M, Spear C, Nicholson W, Radhika A, Shiota M, Charron MJ, Wright CVE, Powers AC. Reduced PDX-1 expression impairs islet response to insulin resistance and worsens glucose homeostasis. Am J Physiol Endocrinol Metab. 2005;288(4):E707–14. doi:10.1152/ajpendo.00252.2004.
- Hayes HL, Moss LG, Schisler JC, Haldeman JM, Zhang Z, Rosenberg PB, Newgard CB, Hohmeier HE. Pdx-1 activates islet alpha- and beta-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol Cell Biol. 2013;33:4017–4029. doi:10.1128/MCB.00469-13.
- Liu S, Edgerton SM, Moore DH 2nd, Thor AD. Measures of cell turnover (proliferation and apoptosis) and their association with survival in breast cancer. Clin Cancer Res. 2001;7:1716–1723.

