

ABSTRACT
Glucagon-Like Peptide-1 (GLP-1) is an important peptide hormone secreted by L-cells in the gastrointestinal tract in response to nutrients. It is produced by the differential cleavage of the proglucagon peptide. GLP-1 elicits a wide variety of physiological responses in many tissues that contribute to metabolic homeostasis. For these reasons, therapies designed to either increase endogenous GLP-1 levels or introduce exogenous peptide mimetics are now widely used in the management of diabetes. In addition to GLP-1 production from L-cells, recent reports suggest that pancreatic islet alpha cells may also synthesize and secrete GLP-1. Intra-islet GLP-1 may therefore play an unappreciated role in islet health and glucose regulation, suggesting a potential functional paracrine role for islet-derived GLP-1. In this review, we assess the current literature from an islet-centric point-of-view to better understand the production, degradation, and actions of GLP-1 within the endocrine pancreas in rodents and humans. The relevance of intra-islet GLP-1 in human physiology is discussed regarding the potential role of intra-islet GLP-1 in islet health and dysfunction.
Introduction
The proglucagon gene GCG is expressed in pancreatic alpha cells, intestinal L-cells, and neurons in the nucleus tractus solitarus.Citation1 Differential processing of proglucagon by Prohormone Convertase 2 (PC2, encoded by the PCSK2 gene) yields glucagon, whereas PC1/3 (encoded by the PCSK1 gene) yields GLP-1 and GLP-2.Citation2,Citation3 It is generally accepted that glucagon production and secretion are largely restricted to the alpha cells within pancreatic islets, whereas GLP-1 production and secretion are confined to the enteroendocrine L-cells and the central nervous system. However, as early as 1985, GLP-1 expression was predicted in both the gastrointestinal tract and pancreas.Citation4 Moreover, GLP-1 expression was documented in human pancreatic extracts and proglucagon-producing pancreatic tumors,Citation5 and early HPLC analysis of human and porcine alpha cell extracts identified glucagon and small quantities of N-terminally extended GLP-1.Citation6 Subsequent papers have identified GLP-1 in the human pancreas where it is co-packaged within the glucagon secretory granules of an alpha cell subpopulation.Citation7–9 Conversely, recent evidence suggests that L-cells can secrete glucagon.Citation10 This relatively unexpected pancreatic source of GLP-1 suggests a potential paracrine role for alpha cell-derived GLP-1 and suggests plasticity in the differential processing of the proglucagon peptide in both alpha cells and L-cells.
The incretin effect is characterized by the glucose-dependent insulinotropic action of gut-derived peptides.Citation11 These peptides include GLP-1 and Glucose-dependent Insulinotropic Polypeptide (GIP), hormones that are released upon nutrient sensing in the gastrointestinal tract.Citation11–13 In the classical incretin model, oral nutrient intake initiates the release of GLP-1 and GIP from L- and K-cells in the gut, respectively. These incretin hormones pass through the portal circulation and reach their respective canonical receptors in the pancreas and other target organ systems. With respect to the pancreas, GLP-1 potentiates insulin secretion in a glucose-dependent manner via its receptors on beta cells.Citation14,Citation15 GIP similarly potentiates beta cell insulin secretion, but GIP also enhances glucagon secretion from alpha cells during hypoglycemia.Citation16 Recent research has led to further refinements of the existing incretin model and these findings now indicate that GLP-1 can act locally in the enteric afferent nervous system, signaling directly to the central nervous system. Central signals are then relayed to the pancreas through the efferent autonomic nervous system, indirectly inducing insulin secretion in response to gastric stimuli.Citation17 Currently, the relative incretin contributions of the recently-described gut-brain-axis versus the well-established gut-portal-pancreas axis are still unclear and are under active investigation.
The importance of the incretin effect on glucose homeostasis has been elegantly demonstrated by the use of double incretin receptor knockout “DIRKO” mice. Mice lacking both GLP-1 and GIP receptors (GLP-1R and GIPR, respectively) have impaired oral glucose tolerance but intact intraperitoneal glucose tolerance.Citation18–20 This confirms that incretin hormones and their receptor-activation are vital for coordinated insulin release in feeding. In addition to potentiating insulin secretion, GLP-1 also plays a crucial role in islet health, with basal GLP-1R activation contributing to maintaining beta cell function and survival, especially under conditions of islet stress and dysfunction.Citation21 Interestingly, GIP’s effects are dampened in metabolic disease, whereas GLP-1 signaling remains intact.Citation1 This has resulted in GLP-1 becoming an effective therapeutic target for managing obesity and Type 2 Diabetes (T2D). In vitro, GLP-1 and GLP-1R agonists protect alpha and beta cell lines from apoptosis.Citation22–24 Drugs that increase the actions of endogenous GLP-1 are also known to promote beta cell survival in isolated human and rodent islets.Citation14,Citation22,Citation25–27 These drugs include synthetic GLP-1 mimetics and inhibitors of GLP-1’s enzymatic breakdown by Dipeptidyl Peptidase 4 (DPP4).
The enzymatic degradation of GLP-1 also plays a significant role in regulating glucose metabolism. In a seminal paper, Kieffer et al.Citation28 demonstrated that DPP4 is the enzyme responsible for inactivating GLP-1 and GIP in vivo. Other groups extended this finding to human serum and consequently laid a foundation for early clinical trials with DPP4 inhibitors.Citation29,Citation30 To use these therapies to their maximum potential, we must also consider the expression of islet-derived GLP-1 and DPP4 in the context of human health and disease.
The mechanisms controlling GLP-1 and GIP secretion from L-cells and K-cells, respectively, and their systemic effects and target organs have been studied and reviewed extensively.Citation31–35 In contrast, the existence and potential physiological roles of intra-islet GLP-1 have only recently begun to be examined more comprehensively. This potentially important aspect of GLP-1 pharmacology has received less attention, perhaps due to the conflicting evidence in the literature.
Herein, we aim to provide a comprehensive review of the evidence for the existence of islet-derived GLP-1 and consider the expression, degradation, and paracrine actions of this localized source of GLP-1 within pancreatic islets. Specifically, we review the literature on the existence of intra-islet GLP-1 in animal models and humans and then discuss the potential underlying signaling pathways that may regulate the processing and secretion of GLP-1 from islet alpha cells. Finally, we consider the role of the GLP-1R on alpha, beta, and delta cells, and GLP-1’s potential contribution as a paracrine signaling factor in the islet.
Evidence for GLP-1 expression in rodent islets
Islet-derived GLP-1 has been documented in several rodent islet models where alpha cells begin to differentially process proglucagon and secrete GLP-1 in response to beta cell destruction.Citation36 Moreover, GLP-1 is produced from alpha cells in rats after several injections of streptozotocin (STZ; 50 mg/kg per day for five days), and its production partially protects the islet from further beta cell loss.Citation37 The sand rat rodent model “Psammomys obesus” develops obesity and insulin-resistance when placed on a regular laboratory rodent chow diet. In these rodents, intra-islet GLP-1 production begins during the development of T2D symptoms and persists with fasting hyperglycemia.Citation38 GLP-1 and PC1/3 expression are elevated during the progression of T2D and hyperglycemia in the db/db mouse model.Citation39 All three rodent models experience cellular stress and beta cell damage before the detection of GLP-1. In a study using non-diabetic C57BL/6 J mice, researchers identified a subpopulation of GLP-1 expressing alpha cells in dispersed islets.Citation40
As these studies used isolated primary islet samples, it has been suggested that islet-isolation and culturing may cause sufficient islet stress to induce GLP-1. This stress-induced GLP-1 production may be a potential mechanism to prevent further islet cell damage. While this type of experimental non-physiological stress may well induce GLP-1 processing from proglucagon, recent studies using genetic mouse models that alter intra-islet GLP-1 production in vivo demonstrate that islet-derived GLP-1 may play an important role in glucose homeostasis in the whole animal.Citation8,Citation41 Therefore, the available evidence indicates that rodent islet alpha cells can process proglucagon to produce and secrete GLP-1. Whether or not this occurs in vivo in non-genetically modified mice remains to be determined conclusively.
Evidence for GLP-1 expression in human islets
For obvious reasons, the existence and roles of intra-islet GLP-1 have been more extensively studied in rodents, although several studies have confirmed that human alpha cells are capable of processing and secreting GLP-1 through the use of isolated, intact human islets.Citation42–44 In this regard, our group reported that isolated human islets secrete ~50-fold more active GLP-1 than mouse islets in culture, despite only possessing approximately 3-4-fold more alpha cells than mouse islets.Citation44 These studies confirm that intact human islets can both synthesize and secrete significant amounts of GLP-1. It should be mentioned that isolated islets may not behave like islets in situ and the possibility remains that the presence of intra-islet GLP-1 may represent an artifact resulting from islet stress induced by their isolation and subsequent culturing. However, our group recently analyzed living-donor biopsy sections that provides direct evidence for GLP-1 production existing in human islets in the absence of cadaveric islet isolation and subsequent islet culturing. Although the biopsy-donors in this study had underlying pancreatic cancer or pancreatitis, the islet sections also contained a substantial subpopulation of GLP-1 positive alpha cells.Citation45 Therefore, it is important to determine whether intra-islet GLP-1 production is a consequence of islet stress or if it is constitutively secreted and plays a physiological role in the healthy adult human pancreas. Future research using a perfused whole human pancreas model from donors without diabetes or pancreatic disease is therefore warranted to help definitively answer this important question. Whether or not alpha cell GLP-1 is secreted in normal human physiology, there is compelling evidence to suggest that human alpha cells are capable of producing and releasing GLP-1 and that its production is elevated in disease-states like diabetes.
Regulation of alpha cell proglucagon processing and GLP-1 secretion
Although induction of the GCG gene is critical for proglucagon production, the subsequent processing of proglucagon into glucagon or glucagon-related peptides (such as GLP-1 and GLP-2) is critically dependent on the relative expression levels of PC2 and PC1/3 (). Greater PC1/3 expression favors GLP-1 production, whereas greater PC2 expression favors glucagon production (). In this regard, PC1/3 is highly expressed in juvenile human islets, although its expression is decreased in adult alpha cells. Instead, mature islets preferentially express the nonfunctional “long-noncoding” PC1/3 gene lncPCSK1.Citation46 Despite its decreased expression in aging, alpha cell PC1/3 expression is still well-documented in adult alpha cells where it correlates positively with BMI.Citation47 Indeed, PC1/3 expression is detectable in FACS-sorted alpha cells from the adult, non-diabetic human pancreas and alpha cell PC1/3 expression is elevated in T2D alpha cells.Citation44,Citation48 This is supported by our own recent research that demonstrates that islets from T2D donors contain a higher proportion of GLP-1 positive alpha cells than islets from non-diabetic donors.Citation44 While there is substantial evidence that the adult human endocrine pancreas can express intra-islet GLP-1, the precise signaling pathways regulating alpha cell PC1/3 expression is an active research area and several potential pathways that regulate alpha cell PC1/3 expression have been identified and are described below.
Figure 1. Alpha cell differential proglucagon processing to glucagon or GLP-1 and the signaling pathways that may regulate this process via PC2 and PC1/3 expression
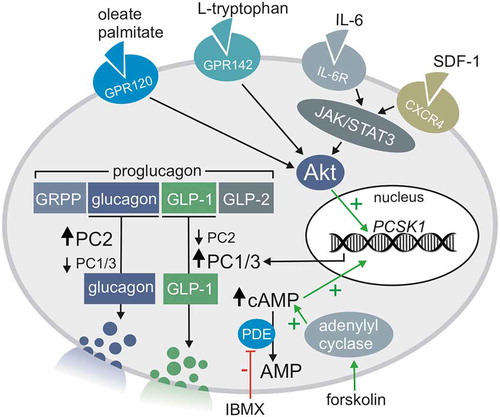
Interleukin-6 (IL-6)
IL-6 is a cytokine with both pro- and anti-inflammatory properties that is released during exercise and low-grade inflammation and has been identified as an important driver of PC1/3 expression in islets.Citation49–51 Systemic IL-6 production is also chronically elevated in the context of obesity and T2D;Citation49 instances where levels of intra-islet GLP-1 are also found to be elevated.Citation44 IL-6 receptors (IL-6 R) are highly expressed on endocrine cells of the gastrointestinal tract and pancreas. In islets, IL-6 R expression is elevated in alpha cells compared to beta or delta cells.Citation49,Citation51 IL-6 R blockade decreases plasma GLP-1 levels in both mice and humans,Citation51 and IL-6 R KO decreases the amount of GLP-1 r eleased from cultured mouse islets.Citation52
Taken together, this evidence reinforces the relevance of IL-6 R signaling to the production and secretion of GLP-1 from intestinal sources, as we would expect the gut to be the most substantial source of circulating GLP-1. However, the contribution of IL-6 signaling in the production of pancreatic GLP-1 is less clear, although the reduction of GLP-1 secretion from isolated islets in the presence of an IL-6 R blockade indicates a strong regulatory role for IL-6 in pancreatic GLP-1 processing. However, IL-6 receptor signaling is not required for normal islet function and development,Citation49 suggesting that this pathway may be activated only under specific physiological or pathophysiological conditions.
IL-6 enhances the induction of the PCSK1, PCSK2, and GCG gene transcription in both alpha cells and L-cells via the IL-6 R/JAK/STAT3 pathway (). Citation24,Citation50,Citation52,Citation53 Although levels of PC1/3 and PC2 are both increased by IL-6, there is no acute increase in glucagon secretion in cultured human and rodent islets. Instead, IL-6 treatment increases proglucagon and GLP-1 levels in acute exposure, while glucagon levels only increase after chronic (24 hours) IL-6 exposure.Citation49,Citation50 Acute increases in islet-derived GLP-1 are thought to play a protective role in beta cells. This protective effect is two-fold as IL-6 directly improves beta cell survival by potentiating autophagic flux,Citation54 and IL-6 promotes beta cell health via intra-islet GLP-1 production.Citation50 IL-6 also protects alpha cells from lipo- and gluco-toxicity, contributing to alpha cell expansion in murine high-fat diet models.Citation49
In addition to its release from active immune cells, IL-6 can also be produced from exercising skeletal muscle, adipocytes, and endocrine tissues.Citation42,Citation51,Citation55 Furthermore, IL-6 is basally expressed in smooth muscle tissue, and the presence of soluble DPP4 can increase its production.Citation56 This may be especially relevant in the islet vasculature, where endothelial cell-derived soluble DPP4 and IL-6 are both readily expressed. Adipose tissue is a primary source of the cytokines IL-6 and IL-1beta in obesity and T2D.Citation50 However, the concomitant increase in adipose-derived IL-1beta may limit IL-6’s beneficial effects and favor a more pro-inflammatory environment. While IL-1beta pretreatment does not impair IL-6 dependent GLP-1 production in cultured islets, IL-1beta treatment alone inhibits GLP-1 secretion.Citation50,Citation52 GLP-1’s sister incretin hormone GIP stimulates IL-6 production in alpha cells and decreases IL-6 production in beta cells,Citation52 leading to an increase in measurable GLP-1 and insulin release from cultured islets.
Taken together, the available evidence strongly supports a key role for IL-6 signaling in the control of proglucagon processing and GLP-1 production from alpha cells. As IL-6 is produced from exercising skeletal muscle,Citation42,Citation49–51 it is tempting to speculate that exercise-derived IL-6 may promote islet health and function through this IL-6/GLP-1 axis. In this regard, Ellingsgaard et al.Citation50 confirm that exercise acutely elevates both plasma IL-6 and GLP-1, although the effects of regular exercise on intra-islet IL-6 and GLP-1 production have yet to be examined.
Metabolite G-Protein Coupled Receptors (GPCRs)
GPCRs that recognize specific metabolites have also been implicated in increasing PC1/3 expression via activation of the pro-survival kinase Akt. Moreover, many GPCRs that induce PC1/3 expression in intestinal L-cells are also present on alpha cells. For example, GPR120 is a candidate for triggering GLP-1 secretion from both alpha and L-cells. This receptor is a member of the rhodopsin-like family of GPCRs that senses oleate, palmitate, and other structurally-similar fatty acid metabolites.Citation24 GPR120 agonists can activate the PCSK1 promoter region in both alpha and L-cell lines, favoring PC1/3 and subsequent GLP-1 production().Citation43
Amino acids are also a potent stimulant of glucagon secretion from alpha cells, and are candidates for triggering alpha cell hyperplasia.Citation57 Another rhodopsin-like GPCR, GPR142, functions as an L-tryptophan sensor. GPR142 signaling increases Akt activation, ultimately increasing PC1/3 expression in both alpha and L-cells ().Citation22 GPR142 agonism augments insulin secretion in a GLP-1R dependent manner,Citation22,Citation58,Citation59 but it is not required for basal GLP-1 secretion. Indeed, GPR142 KO and control littermates have no apparent differences in glucose tolerance and exhibit similar insulin secretion profiles in response to intraperitoneal and oral glucose.Citation59 This latter finding argues against a major physiological role for GPR142 alone, although this receptor may act in concert with other GPCRs to fine-tune islet cell function. While it is not explicitly shown to regulate PC1/3 expression, GPR119, a monoacylglycerol sensing GPCR, can potentiate glucagon granule release from alpha cells in hypoglycemia,Citation60 and its activation elevates intracellular cAMP.Citation61 When considering the co-packaging of glucagon and GLP-1 in secretory granules, it is plausible that GPR119 activation may regulate GLP-1 secretion.
The ligands for these GPCRs are all nutrient metabolites that are readily available in the gut, but they may come from the bloodstream or more localized sources in the islet. For example, aromatic amino acids sensed by GPR142 may be produced by the autophagy of nearby cells, while monoacylglycerols and long-chain fatty acids are released from localized lipolysis.Citation61 Taken together, these findings indicate that proglucagon synthesis and its differential processing and secretion may be under the control of certain nutrients in the local islet microenvironment. Furthermore, the reported effects of GPR119 and 120 agonism on PC1/3 expression and GLP-1 processing in L-cells may complement the increased demand for GLP-1 secretion that is also observed when these GPCRs are activated,Citation62 and the same may be true for islet alpha cells, although this remains to be tested experimentally.
Stromal cell-derived factor-1 alpha (SDF-1α)
The chemokine SDF-1α augments PC1/3 expression in alpha cells in response to beta cell stress or injury (). In the healthy adult rodent islet, SDF-1α is restricted to vascular endothelial and stromal cells and is not expressed in the beta cells.Citation63 However, beta cell injury is proposed to activate the SDF-1α/CXCR4 axis and initiate tissue regeneration.Citation63 Using MIN6 and INS-1 beta cell lines, Liu et al.Citation63 show that cellular stressors such as cytokines, thapsigargin, and STZ can induce SDF-1α expression. These findings demonstrate that inflammation, ER stress, and beta-cell toxins activate the SDF-1α/CXCR4 axis.Citation63 Furthermore, SDF-1α treatment of the alpha cell line αTC1 and mouse islets induced GLP-1 production and secretion. GLP-1 and SDF-1α are proposed to act synergistically to increase beta cell survival and are thought to protect beta cells from further injury.Citation64
The expression of SDF-1α in the beta cells of adult islets appears to be an initiation of a fetal developmental program.Citation65 In support of this concept, Kayali et al.Citation65,Citation66 found that SDF-1α and CXCR4 are expressed in the fetal pancreas, and that inhibition of the CXCR4 receptor in islet-like clusters with the CXCR4 inhibitor AMD3100 inhibits islet development. As adult alpha and beta cells express the CXCR4 receptor, the proposed model for SDF-1α induced GLP-1 secretion in islets involves activation of the CXCR4 receptor on both cell types. SDF-1α can activate the CXCR4 receptor on beta cells in an autocrine manner to increase expression of SDF-1α. The increase in SDF-1α may also induce PC1/3 expression in the nearby alpha cells via paracrine signaling. Upon binding the CXCR4 receptor on the alpha cell, SDF-1α can initiate signaling through the JAK/STAT pathway, eventually activating Akt to induce PC1/3 expression and the subsequent processing and secretion of GLP-1.Citation63,Citation67
Other factors that may mediate alpha cell PC1/3 expression
There is evidence that PC1/3 expression in alpha cells may be induced by conditions and cellular mechanisms in addition to IL-6, metabolite sensing GPCRs, and SDF-1α. For example, hyperglycemia induces PC1/3 expression in alpha cells,Citation24 although the precise mechanisms underlying this effect are unclear. Furthermore, increased PC1/3 and GLP-1 expression is associated with hyperglycemia in rats treated with multiple low dose STZ ().Citation37 Indeed, individuals suffering from chronic hyperglycemia express elevated PC1/3 in alpha cells compared to non-diabetic alpha cells.Citation48 The expression of PC1/3 in alpha cells can be experimentally induced through the sustained elevation of cAMP with forskolin and IBMX ().Citation43 Therefore, signaling pathways that involve cAMP also have the potential to induce PC1/3 expression, and additional undiscovered cAMP-mediated pathways may exist in alpha cells.
Our group has recently examined biopsies from donors with pancreatitis or pancreatic cancer for GLP-1 expression and has identified a large subpopulation of GLP-1 expression alpha cells.Citation44 It may be the case in these donor samples that sustained chronic low-grade inflammation results in the local production and release of IL-6 and SDF-1α, thereby enhancing alpha cell PC1/3 expression. Tissue damage, local lipolysis, and cellular autophagy may also be enhanced in these disease-states, in turn, activating key metabolite GPCRs that are also implicated in alpha cell PC1/3 expression.
In summary, the production of GLP-1 is dependent on the induction of the PCSK1 gene and subsequent PC1/3 expression in islet alpha cells. Factors that increase PC1/3 expression commonly function through the downstream activation of Akt. Examples include activation of the JAK/STAT pathway by IL-6 or SDF-1α and activation of Akt by Rhodopsin-like GPCRs. The signaling molecules that can promote PC1/3 expression via receptor-mediated signaling are diverse and include cytokines (e.g., IL-6, SDF-1), fatty acids (e.g., palmitate, oleate), and amino acids (e.g., L-tryptophan). Therefore, it is likely that these molecules act in concert to control the glucagon/GLP-1 secretory phenotype of alpha cells depending on factors such as the nutrient or inflammatory state of the localized islet environment (). The resulting increase in GLP-1 secretion may protect islet cells by promoting survival under stress and maintaining insulin secretion in the face of increased insulin resistance.
Degradation of incretins by intra-islet dipeptidyl peptidase 4 (DPP4)
DPP4 is a serine exopeptidase that cleaves the two N-terminal residues from GLP-1, GIP, SDF-1α, and other various peptide hormones and cytokines.Citation25,Citation29,Citation30 It is generally accepted that the resultant truncated peptides are largely inactive at their respective canonical receptors and DPP4 functions to regulate the actions of GLP-1 in the blood, contributing to its short circulating half-life. Indeed, DPP4 inactivates considerable amounts of GLP-1 within 1–2 minutes of its release.Citation1 One of the primary challenges to the classical incretin model is that the short half-life and low circulating concentrations of GLP-1 (<10 pM) may limit its effectiveness to activate GLP-1Rs on beta cells. This is further complicated by the widespread expression of DPP4 throughout the body.
Significant sources of DPP4 include endothelial cells, haematopoietically derived Tei2+ cells, enterocytes, and hepatocytes, although DPP4 expression is ubiquitous.Citation56,Citation68 DPP4 can exist in soluble and membrane-bound forms, and, depending on its origin, DPP4 shows substrate preference. In the presence of denosumab, a human monoclonal antibody used in the treatment of osteoporosis, levels of circulating DPP4 are reduced, whereas levels of GLP-1 are increased.Citation69 This indicates that osteoclasts represent a large pool of DPP4-producing cells and may substantially impact glucoregulation. By creating tissue-specific DPP4 KO models, researchers have identified hematopoietic cell-derived DPP4 as vital for GIP inactivation, while endothelial cell-derived DPP4 is vital for the glucoregulatory effects of DPP4 inhibitors.Citation19
Endothelial cell-derived DPP4 is involved in modulating cytokine levels and immune cell activity. DPP4 inhibition by sitagliptin significantly alters plasma cytokine levels yet favors an anti-inflammatory environment.Citation68 Interestingly, the modulation of immune cells by DPP4 may be both catalytically dependent and independent, while its effects on existing cytokines may be strictly enzymatic. This is confirmed by experiments outlining that hematopoietic cell-derived DPP4 can still reduce levels of pro-inflammatory cytokines in the presence of a DPP4 inhibitor. Indeed, DPP4 can interact with the extracellular matrix proteins of various cell types to induce downstream signaling, independent of its peptidase function.Citation69 This is a likely mechanism for the reduction of pro-inflammatory cytokines with the use of sitagliptin.
Within the human endocrine pancreas, DPP4 expression seems to be highly enriched in alpha cells. Consequently, DPP4 expression in human islets has been used as a surface marker to sort for enriched alpha cell populations by FACS, while DPP4 gene expression has been considered a signature gene to identify alpha cells in transcriptomic studies.Citation70 Indeed, DPP4 is found in human alpha cell multivesicular bodies,Citation46,Citation55,Citation71–73 and these compartments are distinct from glucagon-containing secretory granules.Citation20 DPP4 is also derived from endothelial cells, therefore, a local source of soluble DPP4 produced in the islet vasculature.Citation74,Citation75 This local source of DPP4 may act to further inactivate any incretin peptides that reach the pancreas after passing through hepatic portal circulation.Citation1 In rodents islets, the available evidence suggests that DPP4 expression is limited mainly to beta cells rather than alpha cells.Citation20 This observation was made using histochemical techniques, and it would be worthwhile confirming these interesting results with other techniques, such as using flow cytometry to analyze dissociated islets, or identifying enriched DPP4 gene expression in rodent islet transcriptomic studies. At this point, the role of this species-specific difference in islet cell DPP4 expression remains unclear.
Overall levels of DPP4 are not impacted by circulating insulin, incretins, or DPP4 inhibitors.Citation8,Citation55 However, DPP4 levels are elevated in obese and high-fat diet rodent models.Citation68,Citation69,Citation71 Indeed, there are well-documented increases in plasma DPP4 documented in high-fat diet rat models, resulting in decreased circulating GLP-1 when compared to rats on a control diet.Citation69 In humans, levels of islet-derived DPP4 are enhanced in obesityCitation20 and chronic hyperglycemia,Citation76 but islet-DPP4 activity is compromised.Citation14,Citation20 Despite decreased islet-specific DPP4 activity, peripheral DPP4 levels and activity is enhanced in chronic hyperglycemia and plasma DPP4 activity is correlated positively with both HbA1c and fasting glycemia.Citation69 These results suggest that intra-islet GLP-1 inactivation may be compromised in T2D, and this may contribute to maintaining islet health and insulin secretion in disease. However, separating the relative effects of L-cell versus alpha cell-derived GLP-1 in humans is experimentally problematic, and this concept is difficult to test directly.
DPP4 inhibition has an array of beneficial anti-diabetic effects on islets as both islet-derived DPP472 and endothelial-cell derived DPP419 are sensitive to DPP4 inhibitors. Since soluble, endothelial cell-derived DPP4 has a greater contribution to systemic inflammation in obesity and chronic hyperglycemia,Citation19,Citation56,Citation68,Citation76 its inhibition may reduce local islet inflammation and promote beta cell survival.Citation26 The DPP4 inhibitor sitagliptin stabilizes active GLP-1 and augments insulin secretion.Citation20,Citation26 Beta cells are also protected against cytokine-induced toxicity; however, this effect is independent of GLP-1 and likely due to DPP4’s actions on other peptide hormones.Citation14,Citation77 Overall, the use of DPP4 inhibitors in the management of T2D improves beta cell health, function, and survival via the anti-inflammatory and incretogenic effects of this widely-used clinical drug class.
DPP4 inhibitors and intra-islet substrates
DPP4 inhibition protects beta cells from lipo- and glucotoxicity,Citation14,Citation26,Citation78 and improves islet survival in culture.Citation26,Citation78 Intra-islet DPP4, whether endothelial or endocrine cell-derived, may reduce GLP-1 concentrations, limiting the GLP-1 mediated protection of beta cells. As discussed above, DPP4 inhibitors have the potential to increase active GLP-1 concentrations in the local islet microenvironment. This may represent an under-recognized mechanism underlying the therapeutic efficacy of DPP4 inhibitors for the treatment of T2D. Furthermore, there is an increased opportunity to enhance GLP-1-mediated paracrine signaling between human alpha and beta cells through islet DPP4 inhibition due to the difference in human versus rodent islet morphology as alpha cells are greater in number and interspersed throughout the human islet when compared to rodent islets.Citation79–81
DPP4 inhibition in islets likely alters paracrine signaling of other islet peptides that are DPP4 substrates, with the potential to increase insulin secretion and islet survival. For example, SDF-1α is another known DPP4 substrate, and it is secreted by islets in beta cell injury.Citation25 SDF-1α may be induced in human islets from donors with diabetes due to chronic beta cell stressors such as low-grade inflammation, hyperglycemia, and lipotoxicity. As such, DPP4 inhibition in the islet microenvironment has the potential to maintain active forms of SDF-1α and promote increases in PC1/3-mediated GLP-1 processing and secretion.
The therapeutic potential of DPP4 inhibitors may also reach beyond the treatment of overt T2D. If intra-islet GLP-1 is relevant to human glucose homeostasis, then the use of DPP4 inhibitors may provide clinical benefit in obesity and prediabetes by increasing intra-islet GLP-1 levels as the islets adapt to metabolic stress. While direct clinical evidence for such an effect is sparse, we have recently demonstrated that patients with T2D who receive the DPP4 inhibitor with metformin at first diagnosis have improved HbA1c levels and are less likely to require insulin therapy when compared to those receiving metformin alone or when a DPP4 inhibitor is prescribed later on in the progression of T2D.Citation82 In their recent review, Trzaskalski et al.Citation69 hypothesize that concomitant treatment with sitagliptin and metformin robustly improves HbA1c levels through 1) stabilizing active incretin peptides and 2) increasing the expression of GLP-1R and GIPR on beta cells. In summary, these findings indicate that there may be a clinical benefit to the use of DPP4 inhibitors at the time of T2D diagnosis. Furthermore, patients with prediabetes might even benefit from DPP4 inhibition.
Concerning autoimmune Type 1 Diabetes (T1D), DPP4 inhibitors may possess clinical utility in islet transplantation to preserve or increase beta cell mass in culture before transplant. Our group treated human islet cultures with sitagliptin and measured an increase in intra-islet GLP-1 levels. Interestingly, higher levels of active GLP-1 in culture correlated with less cell death in these human islet preparations. This finding suggests that sitagliptin, and other DPP4 inhibitors, may preserve healthy beta cell mass during the pre-transplant culture period and increase beta cell survival immediately post-transplant.Citation78 Finally, DPP4 inhibitors may increase GLP-1 secretion from the remaining alpha cells in T1D, as many T1D patients are identified with residual functional beta cell mass.Citation83 These patients may benefit from increased active GLP-1 levels local to the beta cell, with potentially less need for exogenous insulin and a reduced risk of hypoglycemia. Although it remains unclear whether the islet-DPP4 axis contributes to whole-body glucose regulation, there is evidence that DPP4 inhibition improves local beta cell function.Citation69 The increased intra-islet GLP-1 as a result of DPP4 inhibition may protect remaining beta cells, even potentially induce de novo beta cell proliferation. As DPP4 inhibitors possess an excellent safety profile, they are perhaps ideal for targeting additional patient populations with prediabetes and even T1D.
GLP-1 receptor signaling in the endocrine pancreas
Researchers have identified a compelling role for islet-derived GLP-1 in postprandial glucose regulation using an alpha cell-specific GCG KO mouse model.Citation41 In these animals, tissue-specific reactivation of GCG results in a variable glucoregulatory phenotype. L-cell GCG reactivation improves oral but not intraperitoneal glucose tolerance.Citation41 This is consistent with the well-characterized incretin effect. Interestingly, while exendin-9 causes glucose intolerance in control mice, mice with intestinal GCG reactivation were not significantly influenced by exendin-9. Instead, alpha cell GCG reactivation made mice sensitive to the negative glucoregulatory influence of exendin-9. Chambers et al.Citation41 concluded that pancreatic but not intestinal GLP-1 expression is vital for proper glucoregulation. The administration of exendin-9 to intact cultured mouseCitation41 and humanCitation44 islets results in impaired glucose-stimulated insulin secretion (GSIS), confirming that GLP-1R signaling is critical in maintaining regular insulin dynamics. Isolated GCG KO islets also exhibit markedly decreased glucose and amino acid-stimulated insulin secretion from lost alpha-to-beta cell communication.Citation12
As glucagon and GLP-1 are both derived from GCG, these effects may be explained by the loss of glucagon production and secretion from these animal models, although the phenotype of GCG null versus GCGR null mice is significantly different with respect to insulin signaling.Citation41 Indeed, GCGR null mice have reduced fasting blood-glucose with no increased risk of hypoglycemia, whereas GCG null mice had a comparable metabolic phenotype to GCGR/GLP-1R double KO mice,Citation41,Citation57 where both GCG null and GCGR/GLP-1R double KO mice exhibit mild fasting hyperglycemia and increases in insulin secretion.Citation40,Citation41 In order to assess the specific effects of GLP-1 production in alpha cells, researchers have used genetic and pharmacological approaches to target PCSK1 and the GLP-1R.
Traub et al.Citation84 demonstrated the importance of islet-derived GLP-1 in adapting to metabolic stress by developing alpha cell PCSK1 KO mice. Mice lacking the ability to produce alpha cell PC1/3 exhibit impaired glucose tolerance in metabolic stress.Citation84 Wideman et al.Citation85 used a different approach to elegantly outline the protective effects of islet-derived GLP-1 in vivo. The xenotransplant of PC1/3 expressing alpha cells into STZ-treated mice partially restores glucose tolerance and preserves existing beta cell mass, whereas PC2 expressing alpha cells have no beneficial effect and promote mild fasting hyperglycemia.Citation85 The protective effect of the PC1/3 expressing alpha cell xenotransplant was attenuated in GLP1R KO mice, confirming that increases in GLP-1 production and signaling play a vital role in this mouse model.Citation85 Taken together, these animal models confirm that proglucagon processing into GLP-1 has a beneficial glucoregulatory effect in mice, and that changes in alpha cell PC1/3 expression can modulate these effects. Presumptively, changes of PC1/3 expression primarily influences proglucagon processing into GLP-1, although the alternative effects of PC1/3 expression in alpha cells have yet to be examined.
GLP-1Rs have been identified on alpha, beta, and delta cells.Citation47,Citation86–88 Interestingly, GLP-1Rs are only detected in a subpopulation of alpha cells, and their relevance to normal physiology remains unclear.Citation88,Citation89 The GLP-1R belongs to the Family B Gαs GPCRs, where ligand binding results in the activation of adenylyl cyclase and elevation of cAMP.Citation12 GLP-1R agonists reduce islet inflammation, while the genetic ablation of GLP-1R results in mild fasting hyperglycemia and defective GSIS.Citation90 GLP-1 acts at multiple receptors: the canonical GLP-1R and the glucagon receptor (GCGR). Using a chimeric GCGR/GLP-1R, researchers identified each receptor’s N-terminal domain as critical for ligand selectivity.Citation91 These findings indicate that the GLP-1R is relatively promiscuous with respect to ligand selectivity, such that it can bind multiple proglucagon-related peptides. These peptides include GLP-1(7–36), GLP-1(9–36), and glucagon.Citation12,Citation92–94 Similarly, the GCGR can bind and interact with both GLP-1 and glucagon, although GLP-1’s precise signaling pathway via this receptor is not well-characterized.Citation95 While glucagon’s actions at the GLP-1R in vivo have noteworthy effects on glycemic control, the effect of GLP-1 at the GCGR may be less significant in vivo due to its lower affinity at this receptor compared to the GLP-1R.Citation57 This aspect of GLP-1R pharmacology has significant implications regarding the biological function of proglucagon-derived peptides, as all of these peptide-ligands are present within the localized islet environment.
GLP-1(7–36)amide is considered the active form of human GLP-1 and has the most well-documented biological actions in beta cells where its signaling augments GSIS.Citation94,Citation96 GLP-1R activation results in elevated levels of cAMP that can interact with CREB elements in the beta cell. This protects beta cells from cytokine-induced apoptosis.Citation54 Furthermore, GLP-1R signaling also includes the Wnt/beta-catenin cascade that promotes beta cell proliferation.Citation53 In rodent islets, GLP-1R activation increases the availability of betacellulin, an EGFR agonist, and activates PI3K to induce beta cell proliferation and insulin biogenesis.Citation97 GLP-1R activation can also induce beta cell autophagy in high glucose conditions through modulation of AMPK.Citation23 Both GLP-1(7–36) and GLP-1(9–36) administration increases the release of insulin and c-peptide in vivo. Upon its binding, GLP-1(9–36) stimulates proinsulin production via PKA signaling, whereas canonical GLP-1(7-36)-induced insulin secretion is mediated by the GTPase exchange factor EPAC (Exchange Protein Activated by cAMP).Citation98 In summary, the GLP-1R orchestrates multiple intracellular signaling cascades, some of which are preferentially activated by different ligands.
Alpha cell signaling
GLP-1Rs have only been detected in a subpopulation of alpha cells.Citation88,Citation89,Citation99 Despite this, alpha cell-specific GLP-1R ablation results in impaired intraperitoneal glucose tolerance.Citation100 Although the GLP-1R may only be present in a subpopulation of alpha cells, its role is significant in coordinating the dynamics of glucagon release. Canonical signaling of GLP-1(7–36) at its receptor results in PKA-dependent inhibition of P/Q-type voltage-gated Ca2+ channels, inhibiting glucagon granule release.Citation101 In hyperglycemia, GLP-1R activation is glucagonostatic, whereas alpha cell granule secretion is stimulated in hypoglycemia.Citation100 Interestingly, isolated alpha cells tend to secrete glucagon in response to elevated glucose levels, implying some sort of paracrine inhibition of glucagon secretion at high glucose in vivo.Citation57 However, in this model, GLP-1 secretion was not assessed. The inhibition of alpha cell granule release is well-characterized; however, its potentiation in hypoglycemia is so-far poorly characterized.
Products of DPP4-mediated breakdown of GLP-1 also exhibit pharmacological activity in alpha cells. GLP-1(9–36) is bioactive at the GLP-1R and inhibits glucagon secretion. Unlike the glucagonostatic effects of GLP-1(7–36), GLP-1(9–36) does not require PKA signaling to inhibit glucagon release.Citation95 The dipeptide fragment released from the N-terminal of GLP-1(7–36) breakdown can potentiate glucagon secretion. This effect is dose-dependent, although the receptor and mechanism of action have not yet been identified.Citation102 In summary, GLP-1(7–36), GLP-1(9–36), and dipeptide fragments have pharmacological action in alpha cells. When considering alpha cell-derived GLP-1, these findings are indicative of an autocrine signaling network within alpha cells. Indeed, GLP-1, DPP4, and metabolites of DPP4-mediated GLP-1 breakdown can modulate the secretion of both glucagon and GLP-1.
It is unclear whether GLP-1Rs are only expressed on a subpopulation of alpha cells or if currently-available detection methods have failed to identify its presence consistently. A recent investigation by Gray et al.Citation103 utilized scRNAseq and validated GLP-1R antibody staining to discern that adult mouse islets do not express Glp1r mRNA or the GLP-1R protein. Researchers also confirmed that the GLP1R promoter is inactive in wild-type murine alpha cells.Citation103 In the absence of a GLP-1R, alpha cells can still respond to the downstream effects of GLP-1 signaling within the islet. So far, the literature suggests that the glucagonostatic effect of GLP-1 can occur directly via the GLP-1R on alpha cells and indirectly via GLP-1-mediated somatostatin secretion at delta cells as well as insulin secretion from beta cells.Citation89,Citation101 Both autocrine and paracrine inputs may be relevant to the coordination of alpha cell hormone release and overall islet function, however, interspecies differences in GLP-1R expression between human and rodent islets may complicate the investigation of these inputs.
Beta cell signaling
Proglucagon-derived hormones are vital for normal beta cell function. In the absence of alpha cell GCG expression, beta cells exhibit markedly decreased glucose and amino acid-stimulated insulin release.Citation12 Indeed, modulation of intracellular cAMP by glucagon and GLP-1(7–36) alters the beta cell’s ability to secrete insulin.Citation12,Citation92–94 Classically, glucagon acts on its canonical receptor on beta cells to potentiate insulin secretion; however, there is strong evidence that glucagon’s binding to the GLP-1R potentiates insulin release.Citation12,Citation57,Citation92 In beta cell-specific GCGR KO mice, the infusion of glucagon can still elicit insulin secretion, but this effect requires an intact GLP-1R.Citation104–106 Glucagon signaling via the GLP-1R on beta cells may have a more meaningful contribution to insulin dynamics than glucagon’s signaling through its canonical receptor. Interestingly, these mice have comparable glycemic excursions and glucose clearance when compared to their control littermates.Citation12 This raises the important question: is GLP-1 required for normal beta cell function, or is GLP-1R activation by an alternate ligand sufficient? Future studies must assess the contribution of glucagon signaling at the GLP-1R in different cell-types, including alpha and delta cells, to determine the relevance or redundancy of endogenous alpha cell GLP-1 production.
The potentiation of insulin release by GLP-1 has several downstream paracrine effects in other islet cell types. Indeed, insulin receptors on alpha cells are vital for glucose tolerance and glucagon dynamics. Alpha cell-specific insulin receptor KO mice exhibit hyperglucagonemia, hyperglycemia, and glucose intolerance.Citation107 These findings indicate that islet-derived GLP-1 has the potential to limit its own release through the downstream effects of insulin release. Insulin release can indirectly inhibit alpha cell hormone release via stimulation of somatostatin secretion from delta cells. Insulin’s effects on delta cells were investigated using a ‘somatostatin-secreting delta cell insulin receptor knockout’ (SIRKO) mice; these mice experience impaired insulin-stimulated somatostatin release and impaired insulin sensitivity.Citation108 Taken together, proglucagon-related peptides can interact with beta cells to induce insulin secretion. Insulin interacts with its receptors on adjacent alpha and delta cells to modulate the islet’s hormonal profile.
Delta cell signaling
In recent years, GLP-1Rs have been characterized in delta cells, where receptor activation potentiates glucose-dependent somatostatin release.Citation89,Citation99 Upon its secretion, somatostatin inhibits insulin and glucagon release via its interactions with the somatostatin receptor SSTR2.Citation87–89 SSTR2 activation results in adenylyl cyclase inhibition and the opening of G-protein coupled K+ channels, ultimately maintaining the alpha or beta cell in a negatively-polarized non-excitable state and suppressing hormone secretion.Citation60 While GLP-1 potentiates insulin and somatostatin release, somatostatin inhibits insulin release.Citation60 Therefore, depending on somatostatin availability, GLP-1 can indirectly inhibit insulin rather than potentiate its release.
Somatostatin may also inhibit the secretion of alpha cell-derived GLP-1, as glucagon and GLP-1 are co-packaged in secretory granules.Citation9 In the absence of somatostatin input, alpha cells release excessive quantities of glucagon.Citation87 This speaks to the critical role of somatostatin dynamics in regulating islet-hormone release. Of relevance to this somatostatin pathway, intra-islet GLP-1 may activate GLP-1Rs on delta cells in a paracrine manner, thus potentiating somatostatin release and indirectly inhibiting glucagon and GLP-1 secretion via SSTR2 signaling. This concept has been tested and confirmed through the use of SSTR2-specific antagonists in perfused rodent pancreas models, although only glucagon levels were investigated.Citation87–89 Co-infusion of GLP-1 and a selective SSTR2 antagonist partially attenuate GLP-1 mediated inhibition of glucagon secretion in cultured rat islets.Citation98 This confirms that somatostatin is important in inhibiting glucagon release, and GLP-1 has a direct and indirect inhibitory effect in alpha cells. The contribution of islet-derived GLP-1 in somatostatin dynamics must be further studied to understand the complexities of this possible paracrine network.
Paracrine networks between islet endocrine cells
Alpha cell-derived peptides potentiate insulin release from beta cells and somatostatin release from delta cells. This occurs primarily via GLP-1 and glucagon signaling at the GLP-1R, and the relevance of beta cell GCGR activation by GLP-1 is unclear due to GLP-1’s relative potency at this receptor. GLP-1 has a bidirectional effect on alpha cells, whereby receptor activation in hyperglycemia inhibits alpha cell hormone release, and GLP-1 potentiates hormone release in hypoglycemia. So far, the GLP-1R has only been identified in a subpopulation of alpha cells. Insulin released from beta cells inhibits alpha cell hormone release and potentiates somatostatin release from delta cells. SSTR2 activation on both alpha and beta cells inhibits further hormone secretion by hyperpolarizing cells into a less-excitable state.
Only considering insulin, somatostatin, and proglucagon-derived peptides, there are multiple possible network interactions. For example, alpha cell-derived GLP-1 can potentiate GSIS, insulin can stimulate delta-cell hormone release, and somatostatin can inhibit both GLP-1 and insulin release from alpha and beta cells, respectively. The effects of glucose and other metabolites further modulate these complex network interactions, as described below. The paracrine action between alpha, beta, and delta cells is essential for the coordination of insulin release in vivo and in the presence of pharmacological agents. Indeed, GLP-1R and GCGR agonists lose their effectiveness in controlling plasma-glucose levels in the absence of intact beta cells, as seen in many STZ treated T1D mouse models.Citation57
Glucose is an essential regulator of islet hormone secretion. Both beta and delta cells rely on glucose-stimulated secretion pathways, where hyperglycemia favors the release of insulin and somatostatin from the islet. In beta and delta cells, glucose entry and metabolism results in the closure of KATP channels, membrane depolarization, elevated intracellular calcium, and hormone secretion. This pathway in delta cells is more dependent on calcium-induced CaCitation2+ release, whereas beta cells depend on the activation of voltage-gated Ca2+ channels.Citation60 The sodium-glucose transporter SGLT2 is expressed in 33–58% of human delta cells, and its current contributes to insulin-induced somatostatin secretion.Citation108 In the presence of SGLT2 inhibitors like dapagliflozin, insulin-stimulated somatostatin secretion is modestly suppressed.Citation108,Citation109 As previously stated, the actions of GLP-1 in alpha cells are also glucose-dependent. In hyperglycemia, GLP-1R activation inhibits alpha cell hormone release, while receptor activation in hypoglycemia potentiates hormone secretion.Citation100
GPR120, a metabolite sensing receptor that can potentiate PC1/3 expression alpha cells, is also expressed in delta cells.Citation61 In the presence of fatty acids, including oleate and palmitate, GPR120 activation can inhibit somatostatin secretion by up to 50%.Citation60 In the presence of localized lipolysis, the availability of GPR120 ligands may favor GLP-1 production and limit somatostatin release. Omega-3 fatty acids and palmitate can activate GPR40 expressed in beta cells,Citation61 and GPR40 activation can potentiate glucose-stimulated insulin release.Citation60 Therefore, the transient elevation of palmitate levels in the islet can increase GLP-1 production, potentiate GSIS and inhibit glucose-dependent somatostatin secretion. In the presence of palmitate, the islet’s local hormonal profile could shift toward a net insulin-secreting profile due to elevated GLP-1 production and decreased somatostatin release.
Summary
This review has summarized and discussed the experimental evidence that GLP-1 can be expressed and secreted from human and rodent alpha cells, and that GLP-1Rs are widespread throughout the endocrine pancreas. The expression of alpha cell PC1/3, and therefore proglucagon processing to GLP-1, can be induced by various cytokines and metabolites, many of which are elevated in T2D and obesity. Intra-islet GLP-1 and DPP4 have functional roles in glucose homeostasis, although their contribution to the clinical effectiveness of DPP4 inhibitors has yet to be investigated. Finally, GLP-1 has been demonstrated to possess significant pharmacological activity in the islet, modulating insulin, glucagon, and somatostatin secretion. Similarly, the hormonal microenvironment of the islet can modulate alpha cell glucagon and GLP-1 release. Taken together, islet-derived GLP-1 has the potential for paracrine and autocrine roles in the endocrine pancreas, where its secretion and metabolism can alter the specific hormonal secretory phenotype in the islet.
Future perspectives
Future studies are therefore warranted to further investigate the paracrine effects of glucagon-derived peptides in human islets. To this end, a concerted effort to accurately identify the GLP-1R in alpha, beta, and delta cells would help immensely to establish the functional role of intra-islet GLP-1 in the pancreas. The nonspecific nature of many commercially available GLP-1R antibodies is well documented and an over-reliance on antibodies for identification of the GLP-1R has fueled controversy over the expression of the receptor in different islet cell types.109 However, new validated specific monoclonal antibodies for the GLP-1R have been developed and should bring a greater level of certainty to detection of GLP-1R protein.Citation110 The use of transcriptomics to study GLP-1R expression promises to aid in the identification of the receptor, and yet researchers should proceed with caution as this technique may not consistently or accurately measure transcripts from genes with low expression.Citation111
The clear and proper identification of proglucagon-derived peptides has also been controversial, largely again because of the reliance of antibodies for identification. The use of mass spectrometry to identify proglucagon-derived peptides would complement antibody-based identification and increase certainty around the detection of important peptides such as active GLP-1. In addition, the recent development of protocols that combine mass spectrometry with imaging for mouse and human pancreatic tissue allows for in situ detection of proteins.Citation112 This exciting alternative to immunohistochemical methods affords great promise and would help to identify and confirm heterogeneity of alpha cell proglucagon-derived peptide expression in the islet.
Given that GLP-1 secretion from human alpha cells is potentially important for islet function, the advent of a method for creating human stem-cell derived alpha cells is exciting for the field.Citation113 Currently, there is no human alpha cell line available to study GLP-1 secretion, and this potentially limitless source of human alpha cells would be amenable to genetic manipulation. For example, CRSPR-Cas9 could be used to knockout or mutate the prohormone convertases with the goal of studying the effect on GLP-1 expression. Furthermore, the signaling pathways for GLP-1 expression and secretion could be examined in this human alpha cell model. From a translational perspective, gene editing technologies may also be employed to generate stem cell derived alpha cells with an enhanced GLP-1 secretory phenotype that could be co-transplanted with beta cells to enhance post-graft cellular survival and function.
The key questions remaining to be answered are whether or not GLP-1 is constitutively expressed in the healthy adult pancreas and what role it may play in vivo rather than in isolated cellular systems? The role of intra-islet GLP-1 in the metabolically stressed islet must also be further characterized in order to provide a more complete understanding of this potential signaling axis in disease states. Several of the most effective recently developed therapeutic strategies for obesity, T1D and T2D target the GLP-1 pathway and it is important that the relevance of intra-islet GLP-1 be determined in order to optimize the effectiveness of these pharmacological agents used in treating these diseases.
Acknowledgments
P.E.L. holds the Dr. Charles A. Allard Chair in Diabetes Research. This review was supported by grants from the Canadian Institutes of Health Research and the Dr. Rod Eidem Diabetes Research Fund (P.E.L.). S.A.C. and J.J. received student graduate awards from the Alberta Diabetes Institute.
Additional information
Funding
References
- Is DD. Is GLP1 a hormone: whether and When? J Diabetes Investig. 2016;7(S1):50–55. doi:10.1111/jdi.12466.
- Rouillé Y, Martin S, Steiner DF. Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J Biol Chem [Internet] 1995; 270(44):26488–26496. http://www.jbc.org/lookup/doi/10.1074/jbc.270.44.26488
- Rouillé Y, Kantengwa S, Irminger J-C, Halban PA. Role of the prohormone convertase pc3 in the processing of proglucagon to glucagon-like peptide 1. J Biol Chem [Internet] 1997; 272(52):32810–32816. http://www.jbc.org/lookup/doi/10.1074/jbc.272.52.32810
- Varndell IM, Bishop AE, Sikri KL, Uttenthal LO, Bloom SR, Polak JM. Localization of glucagon-like peptide (GLP) immunoreactants in human gut and pancreas using light and electron microscopic immunocytochemistry. J Histochem Cytochem [Internet] 1985; 33(10):1080–1086. doi:10.1177/33.10.3900195.
- Lo Uttenthel, Ghiglione M, SK George, AE Bishop, JM Polak, SR Bloom. Molecular forms of glucagon-like peptide-1 in human pancreas and glucagonomas. J Clin Endocrinol Metab [Internet] 1985; 61:472–479. https://academic.oup.com/jcem/article-lookup/doi/10.1210/jcem-61-3-472
- Holst JJ, Bersani M, Johnsen AH, Kofod H, Hartmann B, Orskov C. Proglucagon processing in porcine and human pancreas. J Biol Chem [Internet] 1994; 269(29):18827–18833. http://www.ncbi.nlm.nih.gov/pubmed/8034635
- Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D’Aleo V, Piro S, Marselli L, et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia [Internet] 2012; 55(12):3262–3272. doi:10.1007/s00125-012-2716-9.
- Song Y, Koehler JA, Baggio LL, Powers AC, Sandoval DA, Drucker DJ. Gut-proglucagon-derived peptides are essential for regulating glucose homeostasis in mice. Cell Metab. 2019;30(5):976–986.e3. doi:10.1016/j.cmet.2019.08.009. .
- Kilimnik G, Kim A, Steiner DF, Friedman TC, Hara M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of β-cell regeneration. Islets [Internet] 2010; 2(3):149–155. http://www.tandfonline.com/doi/abs/10.4161/isl.2.3.11396
- Lund A, Knop FK. Extrapancreatic glucagon: present status. Diabetes Res Clin Pract [Internet]. 2019;147:19–28. doi:10.1016/j.diabres.2018.06.013. .
- Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia [Internet] 2000; 43(8):1012–1019. http://link.springer.com/10.1007/s001250051484
- Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, Jaffe JL, Coch RW, Haldeman JM, MacDonald PE, et al. β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight [Internet] 2019; 4(5):1–15 https://insight.jci.org/articles/view/126742
- Gribble FM, Reimann F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat Rev Endocrinol [Internet]. 2019;15(4):226–237. doi:10.1038/s41574-019-0168-8.
- Bugliani M, Syed F, Paula FMM, Omar BA, Suleiman M, Mossuto S, Grano F, Cardarelli F, Boggi U, Vistoli F, et al. DPP-4 is expressed in human pancreatic beta cells and its direct inhibition improves beta cell function and survival in type 2 diabetes. Mol Cell Endocrinol [Internet] 2018; 473:186–193. doi:10.1016/j.mce.2018.01.019.
- Kawai K, Yokota C, Ohashi S, Watanabe Y, Yamashita K. Evidence that glucagon stimulates insulin secretion through its own receptor in rats. Diabetologia [Internet] 1995; 38(3):274–276. http://link.springer.com/10.1007/BF00400630
- El K, Campbell JE. The role of GIP in α-cells and glucagon secretion. Peptides [Internet]. 2020;125:170213. doi:10.1016/j.peptides.2019.170213.
- Donath MY, Burcelin RGLP-1. GLP-1 effects on islets: hormonal, neuronal, or paracrine? Diabetes Care [Internet] 2013; 36(Supplement_2):S145–8. http://care.diabetesjournals.org/cgi/doi/10.2337/dcS13-2015
- Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K, Seino Y, Holst JJ, Schuit F, Drucker DJ Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of dpp-iv inhibitors. Diabetes [Internet] 2004; 53(5):1326–1335. doi:10.2337/diabetes.53.5.1326
- Mulvihill EE, Varin EM, Gladanac B, Campbell JE, Ussher JR, Baggio LL, Yusta B, Ayala J, Burmeister MA, Matthews D et al. Cellular sites and mechanisms linking reduction of dipeptidyl peptidase-4 activity to control of incretin hormone action and glucose homeostasis. Cell Metab [Internet]. 2017;25(1):152–165. doi:10.1016/j.cmet.2016.10.007
- Omar BA, Liehua L, Yamada Y, Seino Y, Marchetti P, Ahrén B. Dipeptidyl peptidase 4 (DPP-4) is expressed in mouse and human islets and its activity is decreased in human islets from individuals with type 2 diabetes. Diabetologia [Internet] 2014; 57(9):1876–1883. http://link.springer.com/10.1007/s00125-014-3299-4
- Masur K, Tibaduiza EC, Chen C, Ligon B, Beinborn M. Basal receptor activation by locally produced glucagon-like peptide-1 contributes to maintaining β-cell function. Mol Endocrinol [Internet] 2005; 19(5):1373–1382. https://academic.oup.com/mend/article-lookup/doi/10.1210/me.2004-0350
- Lin HV, Wang J, Wang J, Li W, Wang X, Alston JT, Thomas MK, Briere DA, Syed SK, Efanov AM. GPR142 prompts glucagon-like Peptide-1 release from islets to improve β cell function. Mol Metab [Internet]. 2018;11:205–211. doi:10.1016/j.molmet.2018.02.008.
- Miao X, Gu Z, Liu Y, Jin M, Lu Y, Gong Y, Li L, Li C. The glucagon-like peptide-1 analogue liraglutide promotes autophagy through the modulation of 5′-AMP-activated protein kinase in INS-1 β-cells under high glucose conditions. Peptides [Internet]. 2018;100:127–139. doi:10.1016/j.peptides.2017.07.006.
- Sancho V, Daniele G, Lucchesi D, Lupi R, Ciccarone A, Penno G, Bianchi C, Dardano A, Miccoli R, Del Prato S. Metabolic regulation of GLP-1 and PC1/3 in pancreatic α-cell line. PLoS One [Internet] 2017; 12(11):e0187836. https://dx.plos.org/10.1371/journal.pone.0187836
- Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev [Internet] 2014; 35(6):992–1019. https://academic.oup.com/edrv/article/35/6/992/2354726
- Shah P, Ardestani A, Dharmadhikari G, Laue S, Schumann DM, Kerr-Conte J, Pattou F, Klein T, Maedler MK. The DPP-4 inhibitor linagliptin restores β-cell function and survival in human isolated islets through glp-1 stabilization. J Clin Endocrinol Metab [Internet] 2013; 98(7):E1163–72. https://academic.oup.com/jcem/article/98/7/E1163/2536729
- Wideman RD, Yu ILY, Webber TD, Verchere CB, Johnson JD, Cheung AT, Kieffer TJ. Improving function and survival of pancreatic islets by endogenous production of glucagon-like peptide 1 (GLP-1). Proc Natl Acad Sci Internet] 2006; 103(36):13468–13473. http://www.pnas.org/cgi/doi/10.1073/pnas.0600655103
- Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology [Internet] 1995; 136(8):3585–3596. https://academic.oup.com/endo/article-lookup/doi/10.1210/endo.136.8.7628397
- MENTLEIN R, GALLWITZ B, WE SCHMIDT. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1 (7-36)amide,peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem [Internet]. 1993;214(3):829–835.
- Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab [Internet]. 1995;80(3):952–957.
- Reimann F, Diakogiannaki E, Moss CE, Gribble FM. Cellular mechanisms governing glucose-dependent insulinotropic polypeptide secretion. Peptides [Internet]. 2020;125:170206. doi:10.1016/j.peptides.2019.170206. .
- Kuhre RE, Wewer Albrechtsen NJ, Deacon CF, Balk-Møller E, Rehfeld JF, Reimann F, Gribble FM, Holst JJ. Peptide production and secretion in GLUTag, NCI-H716, and STC-1 cells: a comparison to native L-cells. J Mol Endocrinol [Internet] 2016; 56(3):201–211. https://jme.bioscientifica.com/view/journals/jme/56/3/201.xml
- Goldspink DA, Lu VB, Billing LJ, Larraufie P, Tolhurst G, Gribble FM, Reimann F. Mechanistic insights into the detection of free fatty and bile acids by ileal glucagon-like peptide-1 secreting cells. Mol Metab [Internet] 2018; 7:90–101. https://linkinghub.elsevier.com/retrieve/pii/S2212877817307500.
- Campbell JR, Martchenko A, Sweeney ME, Maalouf MF, Psichas A, Gribble FM, Reimann F, Brubaker PL. Essential role of syntaxin-binding protein-1 in the regulation of glucagon-like peptide-1 secretion. Endocrinology [Internet] 2020; 161(5):1–13. https://academic.oup.com/endo/article/doi/10.1210/endocr/bqaa039/5788420
- Pais R, Gribble FM, Reimann F. Signalling pathways involved in the detection of peptones by murine small intestinal enteroendocrine L-cells. Peptides [Internet]. 2016;77:9–15. doi:10.1016/j.peptides.2015.07.019. .
- MACLEAN N, Ogilvie RF. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes [Internet] 1955; 4(5):367–376. http://diabetes.diabetesjournals.org/cgi/doi/10.2337/diab.4.5.367
- Nie Y, Nakashima M, Brubaker PL, Li Q-L, Perfetti R, Jansen E, Zambre Y, Pipeleers D, Friedman TC. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest [Internet] 2000; 105(7):955–965. http://www.jci.org/articles/view/7456
- Hansen AMK, Bödvarsdottir TB, Nordestgaard DNE, Heller RS, Gotfredsen CF, Maedler K, Fels JJ, Holst JJ, Karlsen AE. Upregulation of alpha cell glucagon-like peptide 1 (GLP-1) in Psammomys obesus—an adaptive response to hyperglycaemia? Diabetologia [Internet] 2011; 54(6):1379–1387. http://link.springer.com/10.1007/s00125-011-2080-1
- O’Malley TJ, Fava GE, Zhang Y, Fonseca VA, Wu H. Progressive change of intra-islet GLP-1 production during diabetes development. Diabetes Metab Res Rev [Internet]. 2014;30(8):661–668. doi:10.1002/dmrr.2534.
- de Souza AH, Tang J, Yadev AK, Saghafi ST, Kibbe CR, Linnemann AK, Merrins MJ, Davis DB. Intra-islet GLP-1, but not CCK, is necessary for β-cell function in mouse and human islets. Sci Rep [Internet] 2020; 10(1):2823. http://www.nature.com/articles/s41598-020-59799-2
- Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, Gutierrez-Aguilar R, Li B, Drucker DJ, D’Alessio DA, et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab [Internet] 2017; 25(4):927–934.e3. doi:10.1016/j.cmet.2017.02.008.
- Linnemann AK, Blumer J, Marasco MR, Battiola TJ, Umhoefer HM, Han JY, Lamming DW, Davis DB. Interleukin 6 protects pancreatic β cells from apoptosis by stimulation of autophagy. Faseb J [Internet] 2017; 31(9):4140–4152. https://onlinelibrary.wiley.com/doi/abs/10.1096/fj.201700061RR
- Whalley NM, Pritchard LE, Smith DM, White A. Processing of proglucagon to GLP-1 in pancreatic α-cells: is this a paracrine mechanism enabling GLP-1 to act on β-cells? J Endocrinol [Internet] 2011; 211(1):99–106. https://joe.bioscientifica.com/view/journals/joe/211/1/99.xml.
- Campbell SA, Golec DP, Hubert M, Johnson J, Salamon N, Barr A, MacDonald PE, Philippaert K, Light PE. Human islets contain a subpopulation of glucagon-like peptide-1 secreting α cells that is increased in type 2 diabetes. Mol Metab [Internet]. 2020;39:101014. doi:10.1016/j.molmet.2020.101014. .
- Ivy JR, Jones NK, Costello HM, Mansley MK, Peltz TS, Flatman PW, Bailey MA. Glucocorticoid receptor activation stimulates the sodium-chloride cotransporter and influences the diurnal rhythm of its phosphorylation. Am J Physiol Renal Physiol. 2019;317(6):F1536–48. doi:10.1152/ajprenal.00372.2019.
- Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. Age-dependent pancreatic gene regulation reveals mechanisms governing human β cell function. Cell Metab [Internet] 2016; 23(5):909–920. https://linkinghub.elsevier.com/retrieve/pii/S155041311630122X.
- Å S, Palasantza A, Eliasson P, Andersson E-M, A-C A, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab Internet] 2016; 24(4):593–607. https://linkinghub.elsevier.com/retrieve/pii/S1550413116304363
- Ramzy A, Asadi A, Kieffer TJ. Revisiting proinsulin processing: evidence that human β-cells process proinsulin with prohormone convertase (PC) 1/3 but not PC2. Diabetes [Internet] 2020; 69(7):1451–1462. http://diabetes.diabetesjournals.org/lookup/doi/10.2337/db19-0276
- Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, Kerr-Conte J, Pattou F, Berney T, Pipeleers D, et al. Interleukin-6 regulates pancreatic -cell mass expansion. Proc Natl Acad Sci [Internet] 2008; 105(35):13163–13168. http://www.pnas.org/cgi/doi/10.1073/pnas.0801059105
- Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med [Internet] 2011; 17(11):1481–1489. http://www.embase.com/search/results?subaction=viewrecord&from=export&id=L51690291%5Cnhttp://dx.doi.org/10.1038/nm.2513%5Cnhttp://sfxhosted.exlibrisgroup.com/sfxtul?sid=EMBASE&=10788956&id=doi:10.1038%2Fnm.2513&atitle=Interleukin-6+enhances+insulin+se
- Ellingsgaard H, Seelig E, Timper K, Coslovsky M, Soederlund L, Lyngbaek MP, Wewer Albrechtsen NJ, Schmidt-Trucksäss A, Hanssen H, Frey WO, et al. GLP-1 secretion is regulated by IL-6 signalling: a randomised, placebo-controlled study. Diabetologia [Internet] 2020; 63(2):362–373. http://link.springer.com/10.1007/s00125-019-05045-y
- Timper K, Dalmas E, Dror E, Rütti S, Thienel C, Sauter NS, Bouzakri K, Bédat B, Pattou F, Kerr-Conte J et al. Glucose-dependent insulinotropic peptide stimulates glucagon-like peptide 1 production by pancreatic islets via interleukin 6, produced by α cells Gastroenterology. 2016;151(1):165–179. doi:10.1053/j.gastro.2016.03.003
- Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17(6):819–837. doi:10.1016/j.cmet.2013.04.008. .
- Linnemann AK, Neuman JC, Battiola TJ, Wisinski JA, Kimple ME, Davis DB. Glucagon-like peptide-1 regulates cholecystokinin production in β-cells to protect from apoptosis. Mol Endocrinol [Internet] 2015; 29(7):978–987. https://academic.oup.com/mend/article/29/7/978/2556543
- Liu L, Omar B, Marchetti P, Ahrén B. Dipeptidyl peptidase-4 (DPP-4): localization and activity in human and rodent islets. Biochem Biophys Res Commun [Internet]. 2014;453(3):398–404. doi:10.1016/j.bbrc.2014.09.096. .
- Mulvihill EE. Dipeptidyl peptidase inhibitor therapy in type 2 diabetes: control of the incretin axis and regulation of postprandial glucose and lipid metabolism. Peptides [Internet]. 2018;100:158–164. doi:10.1016/j.peptides.2017.11.023.
- Finan B, Capozzi ME, Campbell JE. Repositioning glucagon action in the physiology and pharmacology of diabetes. Diabetes. 2020;69(4):532–541. doi:10.2337/dbi19-0004.
- Ueda Y, Iwakura H, Bando M, Doi A, Ariyasu H, Inaba H, Morita S, Akamizu T. Differential role of GPR142 in tryptophan-mediated enhancement of insulin secretion in obese and lean mice. PLoS One [Internet] 2018; 13:e0198762. https://dx.plos.org/10.1371/journal.pone.0198762.
- Rudenko O, Shang J, Munk A, Ekberg JP, Petersen N, Engelstoft MS, Egerod KL, Hjorth SA, Wu M, Feng Y, et al. The aromatic amino acid sensor GPR142 controls metabolism through balanced regulation of pancreatic and gut hormones. Mol Metab [Internet] 2019; 19:49–64. doi:10.1016/j.molmet.2018.10.012.
- Noguchi GM, Huising MO. Integrating the inputs that shape pancreatic islet hormone release. Nat Metab [Internet]. 2019;1(12):1189–1201. doi:10.1038/s42255-019-0148-2. .
- Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab [Internet]. 2017;25(4):777–796. doi:10.1016/j.cmet.2017.03.008.
- Moss CE, Glass LL, Diakogiannaki E, Pais R, Lenaghan C, Smith DM, Wedin M, Bohlooly-Y M, Gribble FM, Reimann F. Lipid derivatives activate GPR119 and trigger GLP-1 secretion in primary murine L-cells. Peptides [Internet]. 2016;77:16–20. doi:10.1016/j.peptides.2015.06.012.
- Liu Z, Stanojevic V, Avadhani S, Yano T, Habener JF. Stromal cell-derived factor-1 (SDF-1)/chemokine (C-X-C motif) receptor 4 (CXCR4) axis activation induces intra-islet glucagon-like peptide-1 (GLP-1) production and enhances beta cell survival. Diabetologia [Internet] 2011; 54(8):2067–2076. http://link.springer.com/10.1007/s00125-011-2181-x.
- Habener JF, Stanojevic V. α-cell role in β-cell generation and regeneration. Islets [Internet] 2012; 4(3):188–198. http://www.tandfonline.com/doi/abs/10.4161/isl.20500
- Kayali AG, Lopez AD, Hao E, Hinton A, Hayek A, King CC, Maedler K. The SDF-1α/CXCR4 axis is required for proliferation and maturation of human fetal pancreatic endocrine progenitor cells. PLoS One [Internet] 2012; 7(6):e38721. http://www.ncbi.nlm.nih.gov/pubmed/14638861
- Kayali AG, Van Gunst K, Campbell IL, Stotland A, Kritzik M, Liu G, Flodstrom-Tullberg M, Zhang Y-Q Y-Q, Sarvetnick N. The stromal cell–derived factor-1α/CXCR4 ligand–receptor axis is critical for progenitor survival and migration in the pancreas. J Cell Biol [Internet] 2003; 163(4):859–869. https://rupress.org/jcb/article/163/4/859/33749/The-stromal-cellderived-factor1αCXCR4
- Fava GE, Dong EW, Wu H. Intra-islet glucagon-like peptide 1. J Diabetes Complications [Internet] 2016; 30(8):1651–1658. https://linkinghub.elsevier.com/retrieve/pii/S1056872716301696
- Varin EM, Mulvihill EE, Beaudry JL, Pujadas G, Fuchs S, Tanti J-F, Fazio S, Kaur K, Cao X, Baggio LL, et al. Circulating levels of soluble dipeptidyl peptidase-4 are dissociated from inflammation and induced by enzymatic DPP4 inhibition. Cell Metab Internet] 2019; 29(2):320–334.e5. https://linkinghub.elsevier.com/retrieve/pii/S1550413118306338
- Trzaskalski NA, Fadzeyeva E, Mulvihill EE. Dipeptidyl peptidase-4 at the interface between inflammation and metabolism. Clin Med Insights Endocrinol Diabetes [Internet] 2020; 13:117955142091297. http://journals.sagepub.com/doi/10.1177/1179551420912972.
- Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol Metab [Internet]. 2016;5(3):233–244. doi:10.1016/j.molmet.2016.01.002.
- Augstein P, Naselli G, Loudovaris T, Hawthorne WJ, Campbell P, Bandala-Sanchez E, Rogers K, Heinke P, Thomas HE, Kay TW et al. Localization of dipeptidyl peptidase-4 (CD26) to human pancreatic ducts and islet alpha cells Diabetes Res Clin Pract [Internet]. 2015;110:291–300. doi:10.1016/j.diabres.2015.10.010
- Busek P, Hrabal P, Fric P, Sedo A. Co-expression of the homologous proteases fibroblast activation protein and dipeptidyl peptidase-IV in the adult human Langerhans islets. Histochem Cell Biol [Internet] 2015; 143(5):497–504. http://link.springer.com/10.1007/s00418-014-1292-0
- Poulsen MD, Hansen GH, Dabelsteen E, Høyer PE, Norén O, Sjöström H. Dipeptidyl peptidase IV is sorted to the secretory granules in pancreatic islet A-cells. J Histochem Cytochem [Internet] 1993; 41(1):81–88. http://journals.sagepub.com/doi/10.1177/41.1.8093256
- Rodriguez-Diaz R, Tamayo A, Hara M, Caicedo A. The local paracrine actions of the pancreatic α-cell. Diabetes [Internet] 2020; 69(4):550–558. http://diabetes.diabetesjournals.org/lookup/doi/10.2337/dbi19-0002
- Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, Leibiger IB, Kenyon NS, Ricordi C, Pileggi A, et al. Paracrine interactions within the pancreatic islet determine the glycemic set point. Cell Metab [Internet] 2018; 27(3):549–558.e4. https://linkinghub.elsevier.com/retrieve/pii/S1550413118300676
- Mannucci E, Pala L, Ciani S, Bardini G, Pezzatini A, Sposato I, Cremasco F, Ognibene A, Rotella CM. Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia [Internet] 2005; 48(6):1168–1172. http://link.springer.com/10.1007/s00125-005-1749-8
- Varin EM, Mulvihill EE, Baggio LL, Koehler JA, Cao X, Seeley RJ, Drucker DJ. Distinct neural sites of glp-1r expression mediate physiological versus pharmacological control of incretin action. Cell Rep [Internet]. 2019;27(11):3371–3384.e3. doi:10.1016/j.celrep.2019.05.055. .
- Campbell SA, Hubert M, Johnson J, Salamon N, Light PE. The DPP4 inhibitor sitagliptin increases active glp-1 levels from human islets and may increase islet cell survival prior to transplantation. OBM Transplant [Internet] 2019; 3(2):1. https://www.lidsen.com/journals/transplantation/transplantation-03-02-069
- Caicedo A. Paracrine and autocrine interactions in the human islet: more than meets the eye. Semin Cell Dev Biol [Internet] 2013; 24(1):11–21. https://linkinghub.elsevier.com/retrieve/pii/S1084952112001723
- Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, Berney T. Unique arrangement of - and -cells in human islets of langerhans. Diabetes [Internet] 2010; 59(5):1202–1210. http://diabetes.diabetesjournals.org/cgi/doi/10.2337/db09-1177
- Almaça J, Caicedo A. Blood flow in the pancreatic islet: not so isolated anymore. Diabetes [Internet] 2020; 69(7):1336–1338. http://diabetes.diabetesjournals.org/lookup/doi/10.2337/dbi20-0016
- Campbell SA, Light PE, Simpson SH Costarting sitagliptin with metformin is associated with a lower likelihood of disease progression in newly treated people with type 2 diabetes: a cohort study. Diabet Med [Internet] 2019:dme.14154. https://onlinelibrary.wiley.com/doi/abs/10.1111/dme.14154
- Oram RA, Jones AG, Besser REJ, Knight BA, Shields BM, Brown RJ, Hattersley AT, McDonald TJ. The majority of patients with long-duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia. 2014;57(1):187–191. doi:10.1007/s00125-013-3067-x.
- Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, Koch N, Dalmas E, Stawiski M, Makshana V, et al. Pancreatic α cell-derived glucagon-related peptides are required for β cell adaptation and glucose homeostasis. Cell Rep [Internet] 2017; 18(13):3192–3203. https://linkinghub.elsevier.com/retrieve/pii/S2211124717303224
- Wideman RD, Covey SD, Webb GC, Drucker DJ, Kieffer TJ. A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted -cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes [Internet] 2007; 56(11):2744–2752. http://diabetes.diabetesjournals.org/cgi/doi/10.2337/db07-0563
- Zhu L, Dattaroy D, Pham J, Wang L, Barella LF, Cui Y, Wilkins KJ, Roth BL, Hochgeschwender U, Matschinsky FM, et al. Intraislet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight [Internet] 2019; 4(10):1–15. https://insight.jci.org/articles/view/127994
- Xu SFS, Andersen DB, Izarzugaza JMG, Kuhre RE, Holst JJ. In the rat pancreas, somatostatin tonically inhibits glucagon secretion and is required for glucose‐induced inhibition of glucagon secretion. Acta Physiol [Internet] 2020; 229(3):1–15. https://onlinelibrary.wiley.com/doi/abs/10.1111/apha.13464
- de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia [Internet] 2008; 51(12):2263–2270. http://link.springer.com/10.1007/s00125-008-1149-y
- Ørgaard A, Holst JJ. The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia [Internet] 2017; 60(9):1731–1739. http://link.springer.com/10.1007/s00125-017-4315-2
- Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab [Internet]. 2018;27(4):740–756. doi:10.1016/j.cmet.2018.03.001.
- Runge S, Wulff BS, Madsen K, Bräuner-Osborne H, Knudsen LB. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br J Pharmacol [Internet]. 2003;138(5):787–794. doi:10.1038/sj.bjp.0705120.
- Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, Finan B, D’Alessio DA, Campbell JE. Glucagon lowers glycemia when β cells are active. JCI Insight [Internet] 2019; 4(16):1–12. https://insight.jci.org/articles/view/129954
- Elahi D, Egan JM, Shannon RP, Meneilly GS, Khatri A, Habener JF, Andersen DK. GLP-1 (9-36) amide, cleavage product of glp-1 (7-36) amide, is a glucoregulatory peptide. Obesity [Internet]. 2008;16(7):1501–1509. doi:10.1038/oby.2008.229.
- Vahl TP, Paty BW, Fuller BD, Prigeon RL, D’Alessio DA. Effects of GLP-1-(7–36)NH 2, GLP-1-(7–37), and GLP-1- (9–36)NH2 on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J Clin Endocrinol Metab Internet] 2003; 88(4):1772–1779. https://academic.oup.com/jcem/article-lookup/doi/10.1210/jc.2002-021479
- Guida C, Miranda C, Asterholm IW, Basco D, Benrick A, Chanclon B, M V C, Harris M, Kellard J, Mcculloch LJ, et al. GLP-1 (9-36) mediates the glucagonostatic effect of GLP-1 by promiscuous activation of the glucagon receptor. 1; 2020.
- Kreymann B, Ghatei MA, Williams G, Bloom SR. Bloom SR. GLUCAGON-LIKE PEPTIDE-1 7-36: a PHYSIOLOGICAL INCRETIN IN MAN. Lancet [Internet] 1987; 330(8571):1300–1304. https://linkinghub.elsevier.com/retrieve/pii/S0140673687911949
- Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic -cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes [Internet] 2003; 52(1):124–132. http://diabetes.diabetesjournals.org/cgi/doi/10.2337/diabetes.52.1.124
- Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, Fritsche A, Gribble F, Grill HJ, Habener JF, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab [Internet] 2019; 30:72–130. doi:10.1016/j.molmet.2019.09.010.
- Holst JJ, Christensen M, Lund A, de Heer J, Svendsen B, Kielgast U, Knop FK. Regulation of glucagon secretion by incretins. Diabetes, Obes Metab [Internet]. 2011;13:89–94. doi:10.1111/j.1463-1326.2011.01452.x. .
- Zhang Y, Parajuli KR, Fava GE, Gupta R, Xu W, Nguyen LU, Zakaria AF, Fonseca VA, Wang H, Mauvais-Jarvis F, et al. GLP-1Receptor in pancreatic α-cells regulates glucagon secretion in a glucose-dependent bidirectional manner. Diabetes [Internet] 2019; 68(1):34–44. http://diabetes.diabetesjournals.org/lookup/doi/10.2337/db18-0317
- Ramracheya R, Chapman C, Chibalina M, Dou H, Miranda C, González A, Moritoh Y, Shigeto M, Zhang Q, Braun M, et al. GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca 2+ channels. Physiol Rep [Internet]. 2018;6(17):e13852. doi:10.14814/phy2.13852.
- Waget A, Cabou C, Masseboeuf M, Cattan P, Armanet M, Karaca M, Castel J, Garret C, Payros G, Maida A, et al. Physiological and pharmacological mechanisms through which the dpp-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology [Internet] 2011; 152(8):3018–3029. https://academic.oup.com/endo/article-lookup/doi/10.1210/en.2011-0286
- Gray SM, Xin Y, Ross EC, Chazotte BM, Capozzi ME, El K, Svendsen B, Ravn P, Sloop KW, Tong J et al. Discordance between GLP-1R gene and protein expression in mouse pancreatic islet cells J Biol Chem [Internet]. 2020;295(33):11529–11541. doi:10.1074/jbc.RA120.014368
- Jun LS, Millican RL, Hawkins ED, Konkol DL, Showalter AD, Christe ME, Michael MD, Sloop KW. Absence of glucagon and insulin action reveals a role for the GLP-1Receptor in endogenous glucose production. Diabetes Internet] 2015; 64(3):819–827. http://diabetes.diabetesjournals.org/lookup/doi/10.2337/db14-1052
- Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, Holst JJ. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep [Internet]. 2018;25(5):1127–1134.e2. doi:10.1016/j.celrep.2018.10.018. .
- Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L, Obici S, Tang B, Holst JJ, Fledelius C, et al. Lower blood glucose, hyperglucagonemia, and pancreatic cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci [Internet] 2003; 100(3):1438–1443. http://www.pnas.org/cgi/doi/10.1073/pnas.0237106100
- Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, Herrera PL, Polonsky KS, McGuinness OP, Kulkarni RN Insulin signaling in α cells modulates glucagon secretion in vivo. Metab C [Internet] 2009; 9(4):350–361. https://linkinghub.elsevier.com/retrieve/pii/S1550413109000667
- Vergari E, Knudsen JG, Ramracheya R, Salehi A, Zhang Q, Adam J, Asterholm IW, Benrick A, Briant LJB, Chibalina MV et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion Nat Commun [Internet]. 2019;10(1):139. doi:10.1038/s41467-018-08193-8
- Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, Streutker C, Holland D, Cao X, Baggio LL, et al. GLP-1Receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male apoE−/− mice. Endocrinology [Internet] 2013; 154(1):127–139. https://academic.oup.com/endo/article/154/1/127/2423344
- Nasteska D, Fine NHF, Ashford FB, Cuozzo F, Viloria K, Smith G, Dahir A, Dawson PWJ, Lai Y-C, Bastidas-Ponce A, et al. PDX1LOW MAFALOW β-cells contribute to islet function and insulin release. Nat Commun [Internet] 2021; 12(1):674. http://www.nature.com/articles/s41467-020-20632-z
- Mawla AM, Huising MO. Navigating the depths and avoiding the shallows of pancreatic islet cell transcriptomes. Diabetes [Internet] 2019; 68(7):1380–1393. http://diabetes.diabetesjournals.org/lookup/doi/10.2337/dbi18-0019
- Prentice BM, Hart NJ, Phillips N, Haliyur R, Judd A, Armandala R, Spraggins JM, Lowe CL, Boyd KL, Stein RW, et al. Imaging mass spectrometry enables molecular profiling of mouse and human pancreatic tissue. Diabetologia [Internet] 2019; 62(6): 1036–1047. http://link.springer.com/10.1007/s00125-019-4855–8
- Peterson QP, Veres A, Chen L, Slama MQ, Kenty JHR, Hassoun S, Brown MR, Dou H, Duffy CD, Zhou Q, et al. A method for the generation of human stem cell-derived alpha cells. Nat Commun [Internet] 2020; 11(1):2241. http://www.nature.com/articles/s41467-020-160493

