
ABSTRACT
We aimed to survey the monogenic causes of disorders of sex development (DSD) and thereby its prevalence in India. This study revealed mutations resulting in androgen insensitivity syndrome, 5α-reductase type 2 deficiency, and gonadal dysgenesis were commonly reported. Intriguingly, AR deficits were the most prevalent (32 mutations) and of 11/26 missense mutations were in exons 4–8 (encoding ligand binding domain). The unique features of SRD5A2 defects were p.R246Q (most prevalent) and p.G196S could be mutational hotspots, dual gene defects (p.A596T in AR and p.G196S in SRD5A2) in a patient with hypospadias and novel 8 nucleotide deletion (exon 1) found in a patient with perineal hypospadias. Deficits in SRY, WT1, DHH, NR5A1, and DMRT1 caused 46,XY gonadal dysgenesis. Notably, mutations in AR, SRD5A2, MAMLD1, WT1, and MAP3K1 have led to hypospadias and only one CYP19A1 mutation caused aromatase deficiency was reported to date. Data mining from various databases has not only reinforced the role of well-established genes (e.g., SRY, WT1, DHH, NR5A1, DMRT1, AR, SRD5A2, MAMLD1) involved in DSD but also provided us 12 more potential candidate genes (ACVR1, AMHR2, CTNNB1, CYP11A1, CYP19A1, FGFR2, FGF9, PRKACA, PRKACG, SMAD9, TERT, ZFPM2), which benefit from a close association with the well-established genes involved in DSD and might be useful to screen owing to their direct gene–phenotype relationship or through direct functional interaction. As more genes have been revealed in relation to DSD, we believe ultimately it holds a better scenario for therapeutic regimen. Despite the advances in translational medicine, hospitals are yet to adopt genetic testing and counseling facilities in India that shall have potential impact on clinical diagnosis.
Abbreviations: 5α-RD2: 5α-Reductase type 2; AIS: androgen insensitivity syndrome; AMH: antimullerian hormone; AMHR: antimullerian hormone receptor; AR: androgen receptor gene; CAH: congenital adrenal hyperplasia; CAIS: complete AIS; CAH: congenital adrenal hyperplasia; CHH: congenital hypogonadotropic hypogonadism; CXORF6: chromosome X open reading frame 6 gene; CYP19A1: cytochrome P450 family 19 subfamily A member 1 gene; DHT: dihydrotestosterone; DMRT1: double sex and mab-3 related transcription factor 1 gene; DSD: disorders of sexual development; GD: gonadal dysgenesis; HGMD: human gene mutation database; IH: isolated hypospadias; MAMLD1: mastermind like domain containing 1 gene; MIS: mullerian inhibiting substance; NTD: N-terminal domain; OT DSD: ovotesticular DSD; PAIS: partial AIS; SOX9: SRY-related HMG-box 9 gene; SRY: sex-determining region Y gene; STAR: steroidogenic acute regulatory protein gene; SRD5A2: steroid 5 alpha-reductase 2 gene; T DSD: testicular DSD; T: testosterone; WNT4: Wnt family member 4 gene; WT1: Wilms tumor 1 gene; Δ4: androstenedione
Introduction
The sexual ambiguity in gender assignment of a newborn elicits medical attention (Shulman Citation2012). A defined battery of investigations, viz., ultrasonography, hormonal, cytogenetic, and molecular genetic analysis, would aid to earmark the cause of this disorder (Sfez-Yaiche and Sulmont Citation2000). Once the clinical cause of sexual ambiguity has been established, sex assignment of the affected could be done in consensus with family members or depending on psycho-sexual orientation of the affected (Paul Citation1965). The term ‘intersex’ potentially pejorative to affected individuals was replaced by a new nomenclature ‘disorders of sexual development’ (DSD). DSD is defined as congenital conditions in which development of chromosomal, gonadal, or anatomical sex is atypical (Hughes et al. Citation2006). Molecular diagnostics aims to establish the genotypic (heritable) cause of DSD in karyotypic male (46,XY) or female (46,XX) patients (). From an Indian perspective, we noted that most of the DSD cases (with monogenic cause) reported so far were of 46,XY DSD and thus will be emphasized below.
Figure 1. Molecular genetics of DSD. Under each subcategory of disorder, candidate gene(s) and OMIM in parenthesis have been itemized. The underlined indicates gene mutations that were also reported from India.
Abbreviations: 5α-reductase type 2 (5α-RD2), androgen insensitivity syndrome (AIS), complete gonadal dysgenesis (CGD), congenital adrenal hyperplasia (CAH), congenital hypogonadotropic hypogonadism (CHH), disorders of sexual development (DSD), gonadal dysgenesis (GD), hypogonadotropic hypogonadism 23 without anosmia (HH23), isolated hypospadias (IH), luteinizing hormone (LH), ovotesticular DSD (OT DSD), partial gonadal dysgenesis (PGD), persistent mullerian duct syndrome (PMDS), and testicular DSD (T DSD).

46,XY DSD are clinically typified by ambiguous or female external genitalia, caused by incomplete intrauterine masculinization and the presence or absence of mullerian structures (Mendonca Citation2009). Hence, these were categorized as defects of testis determination, defects in androgen synthesis or its action and other causes that include maternal exposure of endocrine disruptors (e.g., vinclozolin) (Vilela et al. Citation2007), syndromic associations of male genital development, defects in antimullerian hormone (AMH)/mullerian inhibiting substance (MIS) synthesis or its AMH receptor (AMHR), isolated hypospadias (chromosome X open reading frame 6 gene (CXORF6) mutation (Ogata et al. Citation2009). Gonadal dysgenesis (GD) is characterized by incomplete or defective formation of the gonads due to either structural or numerical anomalies of sex chromosome or mutations in the genes involved in the gonadal development (McCann-Crosby et al. Citation2014). 46,XY gonadal dysgenesis can be evaluated by presence of streak gonads and estimating the levels of circulating AMH and sex steroids which aid to score the severity (complete/partial). Mutations in sex-determining region Y gene (SRY) (<20% of cases) and NR5A1 (also known as SF1) accounted for complete and partial 46,XY gonadal dysgenesis, respectively. In addition, mutations in genes involved in gonadal determination, e.g., underexpression of Wilms tumor 1 gene (WT1), NR5A1, SRY-box 9 gene (SOX9), double sex and mab-3-related transcription factor 1 gene (DMRT1), DMRT2; overexpression of DAX1, Wnt family member 4 gene (WNT4) cause 46,XY gonadal dysgenesis associated syndromic phenotype. Defects in dihydrotestosterone (DHT) synthesis or androgen action on the target tissues share common dysmorphic features. The former is referred as steroid 5α-reductase type 2 (5α-RD2) deficiency and the latter as partial AIS (PAIS). Estimation of serum testosterone (T) to androstenedione (Δ4) (T/Δ4 < 0.8) facilitates biochemical diagnosis of a rare disorder of 17β-hydroxysteroid dehydrogenase 3 deficiency; similarly, serum T/DHT ratio (>30) for 5α-RD2 deficiency. Hormonal analysis may help to make an accurate diagnosis in proband(s) with classical disorders but may have a low conclusive value in the patients at their early stage of development or if there is partial defect. The mist of these differentials could be cleared by mutational analysis of candidate gene(s) and thus would aid in treatment modality of the disorder.
One of the deleterious factors for rise of heritable causes of reproductive health issues in India is the prevalence of consanguineous marriages (Hamamy Citation2012; Fareed et al. Citation2017). This could be due to family ties, integrity of estates, illiteracy, and religious practices (Rao et al. Citation2009) and has led to surge in homozygosity. Hospitals need to meticulously adopt medical genetic testing and counseling facilities to alleviate/erase such health issues in our country. Unfortunately, reproductive health issues are considered social stigma in India and thus a psychological barrier to patients with DSD reaching clinical setup/follow-up cases too and could also be the predicted causes for sparse mutational data. In light of this, we thought surveying the molecular genetic data shall reveal the prevalence (and phenotypic spectra) of DSD and its fecundity for research in Indian ethnicity. By and large the current survey, a first of its kind, will not only aid to gain quick access to all gene deficits causing DSD reported to date but also will be stimulable urge to all Indian medical practitioners to join hands with geneticist, and to aim for translational medicine (especially in case(s) of consanguineous couples with family history of DSD) as an integral in clinical diagnosis.
AR mutations
A defective androgen receptor (NR3C4: a member of nuclear receptor super family, 919 amino acids) gene (AR) [cytogenetic location: Xq11-12, comprising of exon 1 code for N-terminal domain (NTD), exons 2 and 3 code for DNA binding domain (DBD), parts of exons 3 and 4 code for hinge region, and exons 4–8 code for ligand binding domain (LBD)] causes end organ resistance to androgens known as androgen insensitivity syndrome (AIS) (OMIM # 300068, X-linked recessive). The affected will be karyotypic male (46,XY) offspring inheriting a defective copy of AR (due to hemizygous state) from carrier mother. Based on phenotypic presentation, it has been classified PAIS (men with minor degree of undervirilization and gynecomastia at time of puberty) and complete AIS (CAIS) (women with clitoromegaly). More than 800 mutations representing over 500 unique AR mutations have been reported from more than 850 patients with AIS (Gottlieb et al. Citation2012) and archived at http://www.androgendb.mcgill.ca. Mutations identified in subjects with AIS range from single base variations, nucleotide insertions or deletions, complete or partial gene deletion to intronic mutations (Bruggenwirth et al. Citation1996; Brinkmann Citation2001). However, amongst the known AR mutations, gene deletion (either partial or complete) and intronic mutations are at low frequency, whereas missense mutations leading to variable phenotypes account for the most common molecular lesion. AR mutations may play a role in the cause of isolated hypospadias (IH), even in the milder forms (Kalfa et al. Citation2013).
Currently, 32 different mutations in AR coding sequences have been documented amongst Indian ethnics (), of which most were in LBD (14 mutations) and the least in Hinge region (2 mutations). Previous reports have shown that Hinge region mutations like p.A645D (MacLean et al. Citation2004; Werner et al. Citation2006) and p.R629W (Deeb Citation2008) have led to AIS. In contrast to the above, AR defects were scored in cases of ovarian failure (Panda et al. Citation2011) and congenital adrenal hyperplasia (CAH) (Sharma et al. Citation2014a), thus expanding the role of AR in 46,XX DSD too. Exon deletion or addition of a premature termination codon of AR has led to varying degree of AIS, while variants of splicing mutations can result in diverse phenotypes and a cause of human infertility diseases (including AIS and polycystic ovary syndrome (Wang F et al. 2015). Trinucleotide repeat expansion (CAG and GGN repeats in exon 1 of AR) has been indicated as a risk for hypospadias (Adamovic and Nordenskjold Citation2012) but was not observed in the current survey. Hitherto, of 20 AR splice mutations (as per literature survey) accounting for AIS only one novel splicing donor site mutation in XY sex reversed female from southern India has been documented (Vasu et al. Citation2012).
Table 1. AR mutations recorded amongst Indian ethnics.
Of all the INDELs in AR recorded in medical literature to date (amongst karyotypic males), single nucleotide deletion leads to the cause of frameshift mutations and this includes two cases from India too (Saranya et al. Citation2016). Intriguingly, two studies documented same 2 bp deletion at codon 472 of AR from Germany (Thiele et al. Citation1999; Holterhus et al. Citation2005) and this probably could be due to founder effect! A rare c.2735_2736delTC AR mutation was shown from Egyptian family (Mazen et al. Citation2014). Albeit, 1–4 nucleotide(s) insertion were the most of insertion mutation documented, while rarely 13 bp (Lobaccaro et al. Citation1995) and 77 bp (Cong et al. Citation2012) INDELs have also been shown. All these frameshift mutations have led to CAIS phenotype. In contrast, a novel c.2369_2370insG (p.Cys669TrpfsX12) was shown in karyotypic female with primary amenorrhea (Sharma et al. Citation2014b). In a broader perspective, coding INDELs generally and frameshifting INDELs precisely have a negative impact on human gene function and possibly account for phenotypic diversity (Mills et al. Citation2011).
SRD5A2 mutations
The steroid 5 alpha reductase 2 gene (SRD5A2) (cytogenetic location: 2p23.1) consists of five exons, expressed at high levels in androgen-sensitive tissue (prostate), encodes a microsomal protein 3-oxo-5α-steroid: acceptor Δ4-oxidoreductase (also referred as steroid 5α-reductase; EC 1.3.99.5) enzyme [converts T → DHT]. Inactivation mutations of the SRD5A2 gene lead to pseudovaginal perineoscrotal hypospadias (OMIM #264600) (Andersson et al. Citation1991), a rare autosomal-recessive (both the parents are carriers) cause of 46,XY DSD, steroid 5α-RD2 deficiency. Currently, 102 mutations (72 missense, 7 splicing, 1 regulatory, 14 small deletions, 2 small insertions, 3 small INDELs, and 3 gross deletions) in SRD5A2 have been documented in the human gene mutation database (HGMD) data accessed on 16 October 2018 (Stenson et al. Citation2017). Amongst Indian ethnics, SRD5A2 mutation is the second most frequently reported amongst the genes causing 46,XY DSD. Exons 1 and 5 of SRD5A2 were the commonly reported site of mutation (). To date, the p.R246Q (g.54676G>A) missense mutation was reported from the northern parts of India (Eunice et al. Citation2008; Sahu et al. Citation2009; Nagaraja et al. Citation2010) and the other ethnicities [Pakistani, African-American, Austrian, Dominican Republic, Brazilian, Egyptian (Thigpen et al. Citation1992a, Citation1992b; Wilson et al. Citation1993), Mexican (Vilchis et al. Citation2000), Chinese (Lee et al. Citation2003; Wang Y et al. Citation2004; Nie et al. Citation2011; Yang et al. Citation2012; Zhu et al. Citation2014; Yuan et al. Citation2017; Jia et al. Citation2018), Korean (Choi et al. Citation2008; Ko et al. Citation2010), and Italian (Nicoletti et al. Citation2005)], thus it could be a mutational hotspot. Functional analysis revealed that p.R246Q mutation decreases the affinity to coenzyme nicotinamide adenine dinucleotide phosphate (reduced form) (NADPH) and leads to lowering of the 5α-reductase activity (Wigley et al. Citation1994). Also, the subjects harboring p.R246Q were either with female or male gender identities (Thigpen et al. Citation1992b; Wang et al. Citation2004, Eunice et al. Citation2008; Sahu et al. Citation2009). Another hotspot of SRD5A2 gene could be p.G196S missense mutation known to reduce enzyme activity (Thigpen et al. Citation1992b), were reported from Indian (two cases) (Kulshreshtha et al. Citation2009a; Nagaraja et al. Citation2010), Swedish (Nordenskjold and Ivarsson Citation1998), Brazilian (Hackel et al. Citation2005), Italian (Bertelloni et al. Citation2007), Turkey (Akcay et al. Citation2014), Bulgarian (Cooke and Ferner Citation1993), and Greek American (Thigpen et al. Citation1992b) ethnics. The association between hypospadias and endogenous endocrine monogenic disorders such as AIS and 5α-RD2 deficiency has been already known, even though these constitute only a minority of cases (Albers et al. Citation1997). Kulshreshtha et al. Citation2009a reported a unique case with dual gene defect – hemizygous p.A596T mutation in AR and heterozygous p.G196S mutation in SRD5A2 and other four affected (comprising the index case) harbored only p.A596T AR mutation in large kindred. Albeit all five affected presented hypospadias and no gynecomastia, but the affected with double mutations also had higher levels of T and was better virilized (as compared to other affected individuals) but appears to be hazy and its inevitable cause could be related to SRD5A2 defect.
Table 2. SRD5A2 mutations (in karyotypic males) recorded amongst Indian ethnics.
Amongst the 14 small deletions of SRD5A2 reported so far (curated at HGMD), 1–3 nucleotide deletion mutations (10 were uninucleotide deletions) were the most to occur and presence of direct repeats (spanning/flanking the deletion site) may be cause deletion mutagenesis. An intriguing study from northern India reported a case of perineal hypospadias that harbored a unique compound heterozygous novel 8 nucleotide deletion (with uninucleotide overhang) and p.R246Q mutation (Nagaraja et al. Citation2010). Presumably, this deletion mutagenesis could be due to co-occurrence of palindrome and short direct repeats, presence of consensus sequence for deletion and polymerase α arrest site might have led to emergence of ‘Cruciform structure’ (secondary structure intermediate) and slipped mispairing. Also, this presumption is in concordance with the mechanisms documented in human deletion mutagenesis (Krawczak and Cooper Citation1991).
SRY mutations
Human sex-determining region Y (SRY) protein (204 amino acids) coded by an intronless gene, SRY (a member of SOX gene family, cytogenetic location-Y chromosome p11.2), consists of three parts, i.e., N-terminus part (1–56 codons), high mobility group (HMG) domain (57 ± 136, 80 codons), and C-terminus part (137 ± 204, 68 codons). This DNA-binding protein is responsible for the initiation of male sex determination. In XY fetus, inactivating SRY mutation fails to activate its target genes, in turn bipotential gonad remains undifferentiated leading to complete GD (CGD) (<20% cases) evidenced by streak gonads, presence of uterine remnants (no AMH production), and female external genitalia (no androgen synthesis). These cases usually present at adolescence due to delayed pubarche and primary amenorrhea or may at later adulthood because of gonadal tumors (unilateral or bilateral dysgerminoma) or other tumors (gonadoblastoma, teratoma, or embryonal carcinoma).
Intriguingly, out of total 98 mutations of SRY (73 missense, 2 regulatory, 10 small deletions, 7 small insertions, 2 small INDELs, 3 gross deletions, and 1 gross insertion), 82 mutations (comprised 71 missense, 5 small deletions, 4 small insertions, 2 small INDELs) (82/98; 83%) were found in the HMG coding sequence, mutations upstream to HMG coding sequence were less frequent (7 missense, 3 small deletions, 2 small insertions) while downstream to HMG coding sequence were least frequent (3 missense, 1 small deletion, 1 small insertion). The majority of these mutations led to either GD (51/98; 52%) or sex reversal (36/98; 36%) (http://www.hgmd.cf.ac.uk accessed on 16 October 2018). The current survey showed SRY mutations were the third most documented amongst genes causing DSD (). From India, of the eight different point mutations reported so far, three point mutations were in upstream to HMG (p. D16Y, p.V21P, and p.M1L), four point mutations in HMG coding sequence (p.Q57R, p.K92M, p.W98X, p.E112L), and one missense mutation in downstream to HMG coding sequence (p.S143C) reported mostly in phenotypic female patients. Upon comparing this survey with that of curated SRY mutations in HGMD (shown above), it appears that mutational frequency in upstream to HMG coding sequence and HMG coding sequence to be comparable from affected Indian ethnicity (although few in number) while most of the SRY mutations were in HMG coding sequence reported globally.
Table 3. Mutations (other than AR and SRD5A2) reported in patients with DSD amongst Indian ethnics.
Additional gene mutations contributing to 46,XY DSD or 46,XX DSD
Mutations in steroidogenic acute regulatory protein gene (STAR), NR5A1, desert hedgehog gene (DHH), MAMLD1, DAX1, DMRT1, and WT1 accounts for a small percent of 46,XY DSD cases (). Mitogen-activated protein kinase kinase kinase 1 gene (MAP3K1) mutation/polymorphisms, viz., p.D806N, p.Q1028Q, p.T428T, and p.942insT, were also reported in the cohort of 10 Indian subjects with 46,XY DSD. Also, Polyphen analysis (in silico approach) revealed p.D806N missense mutation might be a cause for hypospadias (in four patients of the cohort) (Das et al. Citation2013). Hypospadias was also documented as one of the dysmorphic features in patients with defective AR (Nagaraja et al. Citation2010; Sharma et al. Citation2014a), SRD5A2 (Eunice et al. Citation2008; Sahu et al. Citation2009; Kulshreshtha et al. Citation2009a; Nagaraja et al. Citation2010; Shabir et al. Citation2015), MAMLD1 (Ratan et al. Citation2016), and partial deletion of chromosome 1q (Paliwal et al. Citation2011). Intriguingly, Verma et al. (Citation2012) described the clinical course of an Indian girl from birth to 16 years. Briefly, she was characterized by obesity, tall stature, delayed bone age, osteoporosis, hyperinsulinemia with acanthosis nigricans, and hypergonadotropic hypogonadism with cystic ovaries and estrogen replacement therapy (ERT) begun at 13.5 years. Albeit, plateauing of height, improvement of bone maturation, and pubertal progression with the disappearance of ovarian cysts but hyperinsulinemia and acanthosis nigricans persisted despite ERT and metformin regimen. Mutational analysis showed a point mutation in cytochrome P450 family 19 subfamily A member 1 gene (CYP19A1) signified aromatase deficiency (causing 46,XX DSD) and the only Indian report to date ().
Mutations causing 46,XY DSD amongst South and Southeast Asians
At this juncture, we were now curious to know whether the prevalent monogenic causes of DSD found in Indian subjects were also concordant in other populous Asian countries (like China and Japan). To validate the above, we summed up all the gene mutations causing DSD with respective ethnicity published in the research articles available at PubMed and HGMD sites for further comparison. Intriguingly, survey indicated AR mutations were the most reported not only in India but also amongst Chinese and Japanese affected subjects (Eggers et al. Citation2016; Zhou J and Fu Citation2018). On the contrary, possibility of funding or penchant could also had an effect as to these number of AR mutations that overshadowed the other gene mutations causing DSD. SRD5A2 mutations were second frequently reported with 1:3.5 from India and China, respectively. However, an international patient cohort study showed NR5A1 and SRD5A2 being the second and third highest number of variants (Eggers et al. Citation2016). Amongst the mutations reported in the sex-determining genes (i.e., SRY, SOX9 and DAX1) with sex reversal disease, SRY mutations were in 1: 3 from India and China, respectively, SOX9 mutations were scanty from China and Japan but none from India and DAX1 variants (20 mutations) were the highest reported from China. Other mutations (that caused defects in steroidogenesis) such as CYP17A1 [from China (15 mutations) and Japan (16 mutations)], HSD17B3 [China (4 mutations)], and CYP11A1 [China (1 mutation) and Japan (2 mutations)] were not reported from India to date. Also, Kallmann syndrome 1 gene (KAL1) mutations were the most reported from China (25 mutations) and Japan (12 mutations) but none from India.
Systems biology perspective to DSD
In an effort to predict potential genes of interest with possible associations to DSD, we have looked for experimentally derived, evidence-based or co-expression of proteins associated with proteins of interest (included in DSD). Looking at the gene phenotype association and genetic/molecular might give us a different perspective on what we know so far. Firstly, data mining was performed from STRING v10 database (Szklarczyk et al. Citation2015) to see for co-expression of proteins, along with proteins of interest. We have taken evidence-based approach wherein we only chose to identify proteins either experimentally proven to be either physically interacting, co-expressing genes or taken from a curated database as high confidence associations. Gene-association studies have mapped genetic variants to their disorders and the molecular mechanisms of specific genes. In majority of complex disorders like DSD, information pertaining to pathways involved, and genetic interaction networks remain largely unknown. For each DSD-associated gene, we constructed a small network where nodes represent proteins and edges represent type of association (). For example, AR and GSK-3B have been shown in several studies to associate. GSK-3B has been shown as a positive regulator of AR transactivation and is co-expressed with AR independent of any hormone stimulation or any pathway. Due to this association, GSK-3B inhibitors have been considered prospective for prostate cancer chemotherapy (Li et al. Citation2015). GSK-3B phosphorylation also has been shown to be triggered by membrane bound androgen receptor (Gu et al. Citation2014). Such associations often raise the question of gene partnerships in disease association and databases that show gene associations have often curated these genes as partners in crime. We have annotated such genes, curated by databases and experimentally shown to be either physically interacting or co-expressing together and built networks (). Co-expression networks generally do not provide conclusive information about disease causality and associations but are often identified as regulatory genes underlying disease patterns and phenotypes (van Dam et al. Citation2018). For each of these nodes (genes), possible associations are listed (). The KEGG (Kanehisa et al. Citation2017) pathways that are enriched with these smaller networks have been mentioned ( and ). In an effort to connect the dots using a combined network of DSD genes reported hitherto were mined from iHOP resource (Hoffmann Citation2007) literature to analyze their relationships and contribution to DSD. Then we have searched for each of the DSD genes and associated genes in OMIM (Amberger et al. Citation2015) to see any potential gene–phenotype relationships exist that might aid to link an associated gene to DSD. Wherever we did not find a connection in terms of physical interaction for a DSD gene with the rest, we further looked for adaptor proteins that could be a bridge (). For example, NR5A1 and SRD5A2 come together through their associations with other potential genes − HSD17B3 (Sata et al. Citation2010; Carmichael et al. Citation2014) and HSD3B2 (Codner et al. Citation2004). Similarly POR (Auchus and Miller Citation2012) connects AR to STAR and FGFR1. These newer genes could also be potential players in DSD phenotype.
Table 4. All the potential interactions of the DSD proteins (from STRING database) have been presented with STRING scores for each type of interaction, with potential KEGG pathway (Kanehisa et al. Citation2017) annotation and gene–phenotype analysis taken from OMIM (Amberger et al. Citation2015).
Figure 2. All the established DSD proteins were searched in STRING database (Szklarczyk et al. Citation2015) for interactions of three kinds (co-expressed proteins, experimentally derived interactions, and well-annotated physical interactions from other databases) with high confidence. These were laid out as a network where edges are types of interactions and central nodes are the ones connected to established DSD proteins. The potential KEGG (Kanehisa et al. Citation2017) pathway predicted for each network is labeled below each network.
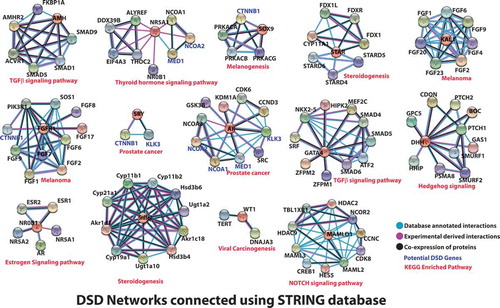
Figure 3. All the DSD proteins were searched in iHOP database (Hoffmann Citation2007), for all the proved physical interactions and a comprehensive network was built. In instances where DSD proteins failed to show physical interactions or literature evidence did not exist, they were connected through an adaptor protein, which might not have a proven DSD phenotype, but could well be a potential DSD gene.
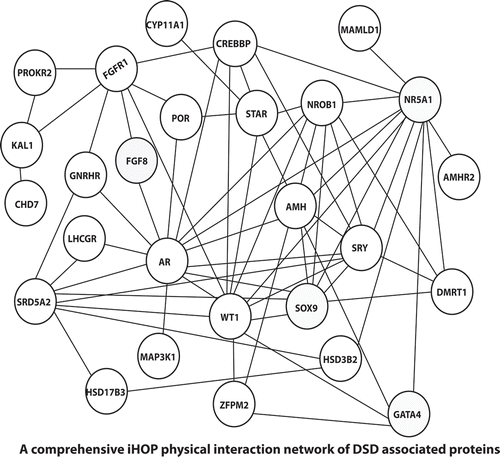
Prediction of protein networks not only provides solidarity to the involvement and function of a known or novel gene in a process (Kotlyar et al. Citation2015) but also has the ability to predict a novel association. Only genes involved in DSD were used for construction of iHOP interaction network and were represented by nodes and their connections by physical interaction represented by edges (). Once the networks were represented, the associated complex’s involvement in a specific KEGG pathway was analyzed. We observed most of the complexes were directly or indirectly related to gonadal development. Also, these complexes were studied as a part of specific cancer signaling pathways, viz., melanoma (Russo et al. Citation2006; Caldarola et al. Citation2010), prostate cancer (Loukola et al. Citation2004; Zhou CK et al. Citation2017), and cellular signaling pathways, viz., estrogen receptor (Pedram et al. Citation2014), transforming growth factor β (Young et al. Citation2015), thyroid hormone receptor (Hernandez Citation2018), melanogenesis (Puverel et al. Citation2016), and steroidogenesis (Sudhakar et al. Citation2018). It has been postulated that probably mutations in the genes resulting in DSD when upregulated or co-expression with the associated complexes resulted in specific pathways.
Since AR mutations have been the most documented amongst DSD, was made the center of reference, and also enjoys the highest number of connections and the observation was that although all the DSD genes can be stitched in to a common network from literature evidence, individual networks exist that do not need the entire complex network to be active (). The already established battery of genes (involved in DSD) is responsible for delineated dysmorphic features. We chose to see if any further clues in the form of gene–phenotype associations can be unveiled from OMIM (Amberger et al. Citation2015), regarding the associated genes that are well connected to a mutated gene. This would aid to trace the faulty crosstalk of mutated gene(s) that leads to DSD and also to screen these defects with atypical presentations ( and Supplemental Table 1). The key aspects of each network are discussed below.
SRD5A2 network
Proteins encoded by SRD5A2 and AKR1D1 [encodes for 3-oxo-5-β-steroid 4-dehydrogenase catalyzes the reduction of steroid hormones (viz., progesterone, androstenedione, 17-α-hydroxyprogesterone, T), and its mutation leads to congenital bile acid synthesis defect type 2 (OMIM 235555)] are associated, although no link to gonadal development has been found till now. Similarly, proteins encoded by SRD5A2 and CYP19A1 are associated and have been implicated in aromatase deficiency and excess syndromes, respectively. Mice deficient in this enzyme have displayed underdeveloped external genitalia and uteri (Fisher et al. Citation1998).
AMH network
AMH mutations are known to cause persistent mullerian duct syndrome type I and are diagnosed by quantifying serum MIS/AMH levels (Rey et al. Citation1999). AMH has been associated with AMHR2 (encodes for AMH receptor type 2), and mutations in the latter also show the obvious phenotype. ACVR1 [encodes for activin A receptor type 1, mutation causes fibrodysplasia ossificans progressiva (OMIM 135100)] is another AMH-associated gene.
SOX9 network
SOX9 mutations have been implicated in campomelic dysplasia with autosomal sex reversal (OMIM 114290), associated with CTNNB1 (encodes for catenin beta 1 protein, mutations cause ovarian cancer), that plays a key role in genetic regulation of testis and ovary development (Eggers et al. Citation2014). Similarly its associations with PRKACA (encodes cAMP-dependent protein kinase catalytic subunit α) implicated in Cushing’s syndrome have proven associations with oocyte maturation (Yoon et al. Citation2018). Also, PRKACG (encodes for cAMP-dependent protein kinase catalytic subunit γ) mutation has been identified as a candidate gene for 46,XY gonadal dysgenesis (Norling et al. Citation2013).
FGF9 and FGFR1 network
Glia-activating factor (encoded by FGF9) is a ligand for fibroblast growth factor receptor 1 protein (encoded by FGFR1). Mutations in FGF9 and FGFR1 cause multiple synostoses syndrome 3 (OMIM 612961) and hypogonadotropic hypogonadism 2 with or without anosmia (OMIM 147950), respectively. Along with DMRT1, FGF9 inactivating mutations and deletions were shown in a cohort of 46,XY patients with gonadal dysgenesis (Machado et al. Citation2012) and FGFR2 mutation has been also implicated in sex reversal (Siggers et al. Citation2014). Other FGFR1-associated genes FGF8 and FGF17 present a straightforward feature of hypogonadotropic hypogonadism.
MAMLD1 network
MAMLD1 shares association and significant homology with MAML2, which is a coactivator for canonical NOTCH signaling (Ogata et al. Citation2009). It is a fact that NOTCH signaling drives sexual development through ovarian follicular formation (Vanorny et al. Citation2014) and regulates luteal progesterone secretion (Wang J et al. 2015) and spermatogenic cycle (Murta et al. Citation2013). Another MAMLD1-associated protein CREB was implicated in testes development and its alternate shorter splice variants were found in cryptorchidism (Xiao et al. Citation2007).
DHH network
Hedgehog interacting protein shows involvement in ovarian development (Wang TR et al. Citation2014). Holoprosencephaly, a developmental defect, has been the feature of DHH-associating proteins, PTCH1 and cell adhesion associated, oncogene, which is known to be a key trait in Trisomy 13 syndrome (Moerman et al. Citation1988) and female 46,XY fetus (Witters et al. Citation2001).
Other networks
WT1 associated with TERT has been implicated in the development of testis (Jaskelioff et al. Citation2011). Also, GATA4 associated with ZFPM2 is shown to be mutated and implicated in anomalies of human testis determination (Fujimoto et al. Citation2013). Proteasome subunit alpha 8 (encoded by PSMA8) specifically expresses in sperms (de Mateo et al. Citation2011) and plays a key role in spermatogenesis (Qian et al. Citation2013).
Homozygous CYP11A1 (encodes cholesterol side-chain cleavage cytochrome P450) mutation (p.V359L) has been implicated in adrenal insufficiency with complete sex reversal (Al Kandari et al. Citation2006). Pulmonary hypertension, primary, 2 (OMIM 615342) caused by heterozygous SMAD family member 9 gene (SMAD9) mutation has also been implicated in follicular initiation and steroidogenesis (Diaz et al. Citation2011; Xu et al. Citation2015).
DMRT1 is one gene for which we could not find high confidence associations using our approach. However, it is gonad-specific and has sexually dimorphic expression profile. It plays a major role in sex determination, and interestingly it was observed that high DMRT1 expression works toward testicular differentiation, and lower expression is attuned toward ovarian differentiation (Smith et al. Citation1999).
KAL1 (mutations cause hypogonadotropic hypogonadism) encoded Anosmin-1 associates with most of the fibroblast growth factors and promote Leydig cell development (Liu et al. Citation2014) and ovarian follicle development (Wang et al. Citation2014). HDAC2 has been shown to regulate transcriptional events during oocyte development (Ma et al. Citation2012). Also, ACVR1 missense (c.772A>G; p.R258G) mutation shown to be involved in gonadal dysgenesis with sex reversal (Kaplan et al. Citation2015) could be a potential gene and can be used in screening efforts of DSD.
From the above analysis, we can state positively about the state of involvement of the potential genes (ACVR1, AMHR2, CTNNB1, PRKACA, PRKACG, FGFR2, FGF9, CYP11A1, TERT, ZFPM2, SMAD9, CYP19A1) in DSD, which have not been reported before for DSD-related mutations and gene–phenotype relations. These genes can be further screened and sequenced in cases with DSD from India and elsewhere.
Conclusions
AR and SRD5A2 mutations were the commonly reported amongst cases of 46,XY DSD from Indian clinics. Mutations in AR, SRD5A2, MAMLD1, WT1, and MAP3K1 often presented hypospadias as one of the or as a soul dysmorphic feature. Deficits in testis-promoting, differentiation, and/or maintenance genes (such as SRY, WT1, DHH, NR5A1, and DMRT1) led to distinct dysmorphic phenotypes causing GD. SRY mutations were reported in patients diagnosed with gonadoblastoma, infertility, and hypogonadism. STAR and DAX1 mutations led to congenital lipoid adrenal hyperplasia and adrenal hypoplasia, respectively. In addition, a rare case of 46,XX DSD harbored CYP19A1 missense mutation that led to aromatase deficiency was reported.
Molecular genetic analysis of a DSD may sometimes fail to determine the causative mutation. This could be due to screening mutation(s) only in exons (and splice junctions) of known gene(s) but dysmorphic presentation could be result of mutation(s) in any one (or more than one) of the other gene involved in sexual development/differentiation pathway. A Systems Biology approach aids to establish a physical network for a known or novel gene and thus would not only reveal molecular pathology (i.e., aberrant interactions) but may also provide futuristic molecular diagnostics. Hence, mutational analysis of all genes in a particular network would scale down the differential diagnosis (if any) in higher percent of DSD cases. Nonetheless, hospitals may want to consider genetic testing and counseling facilities to establish the molecular cause of genetic defects and educate the patients (and family members), respectively; in the long run these measures will be instrumental to manage/prevent genetic disorders and in particular DSD.
Supplemental Material
Download MS Word (229.4 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Notes on contributors
MR Nagaraja
Conceived review article: MRN, RA; Data mining and network construction: SPG, RA; Mutational survey: MRN, RA, CRWDS; Systems biology gene interaction phenotype studies: RA; Wrote, critically reviewed, and finally approved the manuscript: MRN, RA.
Related Research Data
References
- Abilash VG, Radha S, Marimuthu KM, Thangaraj K, Arun S, Nishu S, Meena J, Anuradha D. 2016. Clinical, cytogenetic and molecular analysis of androgen insensitivity syndromes from south Indian cohort and detection and in-silico characterization of androgen receptor gene mutations. Clin Chim Acta. 453:123–130.
- Adamovic T, Nordenskjold A. 2012. The CAG repeat polymorphism in the androgen receptor gene modifies the risk for hypospadias in Caucasians. BMC Med Genet. 13:109.
- Akcay T, Fernandez-Cancio M, Turan S, Guran T, Audi L, Bereket A. 2014. AR and SRD5A2 gene mutations in a series of 51 Turkish 46,XY DSD children with a clinical diagnosis of androgen insensitivity. Andrology. 2(4):572–578.
- Akella RR. 2017. Mutational analysis of androgen receptor gene in two families with androgen insensitivity. Indian J Endocrinol Metab. 21(4):520–523.
- Al Kandari H, Katsumata N, Alexander S, Rasoul MA. 2006. Homozygous mutation of P450 side-chain cleavage enzyme gene (CYP11A1) in 46, XY patient with adrenal insufficiency, complete sex reversal, and agenesis of corpus callosum. J Clin Endocrinol Metab. 91(8):2821–2826.
- Albers N, Ulrichs C, Gluer S, Hiort O, Sinnecker GH, Mildenberger H, Brodehl J. 1997. Etiologic classification of severe hypospadias: implications for prognosis and management. J Pediatr. 131(3):386–392.
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. 2015. OMIM.org: online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43(Database issue):D789–798.
- Andersson S, Berman DM, Jenkins EP, Russell DW. 1991. Deletion of steroid 5 alpha-reductase 2 gene in male pseudohermaphroditism. Nature. 354(6349):159–161.
- Auchus RJ, Miller WL. 2012. Defects in androgen biosynthesis causing 46,XY disorders of sexual development. Semin Reprod Med. 30(5):417–426.
- Bashamboo A, Rahman MM, Prasad A, Chandy SP, Ahmad J, Ali S. 2005. Fate of SRY, PABY, DYS1, DYZ3 and DYZ1 loci in Indian patients harbouring sex chromosomal anomalies. Mol Hum Reprod. 11(2):117–127.
- Bertelloni S, Scaramuzzo RT, Parrini D, Baldinotti F, Tumini S, Ghirri P. 2007. Early diagnosis of 5alpha-reductase deficiency in newborns. Sex Dev. 1(3):147–151.
- Brinkmann AO. 2001. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 179(1–2):105–109.
- Bruggenwirth HT, Boehmer AL, Verleun-Mooijman MC, Hoogenboezem T, Kleijer WJ, Otten BJ, Trapman J, Brinkmann AO. 1996. Molecular basis of androgen insensitivity. J Steroid Biochem Mol Biol. 58(5–6):569–575.
- Caldarola G, Battista C, Pellicano R. 2010. Melanoma onset after estrogen, thyroid, and growth hormone replacement therapy. Clin Ther. 32(1):57–59.
- Carmichael SL, Witte JS, Ma C, Lammer EJ, Shaw GM. 2014. Hypospadias and variants in genes related to sex hormone biosynthesis and metabolism. Andrology. 2(1):130–137.
- Chauhan V, Jyotsna VP, Jain V, Khadgawat R, Dada R. 2017. Novel heterozygous genetic variants in patients with 46,XY gonadal dysgenesis. Horm Metab Res. 49(1):36–42.
- Choi JH, Kim GH, Seo EJ, Kim KS, Kim SH, Yoo HW. 2008. Molecular analysis of the AR and SRD5A2 genes in patients with 46,XY disorders of sex development. J Pediatr Endocrinol Metab. 21(6):545–553.
- Codner E, Okuma C, Iniguez G, Boric MA, Avila A, Johnson MC, Cassorla FG. 2004. Molecular study of the 3 beta-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. J Clin Endocrinol Metab. 89(2):957–964.
- Cong P, Ye Y, Wang Y, Lu L, Yong J, Yu P, Joseph KK, Jin F, Qi M. 2012. A large deletion/insertion-induced frameshift mutation of the androgen receptor gene in a family with a familial complete androgen insensitivity syndrome. Gene. 500(2):220–223.
- Cooke MW, Ferner RE. 1993. Chemical burns causing systemic toxicity. Arch Emerg Med. 10(4):368–371.
- Das DK, Rahate SG, Mehta BP, Gawde HM, Tamhankar PM. 2013. Mutation analysis of mitogen activated protein kinase 1 gene in Indian cases of 46,XY disorder of sex development. Indian J Hum Genet. 19(4):437–442.
- Das DK, Sanghavi D, Gawde H, Idicula-Thomas S, Vasudevan L. 2011. Novel homozygous mutations in desert hedgehog gene in patients with 46,XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet. 54(6):e529–534.
- de Mateo S, Castillo J, Estanyol JM, Ballesca JL, Oliva R. 2011. Proteomic characterization of the human sperm nucleus. Proteomics. 11(13):2714–2726.
- Deeb A, Jaaskelainen J, Dattani M, Whitaker HC, Costigan C, Hughes IA. 2008. A novel mutation in the human androgen receptor suggests a regulatory role for the hinge region in amino-terminal and carboxy-terminal interactions. J Clin Endocrinol Metab. 93(10):3691–3696.
- Diaz FJ, Anthony K, Halfhill AN. 2011. Early avian follicular development is characterized by changes in transcripts involved in steroidogenesis, paracrine signaling and transcription. Mol Reprod Dev. 78(3):212–223.
- Eggers S, Ohnesorg T, Sinclair A. 2014. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol. 10(11):673–683.
- Eggers S, Sadedin S, van Den Bergen JA, Robevska G, Ohnesorg T, Hewitt J, Lambeth L, Bouty A, Knarston IM, Tan TY, et al. 2016. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 17(1):243.
- Eunice M, Philibert P, Kulshreshtha B, Audran F, Paris F, Khurana ML, Pulikkanath PE, Kucheria K, Sultan C, Ammini AC. 2008. Molecular diagnosis of 5alpha-reductase-2 gene mutation in two Indian families with male pseudohermaphroditism. Asian J Androl.. 10(5):815–818.
- Fareed M, Kaisar Ahmad M, Azeem Anwar M, Afzal M. 2017. Impact of consanguineous marriages and degrees of inbreeding on fertility, child mortality, secondary sex ratio, selection intensity, and genetic load: a cross-sectional study from Northern India. Pediatr Res. 81(1–1):18–26.
- Fisher CR, Graves KH, Parlow AF, Simpson ER. 1998. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U.S.A. 95(12):6965–6970.
- Fujimoto Y, Tanaka SS, Yamaguchi YL, Kobayashi H, Kuroki S, Tachibana M, Shinomura M, Kanai Y, Morohashi K, Kawakami K, et al. 2013. Homeoproteins six1 and six4 regulate male sex determination and mouse gonadal development. Dev Cell. 26(4):416–430.
- Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. 2012. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 33(5):887–894.
- Gu S, Honisch S, Kounenidakis M, Alkahtani S, Alarifi S, Alevizopoulos K, Stournaras C, Lang F. 2014. Membrane androgen receptor down-regulates c-src-activity and beta-catenin transcription and triggers GSK-3beta-phosphorylation in colon tumor cells. Cell Physiol Biochem. 34(4):1402–1412.
- Hackel C, Oliveira LE, Ferraz LF, Tonini MM, Silva DN, Toralles MB, Stuchi-Perez EG, Guerra-Junior G. 2005. New mutations, hotspots, and founder effects in Brazilian patients with steroid 5alpha-reductase deficiency type 2. J Mol Med. 83(7):569–576.
- Hamamy H. 2012. Consanguineous marriages: preconception consultation in primary health care settings. J Community Genet. 3(3):185–192.
- Hernandez A. 2018. Thyroid hormone role and economy in the developing testis. Vitam Horm. 106:473–500.
- Hoffmann R. 2007. Using the iHOP information resource to mine the biomedical literature on genes, proteins, and chemical compounds. Curr Protoc Bioinf. 1:20:1.16.1-1.16.16.
- Holterhus PM, Werner R, Hoppe U, Bassler J, Korsch E, Ranke MB, Dorr HG, Hiort O. 2005. Molecular features and clinical phenotypes in androgen insensitivity syndrome in the absence and presence of androgen receptor gene mutations. J Mol Med. 83(12):1005–1013.
- Hughes IA, Houk C, Ahmed SF, Lee PA, Group LC, Group EC. 2006. Consensus statement on management of intersex disorders. Arch Dis Child. 91(7):554–563.
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, et al. 2011. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 469(7328):102–106.
- Jia W, Zheng D, Zhang L, Li C, Zhang X, Wang F, Guan Q, Fang L, Zhao J, Xu C. 2018. Clinical and molecular characterization of 5alpha-reductase type 2 deficiency due to mutations (p.Q6X, p.R246Q) in SRD5A2 gene. Endocr J. 65(6):645–655.
- Joshi R, Das D, Tamhankar P, Shaikh S. 2014. Phenotypic variability in congenital lipoid adrenal hyperplasia. Indian Pediatr. 51(5):399–400.
- Kalfa N, Philibert P, Werner R, Audran F, Bashamboo A, Lehors H, Haddad M, Guys JM, Reynaud R, Alessandrini P, et al. 2013. Minor hypospadias: the “tip of the iceberg” of the partial androgen insensitivity syndrome. PLoS ONE. 8(4):e61824.
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45(D1):D353–D361.
- Kaplan FS, Kobori JA, Orellana C, Calvo I, Rosello M, Martinez F, Lopez B, Xu M, Pignolo RJ, Shore EM, et al. 2015. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (ACVR1 c.772G>A; R258G): A report of two patients. Am J Med Genet A. 167A(10):2265–2271.
- Khadilkar VV, Mangtani HR, Jahagirdar RR, Khatod KA, Phadke ND, Deepa PS, Khadilkar AV. 2013. Entire DAX1 gene deletion in an Indian boy with adrenal hypoplasia congenita. Indian J Pediatr. 80(8):631–635.
- Ko JM, Cheon CK, Kim GH, Kim SH, Kim KS, Yoo HW. 2010. Clinical characterization and analysis of the SRD5A2 gene in six Korean patients with 5alpha-reductase type 2 deficiency. Horm Res Paediatr. 73(1):41–48.
- Kota SK, Gayatri K, Kota SK, Jammula S. 2013. Genetic analysis of a family with complete androgen insensitivity syndrome. Indian J Hum Genet. 19(3):355–357.
- Kotlyar M, Pastrello C, Pivetta F, Lo Sardo A, Cumbaa C, Li H, Naranian T, Niu Y, Ding Z, Vafaee F, et al. 2015. In silico prediction of physical protein interactions and characterization of interactome orphans. Nat Methods. 12(1):79–84.
- Krawczak M, Cooper DN. 1991. Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum Genet. 86(5):425–441.
- Kulshreshtha B, Philibert P, Eunice M, Audran F, Paris F, Khurana ML, Ammini AC, Charles S. 2009a. Phenotype, hormonal profile and genotype of subjects with partial androgen insensitivity syndrome: report of a family with four adult males and one child with disorder of sexual differentiation. Andrologia. 41(4):257–263.
- Kulshreshtha B, Philibert P, Eunice M, Khandelwal SK, Mehta M, Audran F, Paris F, Sultan C, Ammini AC. 2009b. Apparent male gender identity in a patient with complete androgen insensitivity syndrome. Arch Sex Behav. 38(6):873–875.
- Lee CY, Lam CW, Shek CC. 2003. Steroid 5alpha-reductase 2 deficiency in two generations of a non-consanguineous Chinese family. J Pediatr Endocrinol Metab. 16(8):1197–1201.
- Li B, Thrasher JB, Terranova P. 2015. Glycogen synthase kinase-3: a potential preventive target for prostate cancer management. Urol Oncol. 33(11):456–463.
- Liu H, Yang Y, Zhang L, Liang R, Ge RS, Zhang Y, Zhang Q, Xiang Q, Huang Y, Su Z. 2014. Basic fibroblast growth factor promotes stem Leydig cell development and inhibits LH-stimulated androgen production by regulating microRNA expression. J Steroid Biochem Mol Biol. 144(Pt B):483–491.
- Lobaccaro JM, Lumbroso S, Poujol N, Georget V, Brinkmann AO, Malpuech G, Sultan C. 1995. Complete androgen insensitivity syndrome due to a new frameshift deletion in exon 4 of the androgen receptor gene: functional analysis of the mutant receptor. Mol Cell Endocrinol. 111(1):21–28.
- Loukola A, Chadha M, Penn SG, Rank D, Conti DV, Thompson D, Cicek M, Love B, Bivolarevic V, Yang Q, et al. 2004. Comprehensive evaluation of the association between prostate cancer and genotypes/haplotypes in CYP17A1, CYP3A4, and SRD5A2. Eur J Hum Genet. 12(4):321–332.
- Ma P, Pan H, Montgomery RL, Olson EN, Schultz RM. 2012. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc Natl Acad Sci U.S.A. 109(8):E481–489.
- Machado AZ, Da Silva TE, Frade Costa EM, Dos Santos MG, Nishi MY, Brito VN, Mendonca BB, Domenice S. 2012. Absence of inactivating mutations and deletions in the DMRT1 and FGF9 genes in a large cohort of 46,XY patients with gonadal dysgenesis. Eur J Med Genet. 55(12):690–694.
- MacLean HE, Ball EM, Rekaris G, Warne GL, Zajac JD. 2004. Novel androgen receptor gene mutations in Australian patients with complete androgen insensitivity syndrome. Hum Mutat. 23(3):287.
- Mazen I, Soliman H, El-Gammal M, Torky A, Mekkawy M, Abdel-Hamid MS, Essawi M. 2014. A novel mutation (c.2735_2736delTC) in the androgen receptor gene in 46,XY females with complete androgen insensitivity syndrome in an Egyptian family. Horm Res Paediatr. 82(6):411–414.
- McCann-Crosby B, Mansouri R, Dietrich JE, McCullough LB, Sutton VR, Austin EG, Schlomer B, Roth DR, Karaviti L, Gunn S, et al. 2014. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol. 2014(1):4.
- Mendonca BB, Domenice S, Arnhold IJ, Costa EM. 2009. 46,XY disorders of sex development (DSD). Clin Endocrinol (Oxf). 70(2):173–187.
- Mills RE, Pittard WS, Mullaney JM, Farooq U, Creasy TH, Mahurkar AA, Kemeza DM, Strassler DS, Ponting CP, Webber C, et al. 2011. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 21(6):830–839.
- Modi J, Modi P, Pal B, Kumar S. 2015. Bilateral Wilms’ tumors in an infant with Denys-Drash syndrome and rarely seen truncation mutation in the WT1 gene-exon 6. J Indian Assoc Pediatr Surg. 20(4):197–198.
- Moerman P, Fryns JP, van der Steen K, Kleczkowska A, Lauweryns J. 1988. The pathology of trisomy 13 syndrome. A study of 12 cases. Hum Genet. 80(4):349–356.
- Murta D, Batista M, Silva E, Trindade A, Henrique D, Duarte A, Lopes-da-Costa L. 2013. Dynamics of Notch pathway expression during mouse testis post-natal development and along the spermatogenic cycle. PLoS ONE. 8(8):e72767.
- Nagaraja MR, Rastogi A, Raman R, Gupta DK, Singh SK. 2009. Mutational analysis of the androgen receptor gene in two Indian families with partial androgen insensitivity syndrome. J Pediatr Endocrinol Metab. 22(12):1169–1173.
- Nagaraja MR, Rastogi A, Raman R, Gupta DK, Singh SK. 2010. Molecular diagnosis of 46,XY DSD and identification of a novel 8 nucleotide deletion in exon 1 of the SRD5A2 gene. J Pediatr Endocrinol Metab. 23(4):379–385.
- Nicoletti A, Baldazzi L, Balsamo A, Barp L, Pirazzoli P, Gennari M, Radetti G, Cacciari E, Cicognani A. 2005. SRD5A2 gene analysis in an Italian population of under-masculinized 46,XY subjects. Clin Endocrinol (Oxf). 63(4):375–380.
- Nie M, Zhou Q, Mao J, Lu S, Wu X. 2011. Five novel mutations of SRD5A2 found in eight Chinese patients with 46,XY disorders of sex development. Mol Hum Reprod. 17(1):57–62.
- Nordenskjold A, Ivarsson SA. 1998. Molecular characterization of 5 alpha-reductase type 2 deficiency and fertility in a Swedish family. J Clin Endocrinol Metab. 83(9):3236–3238.
- Norling A, Linden Hirschberg A, Iwarsson E, Persson B, Wedell A, Barbaro M. 2013. Novel candidate genes for 46,XY gonadal dysgenesis identified by a customized 1 M array-CGH platform. Eur J Med Genet. 56(12):661–668.
- Ogata T, Laporte J, Fukami M. 2009. MAMLD1 (CXorf6): a new gene involved in hypospadias. Horm Res. 71(5):245–252.
- Paliwal P, Sharma A, Birla S, Kriplani A, Khadgawat R, Sharma A. 2011. Identification of novel SRY mutations and SF1 (NR5A1) changes in patients with pure gonadal dysgenesis and 46,XY karyotype. Mol Hum Reprod. 17(6):372–378.
- Panda B, Rao L, Tosh D, Dixit H, Padmalatha V, Kanakavalli M, Raseswari T, Deenadayal M, Gupta N, Chakrabarty B, et al. 2011. Germline study of AR gene of Indian women with ovarian failure. Gynecol Endocrinol. 27(8):572–578.
- Paul R. 1965. Problems of psycho-sexual orientation. Guy’s Hosp Rep. 114(3):333–336.
- Pedram A, Razandi M, Lewis M, Hammes S, Levin ER. 2014. Membrane-localized estrogen receptor alpha is required for normal organ development and function. Dev Cell. 29(4):482–490.
- Puverel S, Kiris E, Singh S, Klarmann KD, Coppola V, Keller JR, Tessarollo L. 2016. RanBPM (RanBP9) regulates mouse c-kit receptor level and is essential for normal development of bone marrow progenitor cells. Oncotarget. 7(51):85109–85123.
- Qian MX, Pang Y, Liu CH, Haratake K, Du BY, Ji DY, Wang GF, Zhu QQ, Song W, Yu Y, et al. 2013. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell. 153(5):1012–1024.
- Rajender S, Gupta NJ, Chakrabarty B, Singh L, Thangaraj K. 2009. Ala 586 Asp mutation in androgen receptor disrupts transactivation function without affecting androgen binding. Fertil Steril. 91(3):933 e923–938.
- Rajender S, Gupta NJ, Chakrabarty B, Singh L, Thangaraj K. 2013. L712V mutation in the androgen receptor gene causes complete androgen insensitivity syndrome due to severe loss of androgen function. Steroids. 78(12–13):1288–1292.
- Rajender S, Pooja S, Gupta NJ, Chakrabarty B, Singh L, Thangaraj K. 2011. G708E mutation in the androgen receptor results in complete loss of androgen function. J Androl. 32(2):193–198.
- Rajender S, Rajani V, Gupta NJ, Chakravarty B, Singh L, Thangaraj K. 2006. SRY-negative 46,XX male with normal genitals, complete masculinization and infertility. Mol Hum Reprod. 12(5):341–346.
- Rao TS, Asha MR, Sambamurthy K, Rao KS. 2009. Consanguinity: still a challenge. Indian J Psychiatry. 51(1):3–5.
- Ratan SK, Sharma A, Kapoor S, Polipalli SK, Dubey D, Mishra TK, Sinha SK, Agarwal SK. 2016. Polymorphism of 3ʹ UTR of MAMLD1 gene is also associated with increased risk of isolated hypospadias in Indian children: a preliminary report. Pediatr Surg Int. 32(5):515–524.
- Rey RA, Belville C, Nihoul-Fekete C, Michel-Calemard L, Forest MG, Lahlou N, Jaubert F, Mowszowicz I, David M, Saka N, et al. 1999. Evaluation of gonadal function in 107 intersex patients by means of serum antimullerian hormone measurement. J Clin Endocrinol Metab. 84(2):627–631.
- Russo D, Blanco M, Falke G, Rocca Rivarola M, Seller R, Puigdevall JC, Bergada C. 2006. Pure gonad dysgenesia or Swyer sindrome. A case report having tumoral development: melanoma. Cir Pediatr. 19(4):244–246.
- Sahu R, Boddula R, Sharma P, Bhatia V, Greaves R, Rao S, Desai M, Wakhlu A, Phadke S, Shukla M, et al. 2009. Genetic analysis of the SRD5A2 gene in Indian patients with 5alpha-reductase deficiency. J Pediatr Endocrinol Metab. 22(3):247–254.
- Saranya B, Bhavani G, Arumugam B, Jayashankar M, Santhiya ST. 2016. Three novel and two known androgen receptor gene mutations associated with androgen insensitivity syndrome in sex-reversed XY female patients. J Genet. 95(4):911–921.
- Sata F, Kurahashi N, Ban S, Moriya K, Tanaka KD, Ishizuka M, Nakao H, Yahata Y, Imai H, Kakizaki H, et al. 2010. Genetic polymorphisms of 17 beta-hydroxysteroid dehydrogenase 3 and the risk of hypospadias. J Sex Med. 7(8):2729–2738.
- Sfez-Yaiche A, Sulmont V. 2000. Evaluation approach in sexual ambiguity: reims experience from a retrospective study. Arch Pediatr. 7(Suppl 2):382s–384s.
- Shabir I, Khurana ML, Joseph AA, Eunice M, Mehta M, Ammini AC. 2015. Phenotype, genotype and gender identity in a large cohort of patients from India with 5alpha-reductase 2 deficiency. Andrology. 3(6):1132–1139.
- Shahid M, Dhillion VS, Jain N, Hedau S, Diwakar S, Sachdeva P, Batra S, Das BC, Husain SA. 2004. Two new novel point mutations localized upstream and downstream of the HMG box region of the SRY gene in three Indian 46,XY females with sex reversal and gonadal tumour formation. Mol Hum Reprod. 10(7):521–526.
- Shahid M, Dhillon VS, Hussain Z, Masa JF, Aslam M, Raish M, Ahmad A, Khan NJ, Prasad S, Batra S, et al. 2008. Analysis of the SRY gene in two sex-reversed XY sisters identifies two new novel point mutations in the high mobility group box domain. Fertil Steril. 90(4):1199e1191–1198.
- Sharma V, Singh R, Thangaraj K, Jyothy A. 2011. A novel Arg615Ser mutation of androgen receptor DNA-binding domain in three 46,XY sisters with complete androgen insensitivity syndrome and bilateral inguinal hernia. Fertil Steril. 95(2):804 e819–821.
- Sharma V, Thangaraj K, Jyothy A. 2014a. A novel androgen receptor gene mutation in a patient with congenital adrenal hyperplasia associated with penoscrotal hypospadias. Transl Res. 164(2):149–152.
- Sharma V, Thangaraj K, Jyothy A. 2014b. A novel insertion-induced frameshift mutation of the androgen receptor gene in a patient with primary amenorrhea. Meta Gene. 2:11–15.
- Shulman ST. 2012. Adolescents and sexual ambiguity. Pediatr Ann. 41(4):129–130.
- Siggers P, Carre GA, Bogani D, Warr N, Wells S, Hilton H, Esapa C, Hajihosseini MK, Greenfield A. 2014. A novel mouse Fgfr2 mutant, hobbyhorse (hob), exhibits complete XY gonadal sex reversal. PLoS ONE. 9(6):e100447.
- Singh R, Shastry PK, Rasalkar AA, Singh L, Thangaraj K. 2006. A novel androgen receptor mutation resulting in complete androgen insensitivity syndrome and bilateral Leydig cell hyperplasia. J Androl. 27(4):510–516.
- Singh R, Singh P, Gupta NJ, Chakrabarty B, Singh L, Thangaraj K. 2010. C601S mutation in the androgen receptor results in partial loss of androgen function. J Steroid Biochem Mol Biol. 122(5):359–363.
- Sinha A, Sharma S, Gulati A, Sharma A, Agarwala S, Hari P, Bagga A. 2010. Frasier syndrome: early gonadoblastoma and cyclosporine responsiveness. Pediatr Nephrol. 25(10):2171–2174.
- Smith CA, McClive PJ, Western PS, Reed KJ, Sinclair AH. 1999. Conservation of a sex-determining gene. Nature. 402(6762):601–602.
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. 2017. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 136(6):665–677.
- Sudhakar DVS, Nizamuddin S, Manisha G, Devi JR, Gupta NJ, Chakravarthy BN, Deenadayal M, Singh L, Thangaraj K. 2018. NR5A1 mutations are not associated with male infertility in Indian men. Andrologia. 50(3):e12931.
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43(Database issue):D447–452.
- Thiele B, Weidemann W, Schnabel D, Romalo G, Schweikert HU, Spindler KD. 1999. Complete androgen insensitivity caused by a new frameshift deletion of two base pairs in exon 1 of the human androgen receptor gene. J Clin Endocrinol Metab. 84(5):1751–1753.
- Thigpen AE, Davis DL, Gautier T, Imperato-McGinley J, Russell DW. 1992a. Brief report: the molecular basis of steroid 5 alpha-reductase deficiency in a large Dominican kindred. N Engl J Med. 327(17):1216–1219.
- Thigpen AE, Davis DL, Milatovich A, Mendonca BB, Imperato-McGinley J, Griffin JE, Francke U, Wilson JD, Russell DW. 1992b. Molecular genetics of steroid 5 alpha-reductase 2 deficiency. J Clin Invest. 90(3):799–809.
- van Dam S, Vosa U, van der Graaf A, Franke L, de Magalhaes JP. 2018. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinformatics. 19(4):575–592.
- Vanorny DA, Prasasya RD, Chalpe AJ, Kilen SM, Mayo KE. 2014. Notch signaling regulates ovarian follicle formation and coordinates follicular growth. Mol Endocrinol. 28(4):499–511.
- Vasu VR, Saranya B, Jayashankar M, Munirajan AK, Santhiya ST. 2012. A novel splice site and two known mutations of androgen receptor gene in sex-reversed XY phenotype. Genet Test Mol Biomarkers. 16(7):749–755.
- Vasudevan L, Joshi R, Das DK, Rao S, Sanghavi D, Babu S, Tamhankar PM. 2013. Identification of novel mutations in STAR gene in patients with lipoid congenital adrenal hyperplasia: a first report from India. J Clin Res Pediatr Endocrinol. 5(2):121–124.
- Verma N, Jain V, Birla S, Jain R, Sharma A. 2012. Growth and hormonal profile from birth to adolescence of a girl with aromatase deficiency. J Pediatr Endocrinol Metab. 25(11–12):1185–1190.
- Vilchis F, Mendez JP, Canto P, Lieberman E, Chavez B. 2000. Identification of missense mutations in the SRD5A2 gene from patients with steroid 5alpha-reductase 2 deficiency. Clin Endocrinol (Oxf). 52(3):383–387.
- Vilela ML, Willingham E, Buckley J, Liu BC, Agras K, Shiroyanagi Y, Baskin LS. 2007. Endocrine disruptors and hypospadias: role of genistein and the fungicide vinclozolin. Urology. 70(3):618–621.
- Wang F, Pan J, Liu Y, Meng Q, Lv P, Qu F, Ding GL, Klausen C, Leung PC, Chan HC, et al. 2015a. Alternative splicing of the androgen receptor in polycystic ovary syndrome. Proc Natl Acad Sci U.S.A. 112(15):4743–4748.
- Wang J, Liu S, Peng L, Dong Q, Bao R, Lv Q, Tang M, Hu C, Li G, Liang S, et al. 2015b. Notch signaling pathway regulates progesterone secretion in murine luteal cells. Reprod Sci. 22(10):1243–1251.
- Wang TR, Yan LY, Yan J, Lu CL, Xia X, Yin TL, Zhu XH, Gao JM, Ding T, Hu WH, et al. 2014. Basic fibroblast growth factor promotes the development of human ovarian early follicles during growth in vitro. Hum Reprod. 29(3):568–576.
- Wang Y, Li Q, Xu J, Liu Q, Wang W, Lin Y, Ma F, Chen T, Li S, Shen Y. 2004. Mutation analysis of five candidate genes in Chinese patients with hypospadias. Eur J Hum Genet. 12(9):706–712.
- Werner R, Holterhus PM, Binder G, Schwarz HP, Morlot M, Struve D, Marschke C, Hiort O. 2006. The A645D mutation in the hinge region of the human androgen receptor (AR) gene modulates AR activity, depending on the context of the polymorphic glutamine and glycine repeats. J Clin Endocrinol Metab. 91(9):3515–3520.
- Wigley CW, Prihoda JS, Mowszowicz I, Mendonca BB, New MI, Wilson JD, Russell DW. 1994. Natural mutagenesis study of the human steroid 5α-reductase 2 isozyme. Biochemistry. 33(5):1265–1270.
- Wilson JD, Griffin JE, Russell DW. 1993. Steroid 5 alpha-reductase 2 deficiency. Endocr Rev. 14(5):577–593.
- Witters I, Moerman P, Muenke M, Van Assche FA, Devriendt K, Legius E, Van Schoubroeck D, Fryns JP. 2001. Semilobar holoprosencephaly in a 46,XY female fetus. Prenat Diagn. 21(10):839–841.
- Xiao PJ, Hu L, Li J, Lin W, Chen X, Xu P. 2007. NSSR1 is regulated in testes development and cryptorchidism and promotes the exon 5-included splicing of CREB transcripts. Mol Reprod Dev. 74(11):1363–1372.
- Xu J, Li J, Wang H, Wang G, Chen J, Huang P, Cheng J, Gan L, Wang Z, Cai Y. 2015. A novel SMAD family protein, SMAD9 is involved in follicular initiation and changes egg yield of geese via synonymous mutations in exon1 and intron2. Mol Biol Rep. 42(1):289–302.
- Yang Y, Wang BA, Guo QH, Dou JT, Lv ZH, Ba JM, Lu JM, Pan CY, Mu YM. 2012. Clinical and genetic analysis of three Chinese patients with steroid 5alpha-reductase type 2 deficiency. J Pediatr Endocrinol Metab. 25(11–12):1077–1082.
- Yoon H, Jang H, Kim EY, Moon S, Lee S, Cho M, Cho HJ, Ko JJ, Chang EM, Lee KA, et al. 2018. Knockdown of PRKAR2B results in the failure of oocyte maturation. Cell Physiol Biochem. 45(5):2009–2020.
- Young JC, Wakitani S, Loveland KL. 2015. TGF-beta superfamily signaling in testis formation and early male germline development. Semin Cell Dev Biol. 45:94–103.
- Yuan S, Meng L, Zhang Y, Tu C, Du J, Li W, Liang P, Lu G, Tan YQ. 2017. Genotype-phenotype correlation and identification of two novel SRD5A2 mutations in 33 Chinese patients with hypospadias. Steroids. 125:61–66.
- Zhou CK, Stanczyk FZ, Hafi M, Veneroso CC, Lynch B, Falk RT, Niwa S, Emanuel E, Gao YT, Hemstreet GP, et al. 2017. Circulating and intraprostatic sex steroid hormonal profiles in relation to male pattern baldness and chest hair density among men diagnosed with localized prostate cancers. Prostate. 77(16):1573–1582.
- Zhou J, Fu BQ. 2018. The research on gene-disease association based on text-mining of PubMed. BMC Bioinformatics. 19(1):37.
- Zhu H, Liu W, Han B, Fan M, Zhao S, Wang H, Lu Y, Pan C, Chen F, Chen M, et al. 2014. Phenotypic and molecular characteristics in eleven Chinese patients with 5alpha-reductase Type 2 deficiency. Clin Endocrinol (Oxf). 81(5):711–720.