
ABSTRACT
The current study describes a successful case of preimplantation genetic diagnosis (PGD) of primary open angle glaucoma (POAG) and verifies the efficiency of next-generation sequencing (NGS)-based haplotyping for PGD of POAG. In this study, we applied NGS as part of PGD to effectively detect POAG prior to embryo implantation and avoid the prospect of pregnancy termination in event of vertical inheritance of POAG. We used the technique of multiple annealing and looping based amplification cycles (MALBAC) to conduct whole genome amplification (WGA) and to reduce the allele dropout (ADO). We also employed Sanger sequencing to directly detect the mutation c.1109 C > T in MYOC and NGS-based single nucleotide polymorphism (SNP) haplotyping to distinguish the chromosomes that carried the mutation. Copy number variation (CNV) analysis was carried out to determine the copy number of embryos’ chromosomes. Of the 4 blastocysts obtained in this study, only 2 (sample 5 and 7) could be successfully amplified by WGA. CNV results indicated that chromosomes of both these samples were balanced (46, XN). Sanger sequencing and NGS-based SNP haplotyping confirmed that sample 7 carried the mutation c.1109 C > T in MYOC, while sample 5 did not. Moreover, no ADO was observed. Thus, blastocyst 5 was transferred into the uterus of the patient, and a healthy baby without the MYOC mutation c. 1109C>T was born 39 weeks after transplantation. Our study suggests that NGS-based SNP haplotyping is an effective technique for the PGD of POAG.
Abbreviations: PGD: preimplantation genetic diagnosis; POAG: primary open angle glaucoma; NGS: next-generation sequencing; MALBAC: multiple annealing and looping based amplification cycles chemistry; WGA: whole genome amplification; ADO: allele dropout; SNP: single nucleotide polymorphism; CNV: copy number variation; MYOC: Myocilin; OPTN: Optineurin; WDR36: WD repeat domain 36; CYP1B1: Cytochrome P450 1 B Chain; ICSI: intracytoplasmic sperm injection; TFNA: testicular fine-needle aspiration; TE: trophectoderm; PCR: polymerase chain reaction
Introduction
Primary open angle glaucoma (POAG), an important subset of glaucoma, is one of the common causes of irreversible vision loss and is considered to be an important global health concern. POAG shows behaviour of familial aggregation. Studies on epidemiology, heredity, and molecular genetics have shown that genetic factors play a large role in the onset of POAG (Gong et al. Citation2007; Tikunova and Churnosov Citation2014). Four genes, namely, myocilin (MYOC), optineurin (OPTN), WD repeat domain 36 (WDR36), and cytochrome P450 1 B Chain (CYP1B1) have been identified as causative genes for POAG (Stone et al. Citation1997; Rezaie et al. Citation2002; Monemi et al. Citation2005; Kim et al. Citation2011). MYOC was the earliest identified and the most investigated gene for the onset of POAG. MYOC disease-causing mutations are widely detected in POAG. In addition, 2%–4% of these mutations have been identified in unselected patients with POAG, and 8%–36% have been identified in patients with POAG and diagnosed before the age of 40 years (Fingert et al. Citation1999; Shimizu et al. Citation2000; Souzeau et al. Citation2013). Treatments for POAG mainly include medication, laser therapy, and surgery. Although, these treatments can preserve the remnants of a patient’s vision, they cannot block the vertical heredity of the pathogenic gene.
Preimplantation genetic diagnosis (PGD) has been used to select unaffected embryos from patients suffering from known genetic diseases. PGD is a more superior method than prenatal diagnosis because the former can avoid the pain of pregnancy termination if the foetus is affected and is also more ethically acceptable. POAG caused by MYOC mutations is an autosomal dominant disorder. To our knowledge, the most efficient method to detect the gene mutation is Sanger sequencing. However, two inherent defects are observed in Sanger sequencing for whole genome amplification (WGA) products from biopsied cells. Firstly, WGA techniques are unable to cover the whole genome, causing a risk of sequencing failure. Secondly, there is the risk of allele dropout (ADO) caused by amplification bias. Currently, multiple annealing and looping based amplification cycle (MALBAC) is applied to WGA to reduce the bias of amplification and cover the whole genome (Bourcy et al. Citation2014; Deleye et al. Citation2015). However, ADO still occurs in some instances. Since 2006, preimplantation genetic haplotyping analysis of polymorphic markers flanking the mutation site via next-generation sequencing (NGS) has been used in PGD to determine ADO and avoid misdiagnosis (Renwick et al. Citation2006, Citation2010; Hao et al. Citation2018).
In the current study, we aimed to apply NGS-based single nucleotide polymorphism (SNP) haplotyping to PGD for a patient with POAG and MYOC mutation, in order to help give birth to a healthy infant without the mutation and risk of POAG.
Results
In vitro fertilization (IVF), embryo culture, TE biopsy, and blastocyst vitrification
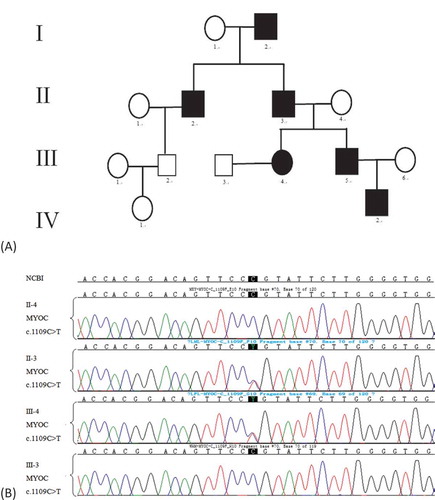
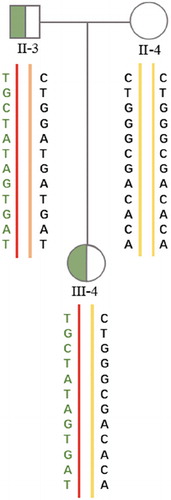
A female patient, who was diagnosed with POAG caused by the MYOC mutation c.1109 C > T, and her husband came to our Assisted Reproductive Technology Centre for PGD. The patient’s father, grandfather, uncle, brother, and her nephew suffered from POAG ().
Figure 1. (A)Pedigree of the primary open angle glaucoma (POGA) family. III-4 represents the female patient. Circles and squares represent females and males, respectively. The black symbols represent patients who suffered POGA. (B)Molecular diagnosis results of parts of the POGA family. Compared with the reference sequence of National Center for Biotechnology Information (NCBI), II-3 and III-4 carried the MYOC mutation c.1109C>T, while II-4 and III-3 did not.
After long-protocol ovulation was promoted, 15 oocytes were collected. Of these, 11 were at metaphase II-stage (MII), 2 were at germinal vesicle stage 2 (GV2), and 2 were at metaphase I-stage (MI). The 11 MII oocytes were inseminated by intracytoplasmic sperm injection (ICSI), and 7 of them developed into 6–8 cells on day 3 post-insemination. The embryos were fertilized, and four blastocysts (1, 2, 5, and 7) were obtained. These blastocysts were all used for the trophectoderm (TE) biopsy on day 5 post-insemination. The biopsied cells of these four blastocysts were used for WGA as part of PGD.
WGA and copy number variation (CNV)
Unfortunately, upon WGA, two of them (1, 2) failed to amplify. The remaining two (5, 7) amplified successfully (). As shown in , the results of CNV indicated that chromosomes of samples 5 and 7 were balanced (46, XN).
Figure 2. Quality control of whole genome amplification (WGA). M, 1, 2, 5, 7, NC, and PC represent markers, sample 1, sample 2, sample 5, sample 7, negative control, and positive control, respectively. Samples 1 and 2 were same as NC that means failed, but samples 5 and 7 were same as PC that means amplified successfully.
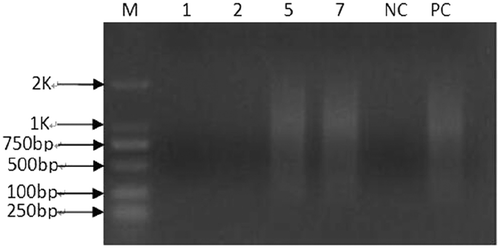
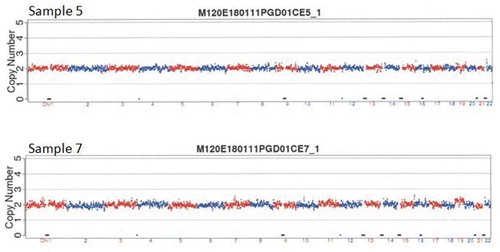
Figure 3. Copy number variation (CNV) results of sample 5 and 7. Compared with the hg19 reference genome through CNV analysis, the sketch maps shown that sample 5 and 7 were both diploid. Both sample 5’s and 7’s chromosome were balanced.
Sanger sequencing and SNP haplotyping
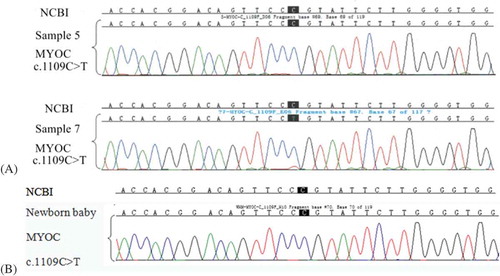
Sanger sequencing results showed that sample 7, but not sample 5, carried the MYOC mutation c.1109C>T ().
Figure 4. (A)Sanger sequencing results of samples 5 and 7. The loci of the mutation c.1109C>T are the nucleotides with a black background. Compared with the reference sequence of NCBI sample 7 carried the MYOC mutation c.1109C>T, while sample 5 did not. (B) Sanger sequencing result of the newborn baby (from sample 5). Compared with the reference sequence of NCBI the newborn baby did not carry the MYOC mutation c.1109C>T. This result is the same as in sample 5.
SNP linkage analysis was conducted by selecting 60 SNPs within 1 Mbp region flanking the mutation site. The gDNA of the patient (III-4) and her parents (father: II-3, mother: II-4) was amplified using these SNP primers to analyze the pedigree haplotypes (). Afterwards, the WGA products were tested in the same way. NGS-based SNP haplotyping was then used to distinguish the chromosomes carrying the mutation. The haplotypes of the samples (5 and 7) combined with that of the patient (III-4) and her husband (III-3) were analyzed. Of the 60 SNPs, 12 informative SNPs confirmed that sample 7 carried the MYOC mutation c.1109C>T, while sample 5 did not ().
Table 1. NGS-based SNP haplotyping results of samples 5 and 7.
Figure 5. Pedigree of haplotype construction of the POGA family. III-4, II-3, and II-4 represent the female patient, her father, and her mother, respectively. The red lines were carrying haplotypes, yellow lines and brown line were normal haplotypes. The carrying haplotype of III-4 was inherited from her father (II-3), and the normal haplotype of III-4 was inherited from her mother (II-3).
The genetic mutation site was directly detected through Sanger sequencing. Preimplantation genetic screening was synchronously conducted to detect the CNV of the embryos’ chromosomes before embryo implantation. Only two samples (5 and 7) were normal and remained to be analyzed, because samples 1 and 2 did not meet the standard of quality control owing to uncontrollable factors, such as sample loss in biopsy, biopsied cell fragmentation, or nuclear loss.
Finally, blastocyst 5 was transferred into the uterus of the patient, and a healthy baby was born 39 weeks after transplantation. Sanger sequencing result of the new-born baby showed that it did not carry the MYOC mutation c.1109C>T ().
Discussion
POAG is a chronic optic neuropathy characterised by the loss of retinal ganglion cells, degenerative cylindraxile, and deficiency of the visual field; in addition to cataracts, glaucoma represents a major cause of vision loss worldwide (Resnikoff et al. Citation2004). The number of cases is expected to reach approximately 60 million by 2020 (Foster et al. Citation2002). The pathogenesis of POAG is complicated and genetic predisposition is by far the most common cause of this autosomal dominant disease.
The current study describes a successful case of NGS-based haplotyping in PGD to avoid POAG. Although PGD is an efficient way to help patients with genetically heritable disorders have a healthy baby, the application of PGD to minimize ADO and determine POAG have not yet been reported. Sanger sequencing has been considered as the gold standard for directly detecting the mutation site in monogenic diseases, but ADO may occur and lead to misdiagnosis. ADO is caused by the amplification bias of two alleles. One of the two alleles is preferentially amplified, and the other allele is amplified at an undetectable level; thus, this condition is the inherent pitfall of single-cell PCR (Chen et al. Citation2015). Therefore, we chose the MALBAC method for WGA to reduce ADO in our study. Bourcy et al. (Citation2014) reported that the inherent bias in amplification in MALBAC is not strongly affected by gain. Huang et al. (Citation2015) also showed that MALBAC method has a low incidence rate of ADO and attributed it to MALBAC’s unique feature of quasilinear amplification, which reduces the sequence-dependent bias exacerbated by exponential amplification. Unfortunately, two of the biopsied cells (sample 1 and 2) failed to amplify. The reason could be the difference in each blastocyst’s growth rate and morphological grading. Blastocyst 1 was grading 3BC, blastocyst 2 was grading 4BB, blastocyst 5 was grading 5CC, and blastocyst 7 was grading 5BC. These indicated that samples 5 and 7 were superior to samples 1 and 2. On the other hand, losses of cells during biopsy and transfer cannot be ruled out. Due to successful amplification of negative control and positive control, we did not regard the WGA method per se to have failed. Further, to avoid risk of ADO, we used NGS-based SNP haplotyping to determine ADO. In addition, SNP haplotyping can distinguish the chromosome carrying the mutation from normal chromosomes. Therefore, SNP haplotyping results can verify Sanger sequencing results and vice versa. Sixty high-frequency SNPs located 1 Mbp upstream and downstream of the MYOC mutation c. 1109C>T were chosen for NGS-based SNP haplotyping. Of these, 12 informative SNPs were used for constructing the haplotype by analysing the genomic DNA of II-3, II-4, and III-4 (). Finally, the haplotypes from the MALBAC products of samples 5 and 7 were analyzed combined with haplotypes from III-3 and III-4. The pathogenic chromosome of sample 7 was successfully distinguished, and no ADO occurred. A healthy baby without the MYOC mutation c. 1109C>T was born from implantation of blastocyst 5 (), suggesting that NGS-based SNP haplotyping is an effective technique for PGD of POAG. Thus, our study highlights the efficiency of using NGS-based SNP haplotyping for efficient PGD and to avoid inheritance of POAG.
Materials and methods
Patient and relatives
A 26-year-old female patient came to our Assisted Reproductive Technology Centre for diagnosis after she was unable to conceive for 3 years. Her 28-year-old husband suffered from severe oligoasthenozoospermia, and the ejaculated sperm could not meet the requirements of ICSI. He underwent diagnostic testicular fine-needle aspiration (TFNA). The couple’s chromosomal karyotypes were normal, and no deletion was observed in the azoospermia factor of the Y chromosome of the male (III-3). The female was diagnosed with POAG caused by the MYOC mutation c.1109 C > T. Thus, the couple was treated with TFNA+ICSI+PGD. The couple signed informed consent documents for PGD. All of the techniques were performed in accordance with the routine protocols of our centre. This study was approved by the Ethics Committee of Northwest Women’s and Children’s Hospital.
IVF, embryo culture, TE biopsy, and blastocyst vitrification
Eleven MII oocytes were inseminated through ICSI with the sperm through TFNA. The oosperms were cultured in fertilisation media in an incubator (Vitrolife). Normal fertilisation was evaluated and confirmed by the presence of two pronuclei and a second polar body at 16–18 h after insemination. For TE biopsy, an 11-µm hole was cut in the pellucida zona of every embryo via laser on day 3 post-insemination. When the embryos developed into blastocysts, the TE biopsy was performed on day 5 post-insemination, and approximately 5–8 cells were extracted from the TE of each blastocyst. The biopsied cells of blastocysts were respectively transferred into 0.2-mL polymerase chain reaction (PCR) tubes with 4.5 µL of buffer (Yikon Genomics) for WGA. The post-biopsied blastocysts were vitrified using a Kitazato vitrification kit (Kitazato Biopharma Co.) and plunged into liquid nitrogen.
WGA and CNV analysis
MALBAC method was applied to conduct WGA of the embryo biopsy samples in accordance with the manufacturer’s standard protocol (Yikon Genomics). The WGA products were used for CNV, Sanger sequencing, and NGS-based SNP haplotyping.
Sequencing libraries were prepared using a gene sequencing library kit (Yikon Genomics), and the libraries were sequenced on an Illumina HiSeq 2500 platform (Illumina) using HiSeq Rapid Cluster Kit V2 (Illumina). CNV analysis was performed as previously described (Zong et al. Citation2012). In brief, after the low-quality bases and adaptors were removed, clean and high-quality reads were compared with the hg19 reference genome (University of California, Santa Cruz Genome Browser; genome.ucsc.edu/) using BWA (Burrows-Wheeler Alignment Tool, version 0.7.12-r1039). Unique mapped reads were calculated, and a reference dataset was obtained to represent the relative copy number.
Sanger sequencing and SNP haplotyping
The mutation site (c.1109C>T) of the WGA products was detected using 2× GoldStar Best MasterMix (Dye) (CoWin Biosciences). MYOC c.1109 front primer (CGCTGAGTCCAGAACTGTCATAAG) and reverse primer (TGCTTCATCCACAGCCAAGTC) were designed using Primer 5.0 software. All PCR amplifications were performed on a 96 Well Thermal Cycler Veriti DX (Life Technologies). The amplification reaction was as following: initial denaturation at 94°C for 5 min; 32 cycles of denaturation at 94°C for 1 min, annealing at 62°C for 1 min, extension at 72°C for 2 min. Sanger sequencing was performed on an Applied Biosystems 3500 platform (Life Technologies) after PCR amplification was conducted. All of the procedures were carried out in accordance with the manufacturer’s protocol.
The WGA products were used to obtain sequencing libraries using the NGS DNA Library Kit for Illumina (CoWin Biosciences) and then sequenced on an Illumina HiSeq 2500 platform (Illumina). All of the procedures were performed in accordance with the manufacturer’s protocols. Using the information of SNPs measured in the embryos and the patient, we could determine whether the embryo carried the chromosome with the mutation. The NGS sequencing data were analysed by Yikon Genomics, Ltd.
Authors’ contributions
Conceived and designed the experiments: BH; Performed the experiments: XJ, ZZ; Analysed the data: XJ, BH; Contributed reagents: JS; Wrote the manuscript: XJ, BH.
Acknowledgments
We would like to express our gratitude to the family for participating in this study; Liu Min of Yikon Genomics, Ltd., for her work; and the embryologist and the geneticists of the Northwest Women’s and Children’s Hospital for contributing to this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bourcy CFAD, Vlaminck ID, Kanbar JN, Wang J, Gawad C, Quake SR. 2014. A quantitative comparison of single-cell whole genome amplification methods. PloS One. 9(8):e105585.
- Chen YF, Jia HT, Chen ZH, Song JP, Liang Y, Pei JJ, Wu ZJ, Wang J, Qiu YL, Liu G, et al. 2015. Mutational spectrum of phenylketonuria in Jiangsu province. Eur J Pediatr. 174(10):1333–1338.
- Deleye L, Coninck DD, Christodoulou C, Sante T, Dheedene A, Heindryckx B, Abbeel EVD, Sutter PD, Menten B, Deforce D, et al. 2015. Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method. Sci Rep. 5:227–228.
- Fingert JH, Héon E, Liebmann JM, Yamamoto T, Craig JE, Rait J, Kawase K, Hoh ST, Buys YM, Dickinson J, et al. 1999. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 8(5):899–905.
- Foster PJ, Buhrmann R, Quigley HA, Johnson GJ. 2002. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol. 86(2):238–242.
- Gong G, Kosoko-Lasaki S, Haynatzki G, Lynch HT, Lynch JA, Wilson MR. 2007. Inherited, familial and sporadic primary open-angle glaucoma. J Natl Med Assoc. 99(5):559–563.
- Hao Y, Chen D, Zhang Z, Zhou P, Cao Y, Wei Z, Xu X, Chen B, Zou W, Lv M, et al. 2018. Successful preimplantation genetic diagnosis by targeted next-generation sequencing on an ion torrent personal genome machine platform. Oncol Lett. 15(4):4296–4302.
- Huang L, Ma F, Chapman A, Lu S, Xie XS. 2015. Single-cell whole-genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet. 16:79–102.
- Kim HJ, Suh W, Park SC, Kim CY, Park KH, Kook MS, Kim YY, Kim CS, Park CK, Ki CS, et al. 2011. Mutation spectrum of CYP1B1 and MYOC genes in Korean patients with primary congenital glaucoma. Mol Vis. 17:2093–2101.
- Monemi S, Spaeth G, Dasilva A, Popinchalk S, Ilitchev E, Liebmann J, Ritch R, Héon E, Crick RP, Child A, et al. 2005. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum Mol Genet. 14(6):725–733.
- Renwick P, Trussler J, Lashwood A, Braude P, Ogilvie CM. 2010. Preimplantation genetic haplotyping: 127 diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod Biomed Online. 20(4):470–476.
- Renwick PJ, Trussler J, Ostad-Saffari E, Fassihi H, Black C, Braude P, Mackie Ogilvie C, Abbs S. 2006. Proof of principle and first cases using preimplantation genetic haplotyping–a paradigm shift for embryo diagnosis. Reprod Biomed Online. 13(1):110–119.
- Resnikoff S1, Pascolini D, Etya’ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP. 2004. Global data on visual impairment in the year 2002. Bull World Health Organ. 82(11):844–851.
- Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Héon E, Krupin T, Ritch R, Kreutzer D, et al. 2002. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 295(5557):1077–1079.
- Shimizu S, Lichter PR, Johnson AT, Zhou Z, Higashi M, Gottfredsdottir M, Othman M, Moroi SE, Rozsa FW, Schertzer RM, et al. 2000. Age-dependent prevalence of mutations at the GLC1A locus in primary open-angle glaucoma. Am J Ophthalmol. 130(2):165–177.
- Souzeau E, Burdon KP, Dubowsky A, Grist S, Usher B, Fitzgerald JT, Crawford A, Hewitt AW, Goldberg I, Mills RA, et al. 2013. Higher prevalence of myocilin mutations in advanced glaucoma in comparison with less advanced disease in an Australasian disease registry. Ophthalmology. 120(6):1135–1143.
- Stone EM, Fingert JH, Alward WLM, Nguyen TD, Polansky JR, Sunden SLF, Nishimura D, Clark AF, Nystuen A, Nichols BE, et al. 1997. Identification of a gene that causes primary open angle glaucoma. Science. 275(5300):668–670.
- Tikunova EV, Churnosov MI. 2014. Genetic studies of primary open-angle glaucoma. Vestnik oftalmologii. 130(5):96–99.
- Zong C, Lu S, Chapman A, Xie X. 2012. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 338(6114):1622–1626.