
Abstract
Despite availability of biologic therapies, limited patient access to many of the most-effective cancer treatments affects overall health outcomes. To address this issue, many governments have enacted legislation for the approval of biosimilars. The term “biosimilar” refers to a biologic product that is developed to be highly similar, as opposed to identical, to a licensed biologic product (the reference or innovator product), such that, per US Food and Drug administration draft guidelines, “no clinically meaningful differences [exist] between the biological product and the reference product in terms of safety, purity, and potency.” This article presents some considerations about the development of biosimilars in cancer treatment through an overview of biosimilars from a clinical perspective. Topics covered include the development requirements and unique regulatory requirements for biosimilars, labeling considerations, potential limitations to the uptake of biosimilars, and review of some biosimilars in development for oncology indications.
Abbreviations
| CI | = | confidence interval |
| EBE | = | European Biopharmaceutical Enterprises |
| EMA | = | European Medicines Agency |
| EPAR | = | European Public Assessment Report |
| FDA | = | US Food and Drug Administration |
| HER2 | = | human epidermal growth factor receptor 2 |
| MBC | = | metastatic breast cancer |
| ORR | = | overall response rate |
| PD | = | pharmacodynamics |
| PK | = | pharmacokinetics |
| SmPC | = | summary of product characteristics |
| WHO | = | World Health Organization |
Introduction
Cancer is the most economically devastating cause of death worldwide.Citation1 The World Health Organization (WHO) estimates that the medical cost associated with treating new cancers in 2010 was $163 billion (calculated in US dollars).Citation2 These estimates do not include the economic impact associated with premature death and disability or the costs of time by caregivers or transportation to treatment facilities, which increase the total annual estimated cost of treating newly diagnosed cancers to US$310 billion in 2010.Citation2 These costs are expected to continue to increase. For example, in the United States alone the annual cost of cancer care for all cancers (not just newly diagnosed cancers) is expected to increase dramatically from an estimated $125 billion in 2010 to a projected $173 billion in 2020.Citation3
Despite regulatory approval of a number of biologic agents in the treatment of various cancers, patient access to treatment is often limited.Citation4,5 Some healthcare systems ration high-cost treatments by limiting the number of treatments or restricting use to specific patient populations even though this might not be the most cost-effective strategy.Citation6 As a result, these systems deny access to many patients who could be helped.Citation6 To date, worldwide access to highly effective cancer treatments remains an unmet medical need in many countries.
Many governments worldwide have enacted legislation to allow regulatory approval of biosimilars.Citation7-10 The term “biosimilar” refers to a biologic product that is developed to be highly similar, as opposed to identical, to an existing licensed biologic product (i.e., the reference or innovator product), such that there are “no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency of the product.”Citation9 Because these reference products are typically large, structurally complex proteins, even minor changes in the manufacturing process can produce post-translational structural differences that can affect the safety and potency of the product.Citation9,11 As a result, biosimilars cannot be considered generic equivalents to the reference product.Citation9,10
Here, we present an overview of biosimilars from a clinical perspective. We discuss the unique developmental processes used for biosimilars, the specific regulatory requirements, labeling considerations (including how labeling may affect clinicians), and potential limitations to the uptake of biosimilars in clinical use. We also provide information about some biosimilars in development for oncology indications.
Developmental and Regulatory Processes for Biosimilars
A critical challenge faced by biosimilar developers is that the manufacturing process for the reference product is proprietary. As a result, the biosimilar developer must analyze the reference product extensively and use a process of reverse engineering to produce a biologic agent that is highly similar to the reference product. This requires substantial knowledge and expertise regarding the development and manufacture of biologics.
Because biologics are relatively large and complex proteins that are difficult to characterize, the regulatory process for biosimilar approval is not the same as that used for small-molecule generics. As a result of the unique considerations related to biosimilars, regulatory agencies have established guidelines specifically for their approval. Regulatory requirements for approval of biosimilars are generally consistent across guidelines of the European Medicines Agency (EMA), Health Canada, WHO, and the draft guidelines from the US Food and Drug Administration (FDA). Although minor differences exist among the agencies (), all require a stepwise approach that includes analytical studies, at least one human pharmacokinetic (PK) study, and generally a minimum of one efficacy and safety study intended to support a demonstration of biosimilarity.Citation7-10 The size and scope of the clinical program depend on the degree of similarity to the reference product demonstrated in non-clinical development, i.e., through comparative analytical and non-clinical data (in vitro or in vivo). Ultimately, regulatory decisions for approval are based on the “totality of the evidence” in support of the biosimilar in comparison to the reference product.Citation7-10
Table 1. Comparisons of regulatory requirements for biosimilars
In order to establish biosimilarity, a stepwise approach is used to develop evidence in support of a potential agent (). First, analytical studies are conducted to confirm that the biosimilar has a foundation of quality based on structural and functional similarity to the reference product (notwithstanding minor differences in clinically inactive components). The complete quality dossier required per current legislation includes extensive state-of-the art characterization studies to demonstrate similarity,Citation12 which, in practical terms, increases both the size (potentially 2 or 3 times greater in content) and complexity of the analyses required. Next, non-clinical studies need to demonstrate that the biosimilar agent acts on the same target or physiologic process (via assessments of the mechanisms of action and functional activity) and has similar toxicity as the reference product.
Figure 1. Demonstrating biosimilarity is built first on a foundation of an analytical comparison of structural and functional similarity to the reference product, supported by non-clinical testing, and clinical evaluation in the intended treatment population. Biosimilarity is considered demonstrated based on the totality of the evidence from all evaluations, with each step supported by the preceding one.
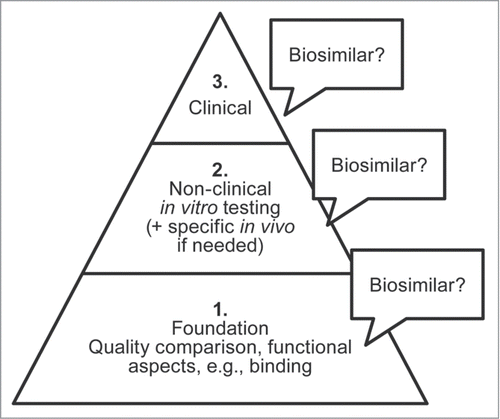
A key component of the biosimilarity exercise is a tailored clinical trial program in which PK, clinical efficacy, clinical safety, and immunogenicity of the biosimilar is compared with that of the reference product.Citation7-10 The goal of the clinical trial program is not to demonstrate patient benefit, as this has already been demonstrated through the clinical studies conducted for the reference product. Rather, the goal is to show the similarity of the potential biosimilar to the reference product. To accomplish this, clinical trials should be specifically designed to resolve any residual uncertainty as to whether the product can be considered a biosimilar.Citation7,9
After non-clinical similarity is established, the first step in clinical evaluation of biosimilarity involves a stepwise procedure that begins with clinical PK comparisons.Citation7-10 When appropriate pharmacodynamic (PD) markers exist, PD parameters are usually investigated as a combined PK/PD study.Citation7-10 The standard equivalence range used to demonstrate PK bioequivalence for biosimilars generally applies the traditional 80% to 125% equivalence range.Citation8,10 A potential biosimilar can be considered comparable to the reference product when the entire confidence interval (CI) falls within the lower and upper limits of this range (i.e., the equivalence margins).Citation8,10,13 PF-05280014, a potential biosimilar for trastuzumab, was compared to trastuzumab products marketed in and sourced from the European Union (trastuzumab-EU) and United States (trastuzumab-US) in a recent PK study. The 90% CIs for the ratios of the key PK parameters were all within the bioequivalence window of 80% to 125%. Bioequivalence was also observed between the 2 reference products as trastuzumab-EU to trastuzumab-US were also within this window. These results therefore, support continued development of PF-05280014.Citation14
After completion of PK and PD analyses, a clinical study (or studies) to demonstrate that the biosimilar produces comparable efficacy within accepted and predefined limits (the equivalence margin) versus the reference product is conducted.Citation7-10 This study also includes evaluation of comparative safety, including immunogenicity assessment, using state of the art methodology and employing appropriate measures of efficacy.
Clinical endpoints employed in clinical trials evaluating biosimilars may be different than those in trials evaluating novel therapeutics because selected endpoints should be sensitive to the detection of product-related differences in efficacy and safety.Citation15 For example, although survival is generally a preferred endpoint in many oncology clinical trials, survival endpoints may not be appropriate in clinical trials evaluating biosimilars for oncology indications because of potential confounding due to tumor burden, disease status, or previous lines of therapy.Citation15-17 Instead, a measure of response may be a better endpoint for clinical evaluation of a potential biosimilar.Citation15-17 The selection of an appropriate response endpoint is complicated by the fact that overall response rate is not always associated with long-term improvements in patient outcome or closely correlate with progression-free survival in patients with metastatic disease.Citation18-20 Novel endpoints (such as total pathologic complete response that is associated with disease-free survival in patients with early-stage breast cancerCitation20) may add supportive evidence for biosimilarity agents and allow for clinical testing in a sensitive and homogenous population, so their inclusion (especially on an exploratory basis) in clinical trials may be useful in establishing comparable efficacy and safety of biosimilars in oncology indications.Citation15
Similar to the clinical PK assessments, evaluation of clinical efficacy and safety in a potential biosimilar is also generally evaluated using equivalence trials.Citation7-10,Citation21 Thus, for 2 therapies to be considered comparable, the same outcome measure for the 2 interventions must fall within a specified range with respect to predefined clinical criteria.Citation22 In these trials, equivalence is usually assessed using 2-sided CIs (typically at the 90% or 95% level) for the difference between treatments.Citation13 Occasionally, non-inferiority designs, in which the aim is to determine if a therapy is no worse than a standard therapy,Citation22 may be considered for biosimilar approvals on an exceptional basis, but the guidelines established by the EMA, Health Canada, WHO, and the FDA draft guidelines prefer the use of equivalence designs for comparison of the proposed biosimilar to the reference product.Citation7-10
In order to conduct appropriate analyses of biosimilar products, the equivalence margins used for the main efficacy endpoint must be justified on historical clinical and statistical grounds, and calculated based on the effect size (including consideration of the magnitude and variability of the effect size for the reference product as derived from historic trials).Citation7-10,Citation21 The use of historical data for establishment of appropriate equivalence margins makes the use of novel pathologic endpoints difficult, but (as noted) could provide supportive efficacy and safety data if used on an exploratory basis. One difficulty in calculating the effect size for the reference product is that most of the data available for this calculation will be from randomized clinical trials. These studies often vary widely in trial design and study population, creating yet another challenge for biosimilar developers, particularly for oncology therapies since these studies can often have small sample sizes and widely varying background-chemotherapy regimens.
Across all phases of the clinical trial program, testing must be done in a sensitive and homogenous patient population so that any differences between the biosimilar and the reference product can be easily detected. For PK evaluations, the study population is often healthy subjects because these individuals lack comorbidity and co-medication concerns. However, results from healthy subjects may not reflect PK parameters in the patient population (e.g., when healthy subjects have reduced target-mediated clearance compared with patients); therefore, PK studies are not always possible or feasible in healthy subjects.Citation7,8 For the confirmatory efficacy and safety trials, study populations representative of approved therapeutic indications of the reference product and sensitive for detecting potential differences between the biosimilar and the reference product should be employed.Citation7
Practical considerations for the selection of an appropriate study population for a single, global Phase 3 study include consideration of the approval status of the reference product for a given indication, as well as the potential for recruiting patients in different countries. Multiple possible choices may exist; therefore, study sponsors must weigh the pros and cons of their choice of specific study populations. In addition, regulatory agencies must agree that a given study population is appropriate to support the application for biosimilar approval. However, the challenges associated with selection of an appropriate study population do not end with agreement with regulatory agencies, since the concept of biosimilarity and the approach to development are novel. As a result, ethics committees not familiar with the concept may raise objections to study designs or selection of study populations based on a misunderstanding of the goals and definitions established to demonstrate biosimilarity. Further education of all stakeholders on this novel regulatory paradigm is necessary.
In summary, although the regulatory paradigm for biosimilars is complex and highly specific, it is abbreviated compared with that of a novel biologic agent. There are several important implications in applying this paradigm to labeling and post-approval safety follow-up. In addition, a number of factors may curtail the uptake of biosimilars and limit patient access to these products.
Labeling Considerations for Clinicians
Consistent policies in product labeling and prescribing information do not exist across the regulatory agencies. WHO guidelines state that the prescribing information label for biosimilars should be as similar as possible to that of the reference product (particularly for posology and safety-related information, including contraindications, warnings, and adverse events) except for product-specific aspects, such as different excipient(s).Citation10 According to WHO guidelines, biosimilar labels potentially may differ from that of the reference product in 2 ways. First, if the biosimilar has fewer indications than the reference product, text related to those indications may be omitted unless information about certain risks is necessary.Citation10 If this information is omitted, the prescribing information should clearly state that the biosimilar is not indicated for use in the specific indication(s) and include the reasons why.Citation10 Second, a national regulatory authority may choose to mention that the product is a biosimilar, the specific studies that have been performed with the biosimilar, or include specific instructions for the physician on biosimilar product use.Citation10
Health Canada has taken a similar approach to the recommendations of WHO. Its guidelines for biosimilar products indicate that the product sponsor will not be allowed to entirely duplicate the product monograph of the reference product for the biosimilar.Citation8 Instead, a product monograph for a biosimilar must include a statement indicating that the product is a biosimilar, key data on which the decision for marketing authorization was made, including tables showing the results of the comparisons between the biosimilar and reference product, and the approved indications for the biosimilar, but make no claims of bioequivalence or clinical equivalence between the biosimilar and the reference product.Citation8
In contrast, EMA, while agreeing that a statement that the product is a biosimilar is included in the label, has indicated that the labeling of biosimilars follows the same principles used for labeling of small-molecule generics and that the label will copy the label of the reference product.Citation23 Specifically, the summary of product characteristics (SmPC) for the biosimilar will be an exact duplicate of the reference product's SmPC; any proposed differences in the biosimilar label need justification.Citation23 Recently, the EMA approved the biosimilar infliximab products, Remsima™ (Celltrion Healthcare Hungary Kft., Budapest, Hungary)Citation24 and Inflectra™ (Hospira UK Limited, Warwickshire, UK).Citation25 The SmPCs for these products duplicate the reference product information with no mention of any clinical trials that compared the biosimilar with the reference product. This means that the evidence used to support approval of the biosimilar, including head-to-head comparisons vs. the reference product, is not provided in the label.Citation26 These data may be located in the European Public Assessment Report (EPAR)Citation27; however, as they are not mentioned at all in the label, the physician may not know that such studies were conducted and are contained in the EPAR. Thus, there is a lack of ready access to key information in the SmPCs that is the primary reference document for physicians.Citation11 The physician may also incorrectly assume that all the clinical data in the label had been generated with the biosimilar product since it is not clear that all data relate to the reference product. Some industry trade associations have highlighted these issues. The European Biopharmaceutical Enterprises (EBE) issued a position paper on the labeling of biosimilars in August 2013.Citation26 More recently, EuropaBio issued a similar statement calling for a transparent approach to labeling and inclusion of data on both the reference product and the biosimilar.Citation28
To date, the FDA has not issued specific guidance regarding labeling of biosimilars, although the draft guidance for biosimilars states that product labels of a proposed biosimilar product should include all the information necessary for a health professional to make prescribing decisions.Citation9 This information should include a clear statement advising that the product is approved as a biosimilar, the specific indications and route of administration, and whether the biosimilar has or has not been determined to be interchangeable (and therefore an option for automatic substitution) with the reference product.Citation9
Post-Approval Considerations
All approved drugs require ongoing safety monitoring.Citation29 In addition, more proactive measures to evaluate long-term safety (such as participation in registries with ongoing evaluation of safety in the clinical setting) are often employed. These long-term, proactive plans are designed to understand the nature and frequency of adverse events and possible identification of risk factors.
Specific requirements for post-approval monitoring vary across regulatory agencies, but these plans are required to be discussed with regulatory authorities or submitted as part of the regulatory approval application.Citation7-10 For example, the EMA requires a description of the pharmacovigilance system and a risk management plan as part of the regulatory submission.Citation7 Similarly, WHO recommends a specific safety and pharmacovigilance plan at the time of submission of the marketing authorization application.Citation10 Health Canada requires the risk management plan be presented prior to the issuance of marketing authorization.Citation8 Although a position and requirement for pharmacovigilance for biosimilars has not yet been specifically defined by the FDA, they note that existing FDA pharmacovigilance guidelines will likely be considered appropriate.Citation30 In the current draft guidance for biosimilars, the FDA encourages discussion with the appropriate regulatory divisions since many aspects of safety monitoring are product-specific.Citation9
In general, the regulatory agencies recommend that pharmacovigilance plans should consider any known safety signals associated with the use of the reference product and its class.Citation7-10 If any additional specific safety monitoring or pharmacovigilance measures are required for the reference biologic or its product class, a biosimilar should apply the same monitoring/pharmacovigilance plan.Citation7,10 In addition, if any novel safety concerns have arisen during evaluation of the biosimilar, these also may be evaluated.Citation7,10 Even though the development plan is expected to include an evaluation of immunogenicity as part of the safety assessment of the biosimilar, as for a novel biologic at the time of regulatory approval, the size of the population evaluated and the duration of exposure are likely not large enough to identify rare, but potentially serious safety events, including immunogenicity. As a result, the regulatory agencies specifically indicate that the on-going post-approval follow-up should specifically monitor immunogenicity.Citation7-10
Potential Limitations to the Uptake of Biosimilars
Potential biosimilars are currently in development for oncology indications; however, to date, published data from clinical trials are available for only some of these products ().
Table 2. Biosimilar products in development for oncology indications with publications
Despite biosimilars providing additional treatment options and having demonstrated similarity to the reference product, it is likely that physicians will not consider biosimilars to be interchangeable with the reference product and, therefore, should not be automatically substituted (i.e., substituted at pharmacy level without a physician's consent). The EMA does not have the legal remit to determine interchangeability; these decisions are determined by policies adopted by national member states. Some of these individual regulatory agencies have enacted policies that prohibit automatic substitution. Health Canada is deferring this decision to the provinces and territories.Citation8 In contrast, the FDA has the legal authority to determine whether a product approved as a biosimilar may attain the higher level of evidence required for approval as an interchangeable biosimilar, but individual state laws will also need to be applied.Citation9 To date, guidance has not yet been issued by the FDA on interchangeability.Citation9
Another key concept in the regulation of biosimilars is the possibility to extrapolate indications, i.e., allowing approval of a biosimilar for use in indications of the reference product even if the biosimilar has not been evaluated in a clinical trial in that specific indication.Citation10 A common misconception about extrapolation is that the focus is on the clinical data alone for making the justification.Citation31 However, since the clinical evidence is only part of the data supporting the regulatory application for a biosimilar, the justification for extrapolation is based on the overall totality of data generated on similarity of the biosimilar to the reference product, including non-clinical data such as analytical and in vitro functional comparisons and the mechanism of action in the indications concerned. Evaluation of a biosimilar for extrapolation to additional indications is conducted on a case-by-case basis and depends on the level of evidence provided by the applicant. Extrapolation of indications is a complex area and, not surprisingly, regulatory agencies are coming to different conclusions on the extent of extrapolation allowed. For example, EMA recently granted approval of the full range of indications of the reference product to the biosimilar infliximab products RemsimaCitation24 and Inflectra,Citation25 whereas Health Canada determined that, because of functional differences between the biosimilar and the reference product, which were also correlated with pertinent differences in the mechanism of action, the data provided did not support extrapolation to Crohn's disease and ulcerative colitis.Citation32,33 A further complicating factor when considering extrapolation is that every biosimilar is a version of the target reference product. Not all biosimilar versions of the same target reference product are the same and therefore, the data presented for each individual product will not be the same. Therefore, regulators need to make case-by-case assessments on the merits of extrapolation. There is no one-size-fits-all approach. The nuanced nature of this assessment may make physician acceptance of this concept more challenging. However, it is important to recognize that the possibility of extrapolating indications is very important for the development of biosimilars because, without this option, the biosimilar developer would have to repeat the entire clinical development program of the reference product to gain approval in all indications. This is contrary to the intent of developing biosimilars and, if this was necessary, development of a potential biosimilar would be more extensive, likely leading to increased development costs and time and potentially reduced patient access to the biosimilar.
Similar to novel biologic therapies, the long-term safety of biosimilars will be limited at the time of approval. As discussed, post-approval safety monitoring will be required as part of pharmacovigilance requirements. These requirements are similar to those required for any approved biologic agent and can often limit initial use of a novel therapy. Because clinicians will initially be unfamiliar with biosimilars as a new type of biological product, improved communication not only to physicians, but also to payers and patients about the labeling and the rigor of oversight for biosimilars, including post-marketing surveillance, is necessary.Citation34
Conclusions
Despite the complex developmental and regulatory processes involved, development of biosimilars to treat cancer should provide additional treatment options that have the potential to generate savings and efficiencies for healthcare systems, which can help free up resources for other healthcare treatments and interventions.Citation4,35,36 This is due, in part, to the projected savings in commercial development associated with the abbreviated approval process for biosimilars compared with novel biologic therapies.Citation37 Biosimilars have already improved access to well-established therapeutic interventions in Europe and other locations where approvals have occurred.Citation4,35
Although there are some limitations to the use of biosimilars due to issues with interchangeability and extrapolation of data, in the long-term, biosimilars should offer a wider array of therapeutic solutions and increase accessibility to effective cancer treatments. Clinicians should understand the importance of biosimilars in clinical practice and how to make informed decisions about their appropriate use. In countries where biosimilars have been approved by regulatory agencies, changes in the practice of medicine have already occurred.Citation4 The on-going development of additional biosimilars should produce further improvements in the care and treatments of patient with cancer due to expanded use of biologic therapies, which may lead to better overall health outcomes and changes in medical guidelines and treatments, allowing more patients access to effective cancer treatments.
Disclosure of Potential Conflicts of Interest
MS has no conflicts of interest to disclose; GC was provided support for a symposium presentation by Pfizer Inc.; IJ, BG, JM, and DT are employees of Pfizer Inc. and hold stock or stock options in Pfizer Inc.
Acknowledgments
Medical writing support was provided by Christina McManus, PhD, of Engage Scientific Solutions and funded by Pfizer Inc.
References
- American Cancer Society, LIVESTRONG. The Global Economic Cost of Cancer. Atlanta, GA: American Cancer Society; 2010. http://www.cancer.org/acs/groups/content/@internationalaffairs/documents/document/acspc-026203.pdf
- Knaul F, Arreola-Ornelas H, Méndez O, Alsan M, Seinfeld J, Marx A, Atun R. The global economic burden of cancer. In: Stewart B, Wild C, eds. World Cancer Report 2014. Lyon, France: International Agency for Research on Cancer (IARC)/World Health Organization (WHO) Press; 2014.
- Mariotto AB, Yabroff KR, Shao Y, Feuer EJ, Brown ML. Projections of the cost of cancer care in the United States: 2010-2020. J Natl Cancer Inst 2011; 103(2):117-28; PMID:21228314; http://dx.doi.org/10.1093/jnci/djq495
- McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther 2012; 91(3):405-17; PMID:22318617; http://dx.doi.org/10.1038/clpt.2011.343
- Pallis A, Tsiantou V, Simou E, Maniadakis N. Pharmacoeconomic considerations in the treatment of breast cancer. Clinicoecon Outcomes Res 2010; 2:47-61; PMID:21935314
- Cornes P. The economic pressures for biosimilar drug use in cancer medicine. Target Oncol 2012; 7 (Suppl 1):S57-67; PMID:22249658; http://dx.doi.org/10.1007/s11523-011-0196-3
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on Similar Biological Medicinal Products Containing Biotechnology-derived Proteins as Active Substance: Non-clinical and Clinical Issues. Draft. London: European Medicines Agency; 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf
- Health Canada, Health Products and Food Branch. Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs) [Internet]. Ottawa Ontario: Health Canada; 2005. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf
- US Food and Drug Administration. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Draft Guidelines [Internet]. Washington, DC: US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2012. A http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
- World Health Organization Expert Committe on Biological Standardization. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs) [Internet]. Geneva, Switzerland: World Health Organization; 2009 [updated WHO Techincal Report Series no. 977-2013 Annex 2]. http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
- Schneider CK. Towards biosimilar monoclonal antibodies pros and cons. EMEA Workshop on Biosimilar Monoclonal Antibodies. Proceedings of the EMEA Workshop on Biosimilar Monoclonoal Antibodies; July 2, 2009; London.
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on Similar Biological Medicinal Products Containing Biotechnology-derived Proteins as Active Substance: Quality Issues (Revision 1) [Internet]. London: European Medicines Agency; 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf
- Chow SC, Liu JP. Statistical assessment of biosimilar products. J Biopharm Stat 2010; 20(1):10-30; PMID:20077246; http://dx.doi.org/10.1080/10543400903280266
- Yin D, Barker KB, Li R, Meng X, Reich SD, Ricart AD, Rudin D, Taylor CT, Zacharchuk CM, Hansson AG. A randomised phase 1 pharmacokinetic trial comparing the potential biosimilar PF-05280014 with trastuzumab in healthy volunteers (REFLECTIONS B327-01). Br J Clin Pharmacol 2014; 78: 1281–90. PMID: 25041377; http://dx.doi.org/10.1111/bcp.12464
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies – Non-clinical and Clinical Issues [Internet]. London: European Medicines Agency; 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
- European Medicines Agency, Oncology Working Party. Guideline on the Evaluation of Anticancer Medicinal Products in Man. Draft. London: European Medicines Agency; 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/01/WC500137128.pdf
- US Food and Drug Administration. Guidance for Industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics. Available at: (accessed July 28, 2014). Washington DC: US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2007. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071590.pdf
- Hotta K, Kiura K, Fujiwara Y, Takigawa N, Oze I, Ochi N, Tabata M, Tanimoto M. Association between incremental gains in the objective response rate and survival improvement in phase III trials of first-line chemotherapy for extensive disease small-cell lung cancer. Ann Oncol 2009; 20(5):829-34; PMID:19221150; http://dx.doi.org/10.1093/annonc/mdp020
- Lee L, Wang L, Crump M. Identification of potential surrogate end points in randomized clinical trials of aggressive and indolent non-Hodgkin's lymphoma: correlation of complete response, time-to-event and overall survival end points. Ann Oncol 2011; 22(6):1392-403; PMID:21266519; http://dx.doi.org/10.1093/annonc/mdq615
- Scappaticci F, Burger H, Bisordi F, Ruiz de Erenchun F, Schreitmueller T. Neoadjuvant-adjuvant treatment of breast cancer: a model for extrapolation of clinical data for trastuzumab biosimilar candidates. Proceedings of the 2013 Annual Meeting of the European Cancer Congress; September 27-October 1, 2013; Amsterdam, The Netherlands.
- Aradi I. European Generic Medicines Association (EGA). Biosimilar Industry Perspective on Clinical Issues - Equivalence versus Non-inferiority 2.2 [Internet]. London: EGA; 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2012/06/WC500128718.pdf
- Dasgupta A, Lawson KA, Wilson JP. Evaluating equivalence and noninferiority trials. Am J Health Syst Pharm 2010; 67(16):1337-43; PMID:20689122; http://dx.doi.org/10.2146/ajhp090507
- European Medicines Agency, Patient Health Protection. QRD General Principles Regarding the SmPC Information for a Generic/Hybrid/Biosimilar Product. EMA/627621/2011; Patient Health Protection [Internet]. London: European Medicines Agency; 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2012/05/WC500127589.pdf
- Celltrion Healthcare Hungary Kft. Summary of Product Characteristics: Remsima (infliximab) European Public Assessment Report [Internet]. Budapest, Hungary: 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf
- Hospira UK LImited. Summary of Product Characteristics: Inflectra (infliximab) European Public Assessment Report [Internet]. Warwickshire, UK: Hospira UK Limited; 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf
- European Biopharmaceutical Enterprises (EBE). Position Paper on Labelling of Biosimilars –Summary of Product Characteristics (SmPC) and Patient Information Leaflet (PIL) – Draft April 2013 (FINAL EBE 21 August 2013) [Internet]. Brussels: European Biopharamceutical Enterprises; 2013. http://www.ebe-biopharma.eu/uploads/Modules/Documents/ebe-position-paper-labelling_3-07-2013.pdf
- European Medicines Agency. European Public Assessment Reports (EPAR). London: European Medicines Agency; 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d125
- EuropaBio. EuropaBio Statement on Labelling of Biosimilars. Brussels, Belgium: EuropaBio; 2014. http://www.europabio.org/positions/europabio-statement-labelling-biosimilars
- Casadevall N, Edwards IR, Felix T, Graze PR, Litten JB, Strober BE, Warnock DG. Pharmacovigilance and biosimilars: considerations, needs and challenges. Expert Opin Biol Ther 2013; 13(7):1039-47; PMID:23527621; http://dx.doi.org/10.1517/14712598.2013.783560
- US Food and Drug Administration. Guidance for Industry: Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment [Internet]. Washington, DC: U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2005. http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126834.pdf
- Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood 2014; PMID:25298038
- Health Canada. Summary Basis of Decision (SBD) for Remsima. Ottawa, Ontario: Health Canada Office of Regulatory Affairs, Biologics and Genetic Therapies Directorate; 2014. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_remsima_160195-eng.php#sbd
- Health Canada. Summary Basis of Decision (SBD) for Inflectra [Internet]. Ottawa, Ontario: Health Canada; 2014. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php
- Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin JH, Weise M, Thirstrup S. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol 2012; 30(12):1179-85; PMID:23222783; http://dx.doi.org/10.1038/nbt.2447
- Gascon P, Tesch H, Verpoort K, Rosati MS, Salesi N, Agrawal S, Wilking N, Barker H, Muenzberg M, Turner M. Clinical experience with Zarzio(R) in Europe: what have we learned? Support Care Cancer 2013; 21(10):2925-32; PMID:23903799; http://dx.doi.org/10.1007/s00520-013-1911-7
- Henry D, Taylor C. Pharmacoeconomics of cancer therapies: considerations with the introduction of biosimilars. Semin Oncol 2014; 41 Suppl 3:S13-20; PMID:24767632; http://dx.doi.org/10.1053/j.seminoncol.2014.03.009
- US Congressional Budget Office. Cost estimate: S. 1695 Biologics Price Competition and Innovation Act of 2007 [Internet]. Washington, DC: United States Federal Government; 2007. http://www.cbo.gov/sites/default/files/cbofiles/ftpdocs/94xx/doc9496/s1695.pdf
- Orlov S, Burdaeva O, Nechaeva M, Kopp M, Kotiv B, Sheveleva L, Gladkov O, Khorinko A, Prokopenko T, Shapovalova J, et al. Pharmacokinetics and safety of BCD-021, bevacizumab biosimilar candidate, compared to Avastin in patients. Abstract e13500. J Clin Oncol 2014; 32(Suppl).
- Grunder B, Costigan L, Johnson K, Kneeland T, Cirelli D, Dufield R, Kitchen R, Porter T, Rouse J, K R. Characterization and similarity assessment of bevacizumab and a proposed biosimilar. 2014 Annual Meeting & Exposition of the American Association of Pharmaceutical Scientists (AAPS); November 2-6, 2014; San Diego, CA. 2014; Abstract W3549.
- Peraza M. Nonclinical development of proposed biosimilars to monoclonal antibodies: trastuzumab and bevacizumab. Proceedings of the Applied Pharmaceutical Toxicology (APT); May 14-16, 2014; Gaithersburg, MD.
- Poddubnaya I, Babicheva L, Kaplanov K, Zaritskey A, Volodicheva E, Alexeev S, Loginov A, Orlova R, Dvornichenko V, Gladkov O, et al. Comparison of pharmacokinetics and pharmacodynamics of BCD-020 with innovator rituximab in patients with indolent non-Hodgkin lymphoma. Abstract e19545. J Clin Oncol 2014; 32(Suppl).
- Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013; 27(5):495-507; PMID:23649935; http://dx.doi.org/10.1007/s40259-013-0036-3
- da Silva A, Kronthaler U, Meyer I, Papandrikopoulou A, Stangler T, Visser J. Target-directed development of a proposed biosimilar rituximab (GP2013): comparability of antibody-dependent cellular cytotoxicity activity and pre-clinical pharmacokinetics and pharmacodynamics with originator rituximab. Arthritis Rheum 2012; 64(Suppl 10): Abstr 2153.
- Yin D, Becker J-C, Melia L, Li R, Gumbiner B, Thomas D, Spencer-Green G, Meng X. A phase I pharmacokinetics trial comparing PF-05280586 (a potential biosimilar) and rituximab in subjects with active rheumatoid arthritis (REFLECTIONS B328-01). Annals of the Rheumatic Diseases 2014; 74 (Suppl 2): 497. http://dx.doi.org/10.1136/annrheumdis-2014-eular.5444
- Becker J, Yin D, Melia L, Li R, Gumbiner B, Thomas D, Spencer-Green G, Meng X. A phase I trial comparing PF-05280586 (a potential biosimilar) and rituximab in subjects with active rheumatoid arthritis. Arthritis Rheumatol 2014; 66(Suppl).
- Yoo D, Park W, Jeka S, Cons Molina F, Hrycaj P, Wiland P, Spieler W, Brzezicki J, Lee E, Medina-Rodriguez F, et al. A randomized, controlled, multicenter, 2-arm, parallel-group, double-blind study to demonstrate the equivalence of CT-P10 to innovator rituximab with respect to pharmacokinetic profile in patients with rheumatoid arthritis [Abstr #1736]. Arthritis Rheumatol 2013; 65(Suppl): S736.
- Yoo D, Park W, Jeka S, Cons Molina F, Hrycaj P, Wiland P, Spieler W, Lee E, Medina-Rodriguez F, Shesternya P, et al. Impact of anti-drug antibody on efficacy and safety over week 24 in both CT-P10 and innovator rituximab treatment groups. Arthritis Rheumatol 2014; 66(Suppl).
- Yoo D, Park W, Jeka S, Cons Molina F, Hrycaj P, Wiland P, Spieler W, Brzezicki J, Lee E, Medina-Rodriguez F, et al. A randomized, controlled, multicenter, 2-arm, parallel-group, double-blind study to demonstrate the equivalence of CT-P10 to innovator rituximab with respect to pharmacokinetic profile in patients with rheumatoid arthritis [Abstr #1736]. Arthritis Rheumatol 2013; 65(Suppl): S736.
- Stenina M, Ignatova E, Frolova M, Burdaeva O, Nechaeva M, Kopp M, Udovitsa D, Kotiv B, Alekseev S, Stroyakovsky D, et al. Pharmacokinetics and safety of BCD-022, trastuzumab biosimilar candidate, compared to Herceptin in patients. Abstract e11576. J Clin Oncol 2014; 32(Suppl).
- Im Y, Krasnozhon D, Bondarenko I, Zvirbule Z, Jung K, Oliynychenko P, Lee M, Im S, Kim H, Tjulandin S, et al. Phase I/IIB clinical trial comparing PK and safety of trastuzumab and its biosimilar candidate CT-P6. Proceedings of the 13th St Gallen International Breast Cancer Conference Primary Therapy of Early Breast Cancer; March 13-16, 2013; Gallen, Switzerland.
- Im Y-H, Odarchenko P, Grecea D, Komov D, Anatoliy C, Gupta S, Shparyk Y, Caguioa P, Makhson A, Krasnozhon D, et al. Double-blind, randomized, parallel group, phase III study to demonstrate equivalent efficacy and comparable safety of CT-P6 and trastuzumab, both in combination with paclitaxel, in patients with metastatic breast cancer (MBC) as first-line treatment. Abstract 629. J Clin Oncol 2013; 31(Suppl); PMID:23569309
- Ewesuedo R, Barker KB, Taylor CT, Jacobs I, on behalf of the REFLECTIONS B327-02 Study Investigators. A phase 3 randomized, double-blind trial comparing PF-05280014 + paclitaxel vs. trastuzumab + paclitaxel for treatment of metastatic breast cancer patients. Proceedings of the 36th Annual Meeting of the San Antonio Breast Cancer Symposium; December 10-14, 2013; San Antonio, TX.
- Jacobs I, Coiro J, Hilton F, Orazem J, Abbas R, Zacharchuk C, on behalf of the REFLECTIONS B327-04 Investigators. A phase 3 randomized, double-blind trial comparing PF-05280014 + docetaxel and carboplatin vs. trastuzumab + docetaxel and carboplatin for neoadjuvant treatment of operable HER2+ breast cancer. Proceedings of the 37th Annual Meeting of the San Antonio Breast Cancer Symposium; December 9 -13, 2014; San Antonio, TX.
- Hurst S, Ryan AM, Ng CK, McNally JM, Lorello LG, Finch GL, Leach MW, Ploch SA, Fohey JA, Smolarek TA. Comparative nonclinical assessments of the proposed biosimilar PF-05280014 and trastuzumab (Herceptin). BioDrugs 2014; 28(5):451-9; PMID:25001079; http://dx.doi.org/10.1007/s40259-014-0103-4