
Abstract
Panitumumab, as a commercially available antibody, is an effective anticancer therapeutic against epidermal growth factor receptor (EGFR), although it exerts weak antibody-dependent cell-mediated cytotoxicity (ADCC) activity owing to its IgG2 nature. Here, we firstly engineered panitumumab by grafting its variable region into an IgG1 backbone. The engineered panitumumab (denoted as Pan) retained binding activity identical to the parental antibody while exhibiting stronger ADCC activity in vitro and more potent antitumor effect in vivo. To further enhance the target selectivity of Pan, we generated Pan-P by tethering an epitope-blocking peptide to Pan via a tumor-specific protease selective linker. Pan-P showed almost 40-fold weaker affinity compared with Pan, but functional activity was restored to a similar extent as Pan when Pan-P was selectively activated by urokinase-type plasminogen activator (uPA). More importantly, targeted localization of Pan-P was observed in tumor samples from colorectal cancer (CRC) patients and tumor-bearing nude mice, strongly indicating that specific activation also existed ex vivo and in vivo. Furthermore, Pan-P also exhibited effective in vivo antitumor potency similar to Pan. Taken together, our data evidence the enhanced antitumor potency and excellent target selectivity of Pan-P, suggesting its potential use for minimizing on-target toxicity in anti-EGFR therapy.
Abbreviations
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| mAb | = | monoclonal antibody |
| EGFR | = | epidermal growth factor receptor |
| uPA | = | urokinase-type plasminogen activator |
| TKI | = | tyrosine kinase inhibitor |
| IgG | = | Immunoglobulin G |
| ELISA | = | enzyme-linked immunosorbent assay |
| SPR | = | surface plasmon resonance |
| CI | = | confidence interval |
| LC | = | light chain |
| HC | = | heavy chain |
| EGFR VIII | = | EGFR Type III Variant |
| CRC | = | colorectal cancer |
| ECD | = | extracellular domain |
| SEC | = | size exclusion chromatography |
| CCK-8 | = | Cell Counting Kit 8Yun |
Introduction
`Aberrant expression of epidermal growth factor receptor (EGFR) has been observed in a variety of solid tumors, and often correlates with a poor clinical outcome for patients.Citation1 These features make EGFR an important target for cancer therapeutics, like monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKI).Citation2-4 Anti-EGFR mAbs panitumumab (Vectibix®) and cetuximab (Erbitux®) are established agents in the treatment of colorectal cancer (CRC).Citation5
By binding to the extracellular domain of EGFR, mAbs mediate direct effector mechanisms, resulting in blockade of ligand binding, receptor downregulation, cell cycle arrest and induction of apoptosis, but also recruitment of immunological activity against cancer cells, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).Citation1,6 Specifically, single anti-EGFR mAb is documented as poor mediator of CDC, whereas combinations of anti-EGFR mAbs can elicit CDC.Citation1,7 However, IgG subclasses of anti-EGFR mAbs do not elicit ADCC equally well. For instance, IgG1 antibodies elicit ADCC more efficiently than IgG2.Citation8-10 Although EGFR is an attractive target for anticancer therapy, it is widely expressed on normal tissue such as the liver, skin and gastrointestinal tract, which usually causes dose-limiting side-effects and characteristic toxicities in patients treated with anti-EGFR therapy.Citation2,11,12
Panitumumab is a human IgG2κ mAb that binds specifically to EGFR with high affinity, and not to the other ErbB family members.Citation13,14 Treatment with panitumumab has been associated with low immunogenicity and low risk of hypersensitivity reactions.Citation15,16 More importantly, cetuximab-resistant patients who harbor the S492R mutation responded to treatment with panitumumab.Citation17 In keeping with the results, Voigt et al.Citation18 revealed that the epitope of panitumumab differs from that of cetuximab, providing a possible explanation for the benefits from panitumumab treatment in some cetuximab-resistant patients. However, as a fully human IgG2 mAb, panitumumab is devoid of natural killer (NK) cell–mediated classical ADCC.Citation9 Recent studies have suggested that the lack of ADCC activity could limit the efficacy of panitumumab in vivo mouse model.Citation19,20 The IgG2 isotype of panitumumab was chosen to minimize potential toxicity to EGFR-expressing normal tissues from ADCC and CDC.Citation15 Consequently, the construction of a novel ADCC-enhanced mAb by engineering panitumumab has not been reported, potentially due to concerns that improving the effector functions of panitumumab might increase the severity of additional side effects.
Side effects like skin toxicities, commonly defined as rash, acne, pruritus and dry skin, have been reported as one of the predominant side effects in cancer patients treated with anti-EGFR antibodies such as panitumumab.Citation21,22 Although the mechanism underlying the skin toxicities caused by EGFR inhibition was still unclear, studies suggest that EGFR signaling in keratinocytes regulates key factors involved in skin inflammation and innate host defense.Citation12,23 Therefore, antibodies recognizing a tumor-specific EGFR epitope, such as EGFR VIII, provide the potential to enhance therapeutic specificity.Citation1 It is noteworthy that a recent study has shown that a novel Probody™ technology platform was able to limit cutaneous side effects in preclinical study.Citation24 As a tumor-specific protease activated EGFR-directed antibody, a Probody™ remains inert in healthy tissues and systemic circulation, but is activated locally at the tumor site.Citation24,25 To some extent, the technology will be an excellent solution to the problem of potent skin toxicities when developing an ADCC-enhanced mAb from panitumumab.
In this study, we developed a novel anti-EGFR mAb, denoted as Pan-P, with the goal of enhancing ADCC and target-selective activity. The enhanced ADCC effect of Pan has been validated in vitro, showing effective antitumor effect in vivo. Furthermore, target selectivity was also shown in vitro and in vivo, even in CRC tissue samples from patients. In conclusion, Pan-P is a novel potential therapeutic candidate that may improve clinical efficacy and limit on-target toxicity in patients treated with anti-EGFR therapy.
Results
Construction and characterization of Pan
The variable region of panitumumab was grafted onto the human IgG1 backbone consisting of light chain κ and heavy chain. The molecular weight of Pan was determined by SDS–PAGE (). Under nonreducing conditions, a single band of approximately 200 kDa for Pan was observed, which showed a slightly higher molecular weight than the parental antibody in both SDS-PAGE and SEC analysis (data not shown). The two neighboring bands of panitumumab demonstrate the heterogeneity of human IgG2 subclass, as previously reported.Citation26 Under reducing conditions, Pan and panitumumab yielded 2 protein bands with molecular mass of 50 kDa (heavy chain) and 25 kDa (light chain), respectively ().
Figure 1. Characterization of Pan. (A) SDS-PAGE analysis of purified Pan under nonreducing (8% polyacrylamide gel) and reducing (12% polyacrylamide gel) conditions. Lane 1, Pan; lane 2, panitumumab; M, Protein Marker. (B) Competitive binding assay. Pan and panitumumab was evaluated in ELISA for their ability to compete with biotin-conjugated panitumumab for binding to coating EGFR-Fc. Points, mean of 3 independent determinations; Bars, SEM. (C) Proliferation of A431 cells grown in the presence of increasing concentrations of panitumumab or Pan. Points, mean of 3 independent CCK-8 assays; Bars, SEM.
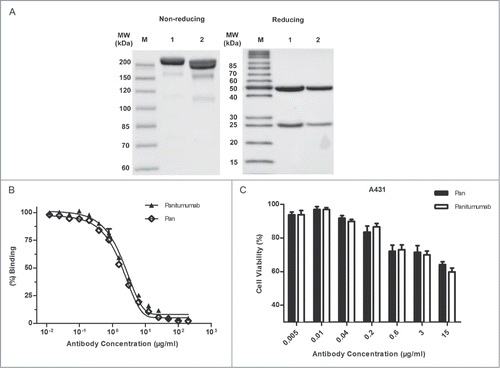
A competitive binding assay was then conducted to examine the relative binding activity of Pan and panitumumab (). It showed that the relative binding activity (mean EC50) of Pan (1.849 μg/mL) was similar to that of panitumumab (2.112 μg/mL). Moreover, we examined the inhibitory effect of Pan on the A431 cell line, which highly expressed EGFR. Consistently, Pan and panitumumab were equally effective in vitro, showing significant inhibition ability (). These results suggested that we successfully constructed Pan, which retains identical binding activity and inhibitory ability with the parental antibody.
Pan exhibits superior ADCC activity compared with panitumumab
Because Pan showed EGFR-binding behavior that was identical with the parental antibody, we investigated whether the ADCC activity of Pan was enhanced. A previous study has demonstrated afucosylated Fc-region carbohydrates of an anti-EGFR mAb increased its affinity for FcγRIIIa and enhanced ADCC activity.Citation27 Accordingly, we investigated the percentage of afucosylation in Pan produced by an engineered Chinese hamster ovary (CHO) cell line, which was stably transfected with the heavy-chain and light-chain expression vectors. Remarkably, Pan contained 33% afucosylation structures in total (generally less than 10% in CHO cell-derived IgG), which may contribute to inducing ADCC activity (; Fig. S1).Citation28
Figure 2. Pan shows superior ADCC activity compared with Panitumumab. (A) Quantitation of N-glycosylation species of Pan, as determined by Mass Biopharmalynx 1.3.3 software. Percentage represents the presence of different glyco-species in total glycan. (B) Activation of NFAT-luciferase reporter in Jurkat cell expressing the FcγRIIIa complex by Pan (1 μg/mL), panitumumab (1 μg/mL) or cetuximab (1 μg/mL). Samples without mAbs were controls. These assays were performed in triplicate, and the data are the mean ± SEM (***, P < 0.001). (C) Jurkat/FcγRIIIa/NFAT-Luc cells were co-incubated in the presence of serially diluted Pan, panitumumab or cetuximab. Luciferase activity (the fold of induction compared to the control sample without mAbs) is represented on the graphs. (D) BALB/c nude mice received subcutaneous injections of A431 cells on day 0. Starting on day 1 (arrow), mice were treated twice weekly by intraperitoneal injections of panitumumab (50 mg/kg), Pan (50 mg/kg), or control IgG (50 mg/kg). Tumors were measured using a caliper and tumor growth was monitored every 3 days for n = 6 mice per group.
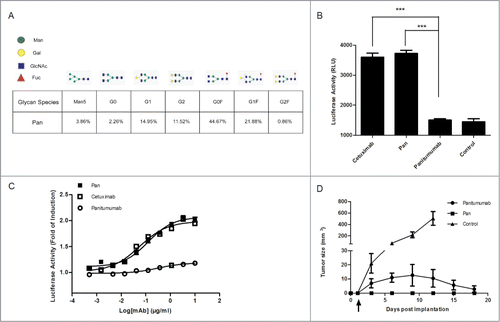
The ADCC reporter gene assay, which is equivalent to LDH ADCC bioassay in testing ADCC activity, was then used for evaluating the pathway activation by therapeutic antibody in an ADCC mechanism of action.Citation29 We chose a Jurkat cell line that stably expresses the FcγRIIIa complex and the luciferase reporter gene under the control of the NFAT response elements as the effector cells. A431 cells were used as target cells. To exert ADCC, FcγRIIIa-expressing effector cells recognized the mAbs that bound to antigen on the surface of target cells.Citation30 This bridging of target and effector cells by the mAb is a critical step for the induction of ADCC, which was quantified with luminescence readout. Our results showed that Pan was approximately 2-fold more potent than the parental antibody at inducing ADCC in the same low concentration (1 μg/mL) (). Furthermore, ADCC assay showed Pan was capable of activating ADCC luciferase reporter signaling in a markedly dose-dependent manner in A431 cell line, which is similar to cetuximab. However, panitumumab only has a minimal concentration-dependent reporter activity compared to cetuximab and Pan ().
We also evaluated the in vivo efficacy of Pan and panitumumab in A431 xenograft model according to a previously reported method.Citation19 Notably, Pan prevented tumor development more effectively than panitumumab in the prophylactic model (). As both antibodies were equally effective in vitro, enhanced ADCC activity in part explained the superior therapeutic activity of Pan. These findings suggested that Pan has superior antitumor potency to panitumumab.
Design and in vitro proteolytic cleavage of Pan-P
We further developed proteolytic processed Pan-P, which was derived from Pan by using previously described techniques.Citation24,25 As shown in , the indicated peptide was fused to the light chain amino terminus of Pan. The sequence consists of blocking peptide (IYPPLLRTSQAM), substrate peptide (LSGRSDNH) and serine–glycine linker peptide (GSSGGSGGSGGSG). The selected blocking peptide, which binds specifically to panitumumab but not to cetuximab, was identified by Vogit et al.Citation18 Protease uPA is known to be up-regulated in a variety of human carcinomas.Citation31 In recent years, it has been widely selected for developing prodrugs, which are inactive until they are converted to active drugs in tumor tissues.Citation32,33
Figure 3. Design and in vitro proteolytic cleavage of Pan-P. (A) Schematic representation of Pan-P showing the blocking peptide, uPA substrate region, flexible peptide linkers and IgG1 backbone. (B) SDS-PAGE analysis of Pan-P before (lane 2) and after proteolytic cleavage with uPA (lane 1). Pan was used as control (lane 3). (C) Validation of sequence-specific cleavage in Pan-P when incubated with uPA by LC/MS analysis.
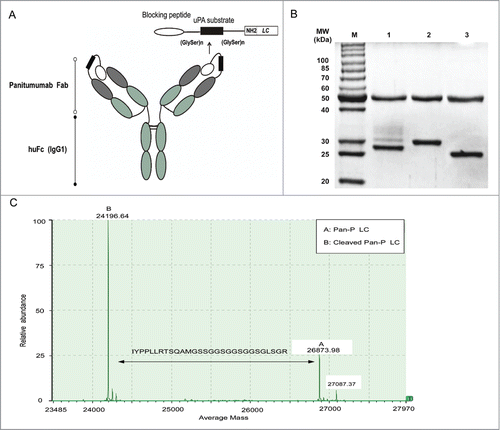
The substrate peptide specificity for uPA, LSGRSDNH, was attached to the blocking peptide via serine–glycine linkers. To determine whether Pan-P was cleaved by uPA, Pan-P was incubated with recombinant uPA in vitro. SDS-PAGE was then conducted to analyze the effect of uPA digestion (). The molecular weight of Pan-P's heavy chain was not affected by addition of uPA, whereas uPA-mediated hydrolysis of the light chain yielded an activated product with a molecular weight lower than the remaining uncleaved light chain. To further characterize the site-specific cleavage of Pan-P by uPA, LC/MS analysis was utilized to verify that the selective cleavage site was between amino acid R and S.Citation32,33 The difference between 2 peaks reflected the expected loss of blocking peptide as shown in . Data shown here indicated that Pan-P has been engineered successfully in structure and could be cleaved by uPA in vitro.
Pan-P has in vitro activity when activated by uPA
To rapidly evaluate the in vitro activity of Pan-P, direct ELISA was utilized to compare the antigen-binding capacity of Pan with Pan-P. Data shown in clearly indicated that EGFR-binding activity of Pan-P was approximately 20-fold weaker than that of Pan. However, uPA-activated Pan-P had binding activity identical with Pan. Surface plasmon resonance (SPR) measurements also showed similar results (; Fig. S2). Pan-P (KD = 2.8 × 10−8 M) showed almost 40-fold decrease in affinity compared with Pan (KD = 7.4 × 10−10 M) or activated Pan-P (KD = 6.9 × 10−10 M). Interestingly, the association rate (Ka) of Pan-P was significantly increased by proteolysis, while the dissociation rate (Kd) was unchanged, demonstrating that the blocking effect is mediated mostly by a reduction in association rate.
Figure 4. In vitro activation of Pan-P. (A) Pan-P exhibited reduced binding to immobilized EGFR by ELISA, whereas digestion of Pan-P with uPA (activated Pan-P) restored binding comparable to Pan. The data are shown as the mean ± SEM. EC50 for Pan, Pan-P, and activated Pan-P are 0.117 μg/mL (95% confidence interval [CI], 0.1039–0.1309 μg/mL), 2.694 μg/mL (95% CI, 2.359–3.075 μg/mL), and 0.1452 (95% CI, 0.1099–0.1918 μg/mL), respectively. (B) Kinetic parameters and affinity determinations of Pan, Pan-P and activated Pan-P for EGFR ECD measured by Biacore T100. (C) Inhibitory effect of increasing concentrations of Pan, Pan-P, or activated Pan-P on the proliferation of A431 cells. The data are shown as the mean ± SEM. (D) CCK-8 assay comparing the effects of Pan, Pan-P and activated Pan-P on DiFi cell proliferation. IC50 for Pan, Pan-P, and activated Pan-P are 0.054 μg/mL (95% CI, 0.048–0.061 μg/mL), 0.53 μg/mL (95% CI, 0.46–0.62 μg/mL), 0.059 μg/ml (95% CI, 0.055–0.063 μg/mL). Bars, SEM.
![Figure 4. In vitro activation of Pan-P. (A) Pan-P exhibited reduced binding to immobilized EGFR by ELISA, whereas digestion of Pan-P with uPA (activated Pan-P) restored binding comparable to Pan. The data are shown as the mean ± SEM. EC50 for Pan, Pan-P, and activated Pan-P are 0.117 μg/mL (95% confidence interval [CI], 0.1039–0.1309 μg/mL), 2.694 μg/mL (95% CI, 2.359–3.075 μg/mL), and 0.1452 (95% CI, 0.1099–0.1918 μg/mL), respectively. (B) Kinetic parameters and affinity determinations of Pan, Pan-P and activated Pan-P for EGFR ECD measured by Biacore T100. (C) Inhibitory effect of increasing concentrations of Pan, Pan-P, or activated Pan-P on the proliferation of A431 cells. The data are shown as the mean ± SEM. (D) CCK-8 assay comparing the effects of Pan, Pan-P and activated Pan-P on DiFi cell proliferation. IC50 for Pan, Pan-P, and activated Pan-P are 0.054 μg/mL (95% CI, 0.048–0.061 μg/mL), 0.53 μg/mL (95% CI, 0.46–0.62 μg/mL), 0.059 μg/ml (95% CI, 0.055–0.063 μg/mL). Bars, SEM.](/cms/asset/f1074fd5-a02f-4f5d-b3bf-6cd44fa001d0/kmab_a_1008352_f0004_b.gif)
To further investigate the functional activity of Pan-P, we compared the inhibitory activity of Pan-P, activated Pan-P and Pan using cell proliferation assay in A431 cell line. Our results indicated that Pan elicited more effective growth inhibition compared with Pan-P in the A431 cell line (). Simultaneously, activated Pan-P showed inhibitory activity equivalent to Pan. We also examined the inhibitory effect using DiFi cells, a high EGFR-expressing colorectal cancer cell line. Results showed the inhibitory activity of Pan-P was reduced by 10-fold relative to that of Pan or activated Pan-P (). Finally, we also verified the equivalent ADCC-eliciting activity of activated Pan-P to that of Pan (Fig. S3). These results suggested that selectively activated Pan-P restored fully functional activity, whereas the inactivated form showed markedly decreased activity.
Ex vivo activation of Pan-P in human tissue sections
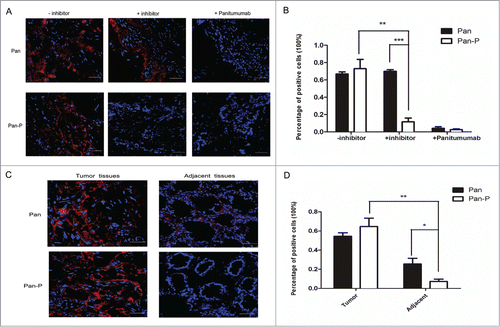
To validate the target-selective activation of Pan-P in a clinical setting, we obtained primary frozen tissue samples from human colorectal cancer (CRC) for testing in situ protease activity. We first screened EGFR (+) human tumor samples from several CRC patients by immunochemistry staining (data not shown). Selected tumor specimens with EGFR (+) were incubated with Alexa Fluor®680 (AF680)-labeled Pan or Pan-P (Table S1). To verify the protease-activated property of Pan-P, a broad spectrum protease inhibitor was preincubated with frozen tissue sections. In contrast to the positive signal of Pan-P in non-pretreated tissues, activation was inhibited in pretreated tissues (). However, the positive signal for Pan was not affected. Moreover, pretreatment with non-labeled panitumumab at high concentration (100 μg/mL) resulted in no positive signal, suggesting that positive staining of Pan-P is dependent on the specific binding to EGFR in tissues (). More importantly, the binding activity of Pan was significantly superior to Pan-P in the adjacent normal colorectal mucosa from the same subject (). Statistical analysis () demonstrated that the percentage of Pan-stained cells in adjacent specimen was 25% ± 5.9% (Mean ± SEM), while the percentage of Pan-P stained cells was only 7% ± 2.4%. To some extent, this evidence demonstrated that the binding activity of Pan-P was weaker than that of Pan in adjacent normal tissues, despite the fluorescent staining that was observed in limited specimens. Consequently, these promising results provided a basis for further evaluating the toxicity of targeting normal skin tissues with Pan-P treatment.
Figure 5. Analysis of Pan-P and Pan binding to human CRC sections using fluorescence microscopy. (A) Representative fluorescent immunostaining showed Pan-P was activated and bound in EGFR-positive colorectal carcinoma specimens. AF680-labeled Pan and Pan-P were incubated with frozen tissue sections of human primary colorectal cancer. After 2 hours of incubation, positive staining (red) validated Pan-P and Pan bound to EGFR in colorectal carcinoma specimens (left panel); however, preincubation of panitumumab blocked the binding positions of Pan and Pan-P (right panel) on EGFR. With preincubation of protease inhibitor, Pan-P binding fluorescent signal was inhibited as Pan binding signal remained unchanged (middle panel). 4’,6-Diamidino-2-phenylindole (DAPI) nuclear staining appears in blue. (B) Statistical analysis of immunoreaction positive cells in colorectal carcinoma specimens was shown when incubated with indicated antibody. (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Data represent mean and SEM of the integrated fluorescence signals from 3 fields for each specimen. Significance is confirmed using unpaired 2-sided Student's t-test (C) Representative micrographs of fluorescent immunostaining showed reduced binding of Pan-P for normal colorectal mucosa adjacent to tumor compared with Pan. Pan-positive staining indicated the presence of EGFR expression in both tumor tissues and adjacent nontumorous mucosa tissues. Pan-P positive staining was observed only in colorectal carcinoma specimens, while no significant signal was detected from adjacent normal colorectal mucosa. (D) Percentage of positive stained cells decreased markedly in adjacent normal colorectal mucosa when incubated with Pan-P. In contrast, Pan remained binding to adjacent section to some extent. (*, P < 0.05; **, P < 0.01). Data are expressed as mean ± SEM of the integrated fluorescence signals from 3 fields for each specimen. Original magnification, ×400. Scale bars, 50 μm.
In vivo activation and distribution of Pan-P
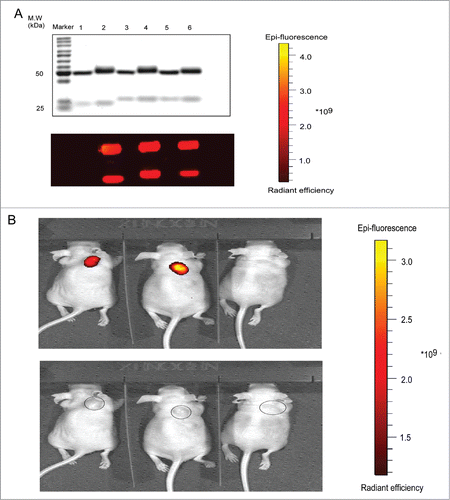
Using the manufacturer's instructions, AF680 was labeled on Pan, Pan-P or Pan-C, which was used as control for the lack of uPA substrate peptide. To evaluate the properties of AF680-labeled antibodies, SDS-PAGE was performed and observed by protein staining and fluorescence. The bands of AF680-labeled mAbs had slightly higher molecular weight than non-labeled controls (). A431 cells express and secrete uPA.Citation34,35 BALB/c nude mice bearing A431 cells were used for the evaluation of in vivo binding selectivity and distribution. The live animal fluorescent images were acquired 72 hours after injection of Pan, Pan-P or Pan-C, with each conjugated to fluorescence probe AF680. The representative results showed accumulation of Pan or Pan-P at the tumor site compared to Pan-C ().
Figure 6. In vivo activation of Pan-P in BALB/c nude mice with A431 tumor xenograft. (A) Validation of AF680 labeled Pan (lane 2), Pan-P (lane 4) and Pan-C (lane 6) by reducing SDS-PAGE (upper: Coomassie brilliant blue staining, lower: fluorescence). Unlabeled Pan (lane 1), Pan-P (lane 3) and Pan-C (lane 5) were used as controls. (B) In vivo fluorescence imaging of representative mice bearing A431 xenografts from intraperitoneal injection of AF680 labeled Pan (left), Pan-P (middle) and Pan-C (right). The lower panel indicated the tumor localization.
In vivo therapeutic efficacy of Pan-P in tumor xenograft model
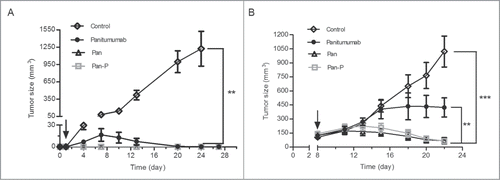
To assess therapeutic efficacy, we subsequently tested the activity of the mAbs against A431 tumor xenografts. Results revealed that Pan-P significantly prevented tumor growth when administered 1 day following tumor cell injection () or when established tumors were treated (), with almost equal efficacy compared to Pan. Moreover, treatment with Pan or Pan-P resulted in a significant benefit over panitumumab in both prophylactic () and established () tumor models. Thus, as a result, Pan-P showed potent inhibitory effect on tumor growth that was in consistent with its in vivo local activation property.
Figure 7. Pan-P inhibits the in vivo growth of A431 cancer cell line. (A, B) Mean tumor volumes of mice xenografted with A431 cells and treated with Pan (50 mg/kg), Pan-P (50 mg/kg), panitumumab (50 mg/kg) or control IgG (50 mg/kg) in prophylactic (A) or established (B) tumor models. There were 6 animals per treatment group. Tumor cells were injected at day 0, and antibody treatment started as indicated in the graphs (black arrow). Error bars show ± SEM. (**P < 0.01; ***P < 0.001).
Discussion
Panitumumab is a human anti-EGFR mAb with a long half-life and low rate of immunogenicity and infusion-related reactions, which include symptoms such as fever, chills and difficulty breathing.Citation15 Although panitumumab and cetuximab have been widely used for the treatment of advanced colorectal cancers, resistance in the anti-EGFR therapy has been commonly found to be conferred by mutations in downstream signaling molecules such as KRAS.Citation36,37 However, clinically relevant extracellular domain mutations of EGFR have been shown to mediate resistance to cetuximab but remain sensitive to panitumumab.Citation17 This mutation may have a role in providing the clinical benefit of panitumumab in a subset of subjects with mCRC who do not respond to treatment with cetuximab.Citation38 Additionally, Voigit et al.Citation18 described a distinct binding site for panitumumab that is different from that of cetuximab. Therefore, panitumumab has unique functional properties in EGFR-targeted therapy, especially for the patients who are refractory to cetuximab.
A previous study demonstrated that panitumumab was only effective in recruiting ADCC by myeloid effector cells (i.e., neutrophils and monocytes).Citation9 However, zalutumumab, as an IgG1 molecule, could induce both NK and myeloid effector cell-mediated ADCC, which shows the advantage of the IgG1 subclass in activating ADCC activity.Citation9 Moreover, Kubach et al.Citation20 provide strong evidence of a novel mechanism of CD8+ T cell-mediated cytotoxicity induced by anti-EGFR mAb with an IgG1 isotype. Recently, Dillon et al.Citation26 revealed that the heterogeneity of human IgG2 antibodies can affect structure and function, although additional studies are required to further characterize how this heterogeneity specifically affects panitumumab and its biological functions.
For these reasons, we constructed the novel antibody Pan, which is composed of the Fv region of panitumumab with human IgG1 heavy and κ light chain constant region. Similarly, Natsume et al.Citation39 generated a comprehensive set of mixed chimeric isotypes of anti-CD20 antibodies by shuffling constant domains of IgG1 and IgG3 to enhance CDC potency. In our study, a band for Pan at above 200 kDa was observed under nonreducing conditions. To our knowledge, disulfide bond formation in mAbs may influence their electrophoretic mobility under nonreducing conditions, which could in part explain why the migration of Pan and panitumumab above 200 kDa in the non-reducing gel is in contrast to the sum of the molecular weights shown in the reducing gel (150 kDa).
Kasper et al.Citation19 demonstrated ADCC indeed provides a therapeutic benefit in a study that compared the in vivo efficacy of panitumumab with that of cetuximab. However, panitumumab is different from cetuximab in many other aspects, such as affinity and inhibitory activity. By comparing panitumumab with Pan in both prophylactic and established tumor models, our results addressed the role of ADCC in anti-EGFR antibody therapy based on the 2 antibodies (panitumumab and Pan) bearing the same variable region. Generally speaking, antibodies that bind to different epitopes on the same protein may have different cytotoxic capabilities (e.g., anti-CD20 mAbs).Citation40 However, our results revealed that cetuximab and Pan exhibited identical ADCC activity in the luciferase reporter assay despite binding to different epitopes on EGFR.
Although the aim of targeted anticancer therapy is to kill tumor cells while minimizing the adverse effect to healthy tissues in the body, improving the effector functions of panitumumab could potentially increase the severity of the side effects. EGFR inhibitors are particularly known for causing a severe rash and skin damage, which sometimes forces patients to prematurely stop their treatments.Citation11 Although the mechanism for causing skin or other toxicities is complex, recent studies showed that widely expressed EGFR in the epiderm was the key regulator of cutaneous immunity.Citation12,41 Attempts to improve the safety of EGFR-targeted therapy included the development of antibodies against EGFRVIII, which is frequently expressed in carcinomas but not in normal tissues.Citation1 However, it is only expressed in a limited number of tumors.
The generation of antibodies with decreased affinity for EGFR provided another solution to overcome relevant adverse effects. It has been concluded that a potential correlation exists between dose-dependent skin toxicity effects and the affinity constant of therapeutic anti-EGFR antibodies.Citation42 Accordingly, nimotuzumab, which shows lower binding affinity than other anti-EGFR antibodies, has not demonstrated any toxic cutaneous effect in numerous clinical trials.Citation43 In another approach, the Probody™ Platform was developed to improve the safety profile of cetuximab.Citation24 The novel ‘Prodrug’ strategy involves tumor-specific activation achieved by ‘mask sequence’ and ‘cleavable protease substrate’.
Utilizing novel technology, we chose uPA as a selective activation protease because it is widely expressed in solid tumors, and we selected blocking peptide sequences that specifically bind to panitumumab, but not to cetuximab.Citation18 Based on the method described above, we successfully constructed Pan-P with target selectivity based on Pan. Upon incubation with normal tissues adjacent to tumor, Pan exhibited moderate binding activity while Pan-P showed no significant binding signals. Our results lead us to speculate that the potential low affinity of Pan-P for binding on normal skin tissues may result in an improved safety profile, which will be assessed in future nonhuman primate experiments.
In conclusion, the data shown here indicate that Pan-P, which is derived from panitumumab and engineered by utilizing Probody™ technology, efficiently enhanced antitumor activity and target selectivity. This newly generated anti-EGFR mAb has the potential to provide an improved clinical benefit/risk ratio in the treatment of colorectal cancer.
Materials and Methods
Cell lines, animals and other materials
The human epithelial carcinoma cell line A431 and the CHO-K1 cell line were obtained from the American Type Culture Collection (ATCC). The human colorectal cancer cell line DiFi was obtained from MD Anderson Cancer Center. The anti-EGFR chimeric antibody cetuximab was purchased from Merck, and the human antibody panitumumab was produced by Zhangjiang Biotech Inc. following reported procedures. Recombinant human uPA was purchased from Sino Biological Inc. Alexa Fluor®680 (AF680) was purchased from Life Technologies Inc. Six-week-old female BALB/c nude mice were obtained from the Shanghai Experimental Animal Center of Chinese Academy of Sciences (Shanghai, China). All animals were treated in accordance with guidelines of the Committee on Animals of the Second Military Medical University.
Construction, expression and purification
The light chain κ and heavy chain of human IgG1 subclass were chosen as the basic framework for Pan. The variable region of Pan was chosen to be identical with that of panitumumab. Pan-P was then constructed according to previous methods.Citation25 Briefly, the indicated peptide comprising blocking peptide, Gly-Ser–rich peptide linkers and uPA substrate sequence was fused to the light chain N terminus of Pan. Additionally, Pan-C, consisting of the same sequence with Pan-P but without uPA cleavable substrate was constructed. The light chain and heavy chain of antibodies mentioned above were each cloned into a pcDNA3.1(+) vector. Then, the heavy chain and light chain expression vectors were co-transfected into CHO-K1 cells. After transfection, the stable transfectants were isolated by limiting dilution in the presence of 1 mg/mL G418. The cell clone producing the highest amount of antibodies was grown in serum-free medium and the recombinant antibody was purified by affinity chromatography on Protein A-Sepharose (GE Healthcare). The concentration of purified mAb was determined by spectrophotometry using the NanoDrop 2000c spectrophotometer (Thermo Scientific). The purified antibodies were analyzed on 12% SDS–PAGE under reducing conditions and on 8% SDS–PAGE under nonreducing conditions, followed by Coomassie Brilliant Blue staining.
LC/MS analysis
The light chain and heavy chain were prepared by reduction of the tested antibody in 50 mM dithiothereitol (DTT), and they were analyzed by reverse LC-MS. A Waters UPLC system was used to generate a gradient flow at 200 μL/min, using typical mobile phases of 0.1% FA in MS-grade water (solvent A) and 0.1% FA in ACN (solvent B). The reverse-phase desalting separation of reduced antibody was performed on a Waters BEH300 C4 Column (2.1 × 50 mm, 1.7 μm) using a 4 min gradient (2–6 min, 25–34% B). The column temperature was maintained at 60°C. Mass spectrometric analysis was performed on a Waters Xevo G2-S QTof MS system, with a positive ion mode. The desolvation gas and source temperature were set to 300°C and 120°C, respectively. The capillary and cone voltages were set at 3,000 and 40 V, respectively. The instrument was calibrated in the m/z range of 500–2500 using NaI. The m/z scan range was set to 500–3,000. An auxiliary pump was used to spray a solution of 100 fmol/mL Glu1-f2ibrinopeptide B (GFP) in 50/50 ACN/water containing 0.1% FA for mass accuracy (lockmass channel), with a flow rate of 10 μL/min and sampling every 1 min. The deconvolution of ESI mass spectra of reduced antibody was performed by Mass BiopharmaLynx 1.3.3 software using Maximum Entropy algorithm.
EGFR-binding assay
Ninety-six–well plates (Nunc) were coated with EGFR-Fc (500 ng/mL; Zhangjiang Biotech Inc.) in phosphate-buffered saline (PBS; pH 6.84) and blocked with PBS containing 10% nonfat dried milk. The plates were incubated with the indicated concentrations of antibodies in PBS for 1 hour at room temperature. The plates were then incubated with horseradish peroxidase (HRP)–conjugated anti-human F(ab’)2 (Sigma) in PBS for 30 min at 37°C, and detection was performed by the addition of 3,3’,5,5 –tetramethylbenzidine (TMB) substrate regent followed by an equal volume of 1 M hydrochloric acid. Absorbance at 450 nm was then measured and reported as optical density (OD 450 nm). Data were fitted using a sigmoidal 4-parameter curve.
Competitive binding assay
Ninety-six-well plates (Nunc) were coated overnight at 4°C with EGFR-ECD (1 μg/well) in PBS buffer. Following blocked with 10% nonfat dried milk for one hour, plates were incubated with biotin-conjugated panitumumab F(ab’)2 (1:200) and indicated amounts of competing unlabeled panitumumab or Pan diluted in PBS buffer in 0.5% BSA for 60 min at 37°C. Plates were then incubated with avidin-conjugated HRP (1:1,000) for 30 min. Detection was performed according to the methods described in EGFR-binding assay. Data were fitted using a sigmoidal 4-parameter curve.
Growth Inhibition Assay
A431 cells (2,000 cells/well) or DiFi cells (5,000 cells/well) in 1% fetal bovine serum containing medium were plated in a 96-well culture plate with 3 replicates per treatment. The next day, the indicated concentrations of anti-EGFR mAbs or control IgGs were added to each well and the cells were incubated for 48 or 72 hours at 37°C. Cell growth inhibition was determined by Cell Counting Kit 8 (Dojindo Molecular Technologies) according to the manufacturer's instructions.
The percentage of cellular viability was calculated using the following formula: [(A450 of experiment - A450 of background) / (A450 of control - A450 of background)] *100
ADCC-reporter gene bioassay
This assay was performed according to previous report.Citation29 A431 cells, which overexpressed EGFR, were seeded at 10,000 cells per well in a 96-well opaque tissue culture plate. Cetuximab, panitumumab or Pan were serially diluted and incubated with the A431 cells for approximately 1 h at 37°C, 5% CO2. Following incubation, Jurkat-expressing FcγRIIIa luciferase reporter cells, were added to the A431/antibody mixture per well. The mixture was incubated for approximately 4 h at 37°C, 5% CO2, and then measured for luciferase production using a luminescent substrate (Promega Bright Glo).
In vivo therapy study
Mice were injected subcutaneously with 0.2 mL of 1 × 106 A431 cells suspended in PBS into the right flank. Treatment with antibodies by intraperitoneal injection started either 1 day after cell inoculation (prophylactic model) or when tumors reached approximately 120-150 mm3 (established model). Antibodies were administered in 200 μL of PBS. Treatment with human IgG was used as control. In the established model, the mice were randomly divided into groups of 6 mice each when tumor volumes reached an average of about 120–150 mm3. In the 2 models mentioned above, mice were treated intraperitoneally with either panitumumab (50 mg/kg), Pan (50 mg/kg), Pan-P (50 mg/kg) or control IgG (50 mg/kg) twice weekly. Tumor size, calculated as length × width2 × 0.5, and body weights were recorded at regular intervals.Citation44 Mice were euthanized with CO2 asphyxiation. All experiments were conducted in accordance with institutional guidelines and under an Institutional Animal Care and Use Committee (IACUC) protocol.
Evaluation of Pan-P localization in vivo by optical imaging
A431 xenograft tumor–bearing mice were injected intraperitoneally with AF680-labeled Pan-P (10 mg/kg), Pan-C (10 mg/kg) or Pan (10 mg/kg). The mice were imaged at 72 hours after AF680-labeled antibody injection with an IVIS® imaging system (Caliper Life Sciences). The images were acquired using a Cy5.5 filter set at excitation and emission wavelengths of 640 and 700 nm and recorded by a built-in CCD camera. During the procedure, the mice were kept under gaseous anesthesia (5% isoflurane) at 37°C.
Ex vivo evaluation with confocal immunofluorescence microscopy (In situ Immunohistology)
The method was established by Desnoyers et al.Citation24 Human colorectal tumor and adjacent normal colorectal mucosa of the same subjects were collected from the patients who underwent surgical resections at the PLA General Hospital (Beijing, China). The samples were frozen with liquid nitrogen within 1 hour of surgical removal. Frozen tissue sections (5 μm) were laid over glass slides. A solution containing AF680-labeled Pan-P (10 μg/mL) and Pan (10 μg/mL) was applied on the tissue and incubated for 2 hours at room temperature in 50 mM Tris-HCl buffer (pH 7.4), containing 150 mM NaCl, 5 mM CaCl2, and 0.05% Tween 20. The tissue was then washed to remove non-bound materials, and detectable label was visualized with a confocal fluorescence microscope (Olympus IX 81) and analyzed by an Imaging Software of FV10-ASW 1.7 Viewer. The project was approved by the Institutional Ethics Committee of PLA General Hospital.
Surface plasmon resonance analysis
The kinetics and affinity of Pan, Pan-P and activated Pan-P were determined by Biacore T100 (GE Healthcare). Anti-human IgG (Fc) antibody (GE Healthcare), diluted to 20 μg/mL in buffer (10 mM sodium acetate, pH 5.0), was immobilized on a Biacore Sensor S Chip CM5 (GE Healthcare). Test antibodies at 2.6 μg/mL were captured on the anti-human IgG Fc chip. The binding of 1 nM to 3,000 nM of the extracellular domain of human EGFR (EGFR-ECD), diluted in HBS-EP (10 mM Hepes, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% Surfactant P20, GE Healthcare), was measured in association and dissociation phases at 30 μL/min. Data were collected at 25°C and evaluated by using BIAevaluation T100 evaluation software (Version 1.1), global fitting was used to determine kinetic parameters that fitted all curves (differing concentrations of EGFR-ECD) within an experiment. Sensorgrams were obtained at each concentration and evaluated with 1:1 binding to determine the association rate constant (Ka) and dissociation rate constant (Kd). The equilibrium dissociation constant (KD) was calculated from the ratio of rate constants Kd/Ka. Concentration of analytes was assessed by using absorbance at A280, molarities were calculated by using the corresponding extinction coefficients.
Statistical analysis
Data are expressed as mean ± SEM. Statistical analysis was conducted by Student's 2 sided unpaired t-test to identify significant differences unless otherwise indicated. Differences were considered significant at P < 0.05 (*). Nonlinear regression analyses were used to fit curves. All statistic calculations were performed using GraphPad Prism 5 Software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Lingxiong Wang, Lei Zhu and Xia Chen for their expert technical assistance.
KMAB_1008352_suppl_table_and_figs.docx
Download MS Word (970 KB)Funding
This work was supported by grants from the Natural Science Foundation of China (81330061), Ministry of Science and Technology of China (973 projects 2010CB833605 and 863 projects 2011AA020114, 2014AA021004), State Key Project for New Drug Development (2013ZX09101021; 2013ZX09401303), Shanghai Commission of Science and Technology (Key Laboratory and Projects 13DZ1930100), and Shanghai Excellent technical leader (13XD1424000).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Klausz K, Berger S, Lammerts van Bueren JJ, Derer S, Lohse S, Dechant M, van de Winkel JG, Peipp M, Parren PW, Valerius T. Complement-mediated tumor-specific cell lysis by antibody combinations targeting epidermal growth factor receptor (EGFR) and its variant III (EGFRvIII). Cancer Sci 2011; 102:1761-8; PMID:21718386; http://dx.doi.org/10.1111/j.1349-7006.2011.02019.x
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008; 358:1160-74; PMID:18337605; http://dx.doi.org/10.1056/NEJMra0707704
- Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol 2008; 26:1742-51; PMID:18375904; http://dx.doi.org/10.1200/JCO.2007.12.1178
- Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol 2003; 21:2787-99; PMID:12860957; http://dx.doi.org/10.1200/JCO.2003.01.504
- You B, Chen EX. Anti-EGFR monoclonal antibodies for treatment of colorectal cancers: development of cetuximab and panitumumab. J Clin Pharmacol 2011; 52(2):128-55; PMID:21427284; 10.1177/0091270010395940
- Peipp M, Dechant M, Valerius T. Effector mechanisms of therapeutic antibodies against ErbB receptors. Curr Opin Immunol 2008; 20:436-43; PMID:18585454; http://dx.doi.org/10.1016/j.coi.2008.05.012
- Dechant M, Weisner W, Berger S, Peipp M, Beyer T, Schneider-Merck T, Lammerts van Bueren JJ, Bleeker WK, Parren PW, van de Winkel JG, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res 2008; 68:4998-5003; PMID:18593896; http://dx.doi.org/10.1158/0008-5472.CAN-07-6226
- Oppenheim DE, Spreafico R, Etuk A, Malone D, Amofah E, Pena-Murillo C, Murray T, McLaughlin L, Choi BS, Allan S, et al. Glyco-engineered anti-EGFR mAb elicits ADCC by NK cells from colorectal cancer patients irrespective of chemotherapy. Br J Cancer 2014; 110:1221-7; PMID:24496456; http://dx.doi.org/10.1038/bjc.2014.35
- Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S, Bleeker WK, Peipp M, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol 2010; 184:512-20; PMID:19949082; http://dx.doi.org/10.4049/jimmunol.0900847
- Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, Parren PW. Crosstalk between human IgG isotypes and murine effector cells. J Immunol 2012; 189:3430-8; PMID:22956577; http://dx.doi.org/10.4049/jimmunol.1200356
- Perez-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol 2005; 23:5235-46; PMID:16051966; http://dx.doi.org/10.1200/JCO.2005.00.6916
- Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, Schrumpf H, Boelke E, Ansari P, Mackenzie C, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med 2013; 5:199ra11; PMID:23966300; http://dx.doi.org/10.1126/scitranslmed.3005886
- Capdevila J, Elez E, Macarulla T, Ramos FJ, Ruiz-Echarri M, Tabernero J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat Rev 2009; 35:354-63; PMID:19269105; http://dx.doi.org/10.1016/j.ctrv.2009.02.001
- Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res 1999; 59:1236-43; PMID:10096554
- Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol 2007; 25:1134-43; PMID:17921999; http://dx.doi.org/10.1038/nbt1337
- Hoy SM, Wagstaff AJ. Panitumumab: in the treatment of metastatic colorectal cancer. Drugs 2006; 66:2005-14; discussion 15-6; PMID:17100412; http://dx.doi.org/10.2165/00003495-200666150-00011
- Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med 2012; 18:221-3; PMID:22270724; http://dx.doi.org/10.1038/nm.2609
- Voigt M, Braig F, Gothel M, Schulte A, Lamszus K, Bokemeyer C, Binder M. Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia 2012; 14:1023-31; PMID:23226096
- Kasper S, Breitenbuecher F, Reis H, Brandau S, Worm K, Kohler J, Paul A, Trarbach T, Schmid KW, Schuler M. Oncogenic RAS simultaneously protects against anti-EGFR antibody-dependent cellular cytotoxicity and EGFR signaling blockade. Oncogene 2012; 32(23):2873-81; PMID:22797062; http://dx.doi.org/10.1038/onc.2012.302
- Kubach J, Hubo M, Amendt C, Stroh C, Jonuleit H. IgG1 anti-epidermal growth factor receptor antibodies induce CD8-dependent antitumor activity. Int J Cancer 2014; 136(4):821-30; PMID:24947844; http://dx.doi.org/10.1002/ijc.29037
- Laux I, Jain A, Singh S, Agus DB. Epidermal growth factor receptor dimerization status determines skin toxicity to HER-kinase targeted therapies. Br J Cancer 2006; 94:85-92; PMID:16306877; http://dx.doi.org/10.1038/sj.bjc.6602875
- Rowinsky EK, Schwartz GH, Gollob JA, Thompson JA, Vogelzang NJ, Figlin R, Bukowski R, Haas N, Lockbaum P, Li YP, et al. Safety, pharmacokinetics, and activity of ABX-EGF, a fully human anti-epidermal growth factor receptor monoclonal antibody in patients with metastatic renal cell cancer. J Clin Oncol 2004; 22:3003-15; PMID:15210739; http://dx.doi.org/10.1200/JCO.2004.11.061
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995; 376:337-41; PMID:7630400; http://dx.doi.org/10.1038/376337a0
- Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med 2013; 5:207ra144; PMID:24132639; http://dx.doi.org/10.1126/scitranslmed.3006682
- Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, Hall SS, Ruoslahti E, Daugherty PS. Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J Control Release 2012; 161:804-12; PMID:22634092; http://dx.doi.org/10.1016/j.jconrel.2012.05.035
- Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem 2008; 283:16206-15; PMID:18339626; http://dx.doi.org/10.1074/jbc.M709988200
- Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, Moessner E, Freytag O, Friess T, Ries CH, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res 2013; 19:1126-38; PMID:23209031; http://dx.doi.org/10.1158/1078-0432.CCR-12-0989
- Chung S, Quarmby V, Gao X, Ying Y, Lin L, Reed C, Fong C, Lau W, Qiu ZJ, Shen A, et al. Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcgamma receptor binding and antibody-dependent cell-mediated cytotoxicity activities. MAbs 2012; 4:326-40; PMID:22531441; http://dx.doi.org/10.4161/mabs.19941
- Parekh BS, Berger E, Sibley S, Cahya S, Xiao L, LaCerte MA, Vaillancourt P, Wooden S, Gately D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs 2012; 4:310-8; PMID:22531445; http://dx.doi.org/10.4161/mabs.19873
- Tada M, Ishii-Watabe A, Suzuki T, Kawasaki N. Development of a cell-based assay measuring the activation of FcgammaRIIa for the characterization of therapeutic monoclonal antibodies. PLoS One 2014; 9:e95787; PMID:24752341; http://dx.doi.org/10.1371/journal.pone.0095787
- Ulisse S, Baldini E, Sorrenti S, D'Armiento M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets 2009; 9:32-71; PMID:19200050; http://dx.doi.org/10.2174/156800909787314002
- Gerspach J, Nemeth J, Munkel S, Wajant H, Pfizenmaier K. Target-selective activation of a TNF prodrug by urokinase-type plasminogen activator (uPA) mediated proteolytic processing at the cell surface. Cancer Immunol Immunother 2006; 55:1590-600; PMID:16636812; http://dx.doi.org/10.1007/s00262-006-0162-6
- Zuluaga MF, Sekkat N, Gabriel D, van den Bergh H, Lange N. Selective photodetection and photodynamic therapy for prostate cancer through targeting of proteolytic activity. Mol Cancer Ther 2013; 12:306-13; PMID:23270928; http://dx.doi.org/10.1158/1535-7163.MCT-12-0780
- Corti A, Nolli ML, Soffientini A, Cassani G. Purification and characterization of single-chain urokinase-type plasminogen activator (pro-urokinase) from human A431 cells. Thromb Haemost 1986; 56:219-24; PMID:3101222
- Stoppelli MP, Tacchetti C, Cubellis MV, Corti A, Hearing VJ, Cassani G, Appella E, Blasi F. Autocrine saturation of pro-urokinase receptors on human A431 cells. Cell 1986; 45:675-84; PMID:3011276; http://dx.doi.org/10.1016/0092-8674(86)90782-8
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:1626-34; PMID:18316791; http://dx.doi.org/10.1200/JCO.2007.14.7116
- Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006; 66:3992-5; PMID:16618717; http://dx.doi.org/10.1158/0008-5472.CAN-06-0191
- Saif MW, Kaley K, Chu E, Copur MS. Safety and efficacy of panitumumab therapy after progression with cetuximab: experience at two institutions. Clin Colorectal Cancer 2010; 9:315-8; PMID:21208847; http://dx.doi.org/10.3816/CCC.2010.n.046
- Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, Wakitani M, Ohta S, Satoh M, Shitara K, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res 2008; 68:3863-72; PMID:18483271; http://dx.doi.org/10.1158/0008-5472.CAN-07-6297
- Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, van Meerten T, Ebeling S, Vink T, Slootstra JW, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol 2006; 177:362-71; PMID:16785532; http://dx.doi.org/10.4049/jimmunol.177.1.362
- Mascia F, Lam G, Keith C, Garber C, Steinberg SM, Kohn E, Yuspa SH. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci Transl Med 2013; 5:199ra10; PMID:23966299; http://dx.doi.org/10.1126/scitranslmed.3005773
- Ramakrishnan MS, Eswaraiah A, Crombet T, Piedra P, Saurez G, Iyer H, Arvind AS. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. MAbs 2009; 1:41-8; PMID:20046573; http://dx.doi.org/10.4161/mabs.1.1.7509
- Allan DG. Nimotuzumab: evidence of clinical benefit without rash. Oncologist 2005; 10:760-1; PMID:16249358; http://dx.doi.org/10.1634/theoncologist.10-9-760
- Shin TH, Sung ES, Kim YJ, Kim KS, Kim SH, Kim SK, Lee YD, Kim YS. Enhancement of the tumor penetration of monoclonal antibody by fusion of a neuropilin-targeting peptide improves the antitumor efficacy. Mol Cancer Ther 2014; 13:651-61; PMID:24435448; http://dx.doi.org/10.1158/1535-7163.MCT-13-0748