
Abstract
The neonatal Fc receptor (FcRn) is expressed by cells of epithelial, endothelial and myeloid lineages and performs multiple roles in adaptive immunity. Characterizing the FcRn/IgG interaction is fundamental to designing therapeutic antibodies because IgGs with moderately increased binding affinities for FcRn exhibit superior serum half-lives and efficacy. It has been hypothesized that 2 FcRn molecules bind an IgG homodimer with disparate affinities, yet their affinity constants are inconsistent across the literature. Using surface plasmon resonance biosensor assays that eliminated confounding experimental artifacts, we present data supporting an alternate hypothesis: 2 FcRn molecules saturate an IgG homodimer with identical affinities at independent sites, consistent with the symmetrical arrangement of the FcRn/Fc complex observed in the crystal structure published by Burmeister et al. in 1994. We find that human FcRn binds human IgG1 with an equilibrium dissociation constant (KD) of 760 ± 60 nM (N = 14) at 25°C and pH 5.8, and shows less than 25% variation across the other human subtypes. Human IgG1 binds cynomolgus monkey FcRn with a 2-fold higher affinity than human FcRn, and binds both mouse and rat FcRn with a 10-fold higher affinity than human FcRn. FcRn/IgG interactions from multiple species show less than a 2-fold weaker affinity at 37°C than at 25°C and appear independent of an IgG's variable region. Our in vivo data in mouse and rat models demonstrate that both affinity and avidity influence an IgG's serum half-life, which should be considered when choosing animals, especially transgenic systems, as surrogates.
Abbreviations
| mAb | = | monoclonal antibody |
| FcRn | = | neonatal Fc receptor |
| rFcRn | = | rat FcRn |
| rIgG | = | rat IgG |
| SPR | = | surface plasmon resonance |
| hFcRn | = | human FcRn |
| hIgG | = | human IgG |
| CFCA | = | calibration-free concentration analysis |
| WT | = | wild-type |
| Rmax | = | maximum binding response |
| RU | = | response units |
| hErbB2 | = | human ErbB2 |
| mFcRn | = | mouse FcRn |
| pI | = | isoelectric point |
| cyFcRn | = | cynomolgus monkey FcRn |
| cyIgG | = | cynomolgus monkey IgG |
| anti-Id | = | anti-idiotypic |
Introduction
Advances in hybridoma methods, display technologies, and protein engineering enable the rapid production of monoclonal antibodies (mAbs) with desirable affinity and specificity for their targeted antigens, generating a demand for therapeutics that exhibit superior biophysical properties such as increased exposure. The central importance of the neonatal Fc receptor (FcRn) in IgG homeostasis has been reviewed elsewhereCitation1 and therapeutic IgGs with moderately enhanced affinity for FcRn have been shown to exhibit extended serum half-lives and efficacy.Citation2 The FcRn/IgG interaction is exquisitely pH dependent, a property believed to endow IgG molecules with a longer serum half-life than other proteins of similar size. Due to the formation of the FcRn/IgG complex at acidic pH (<pH 6.5), which serves to rescue an IgG from lysosomal degradation, followed by its dissociation at neutral pH (or higher) in the blood, FcRn mediates the efficient recycling of an IgG back to the circulation. It has been shown that FcRn/IgG binding affinity is linearly correlated with pH,Citation3 such that an engineered IgG with enhanced affinity at acidic pH exhibits a concomitant affinity increase at neutral pH. Designing therapeutic antibodies with extended serum exposure therefore presents substantial challenges in fine-tuning an IgG's interaction with FcRn to exhibit appropriate affinities at both acidic and neutral pH values to allow an optimum balance between lysosomal rescue and efficient release at neutral pH.
The binding mechanism of the FcRn/IgG interaction has been debated due, in part, to a crystal structure for the complex that revealed a repeating arrangement of rat FcRn (rFcRn) dimers bridging rat IgG2a (rIgG2a) Fc homodimers.Citation4 This suggested the possibility of either a 1:1 or 2:1 FcRn/IgG binding stoichiometry, and both hypotheses were supported by conflicting gel filtration data, since FcRn/Fc complexes studied by gel filtration under non-equilibriumCitation5,6 or equilibrium conditionsCitation7-9 showed apparent binding stoichiometries of 1:1 or 2:1, respectively. Two distinct 2:1 FcRn:Fc complexes were observed in rFcRn/rIgG2a Fc crystals: (1) an asymmetrical arrangement in which a dimer of FcRn molecules interacts with only one side of an Fc (FcRn:FcRn:Fc) and (2) a symmetrical arrangement in which an Fc homodimer is sandwiched between 2 FcRn molecules (FcRn:Fc:FcRn). Data from surface plasmon resonance (SPR) biosensors have been used to support a hypothesis that FcRn dimerization (as inferred from the asymmetrical FcRn/Fc complex) is required for high-affinity binding of IgG,Citation10 yet no FcRn dimers were observed in the recently determined crystal structure of human FcRn (hFcRn) when complexed with a high affinity mutant of human IgG1 (hIgG1) FcCitation11 or when the rFcRn/rIgG2a complex was studied by in vitro column binding assays,Citation7,12 consistent with the symmetrical FcRn/Fc complex.
To characterize the binding affinity and stoichiometry of the rFcRn/rIgG2a interaction, recombinant rIgG2a Fc heterodimer and homodimer fragments bearing one or 2 functional FcRn-binding sites, respectively, were produced previously and analyzed by SPR.Citation12
The authors reported discordant affinity values when the interaction of rFcRn with a monovalent rIgG2a Fc heterodimer was studied via amine-coupling in opposing assay orientations on a Biacore CM5 sensor chip. They reported an apparent equilibrium dissociation constant (KD) of 87 nM when flowing rIgG2a Fc heterodimer over immobilized rFcRn, whereas flowing rFcRn over immobilized rIgG2a Fc heterodimer gave a KD of 480 nM. Furthermore, rIgG2a Fc homodimer flowed over immobilized rFcRn resulted in heterogeneous binding responses that deviated from a simple model and could only be fit to more complex binding models. One of these models, the bivalent analyte model, calculated affinity constants of KD1 = 79 nM and KD2 = 430 nM, which appeared to recapitulate those stated above. The authors concluded that an IgG offers 2 non-equivalent binding sites for FcRn, namely a “high affinity” site, exposed only when rFcRn was immobilized (signifying FcRn dimers on the cell membrane), and a “low affinity” site. Acceptance of this hypothesis has resulted in studies that report divergent affinity constantsCitation9,13-24 and confound engineering efforts. The view that an IgG homodimer supports 2 non-identical binding processes hinders efforts to correlate an IgG's FcRn-binding affinity with its in vivo clearance, and the lack of agreement in the affinity constants across the literature makes it difficult to evaluate new therapeutic mAbs when reporting data to the FDA.
We analyzed the FcRn/Fc interaction utilizing advances in biosensor instrumentation, chip design, and software, and here present data supporting an alternate hypothesis, that 2 FcRn molecules saturate an IgG homodimer with identical affinities at independent sites, consistent with the symmetrical arrangement of the FcRn/Fc complex observed in the crystal structures.Citation4,11 We show that the discordant affinity values obtained when the FcRn/Fc interaction is studied by SPR biosensors in opposing assay orientations can be attributed to immobilization artifacts. We also demonstrate that the complex binding behavior observed when Fc homodimer is flowed over immobilized FcRn can be eliminated by removing avidity artifacts, and does not imply the existence of 2 non-equivalent binding sites.Citation25 Indeed, complex binding is also observed in avidity-prone assay orientations for any bivalent analyte that presents 2 independent binding sites with identical affinities. This study applies current SPR biosensor methodology, including various chip types, the use of gentle immobilization methods, and the routine determination of each analyte's active concentration toward a specific immobilized partner under diffusion-limited (or mass transport-limited) conditions using Biacore's calibration-free concentration analysis (CFCA). The data show that the FcRn/IgG interaction can be studied in opposing assay orientations under conditions that minimize surface artifacts, and yield affinity constants that are in close agreement with solution measurements.
Since native FcRn is expressed on the cell surface, we explored the biological role of both affinity and avidity in influencing an IgG's in vivo clearance. By dosing mice and rats with a panel of human IgG1 homodimers and their corresponding monovalent heterodimers engineered to contain only one FcRn-functional binding site, we show that avidity can extend an IgG's half-life, but increasing an IgG's affinity for FcRn beyond a certain threshold value can result in faster clearance, presumably by impairing release of IgG at neutral pH.
Results
Two rat FcRn molecules saturate a rat IgG2a homodimer with identical affinities at independent binding sites
It is generally accepted that the affinity of the FcRn/IgG interaction varies both across species and IgG subtype,Citation26-28 but their affinity constants are inconsistent in the literature. No crystal structure has been determined for human FcRn (hFcRn) complexed with wild-type (WT) human IgG, but one was reported recently for a mutant form of hIgG1 Fc (M252Y+S254T+T256E) with enhanced affinity for hFcRn.Citation11 The relatively high affinity of the rFcRn/rIgG2a interaction has facilitated its use as a surrogate model system and so, to provide historical context, we used it to explore the sources of common artifacts in biosensor experiments that result in inconsistent affinity reports.
To demonstrate that flowing an IgG homodimer over immobilized FcRn can give complex binding kinetics due to avidity artifacts, we chose to employ low and high immobilization levels on a Biacore C1 chip, as the planar nature of the C1 chip minimizes avidity effects compared to sensor chips with flexible hydrogels such as Biacore CM5 or CM4 chips. When rIgG2a was flowed over high capacities of immobilized FcRn, the binding responses were markedly heterogeneous and were poorly-described by a simple 1:1 Langmuir binding model, as shown by the large and systematically deviated residuals (). In contrast, flowing rIgG2a over sparse capacities of immobilized FcRn removed avidity artifacts, yielding homogeneous binding responses that were well-described by a simple model, as shown by the low and randomly scattered residuals (). Reversing the assay orientation by flowing rFcRn over rIgG2a that was captured via its specific antigen coated on a neutravidin (ProteOn NLC) chip () not only gave homogeneous binding responses that fit very well to a simple model, but the apparent KD value was identical, within the error of the replicates, to that obtained using the opposite assay orientation with low FcRn immobilization levels. Furthermore, the maximum binding response (Rmax value) of 51 RU (response units) that was obtained experimentally using 92 RU of antigen-captured rIgG2a matched the theoretical value expected for 2 rFcRn molecules saturating a single rIgG2a homodimer, assuming molecular masses of 41.4 kDa and 150 kDa respectively, which further confirms a 2:1 FcRn/IgG binding stoichiometry ().
Figure 1. Affinity determination of the rFcRn/rIgG2a interaction at pH 5.8 using various assay orientations. Kinetic analysis of rIgG2a flowed over biotinylated rFcRn that was captured at (A) high or (B) low capacities via amine-coupled neutravidin on a Biacore C1 chip; kinetic analysis of rFcRn flowed over (C) antigen-captured rIgG2a on a ProteOn NLC chip or (D) amine-coupled rIgG2a on a Biacore CM5 chip; and (E) solution affinity using a high capacity of amine-coupled rIgG2a on a Biacore CM5 chip to probe for free rFcRn in equilibrated mixtures with titrating levels of rIgG2a. Analytes were injected as a 3-fold dilution series with top at 500 nM (A and B) or as both a 3-fold (green) and 5-fold (red) dilution series with top at 1000 nM (C) or 3000 nM (D); the 3000 nM curves have been excluded from D. Panel E shows the titration of 17 nM rFcRn with 2 unrelated mAbs of subtype rIgG2a (distinguished by the solid or open symbols). All samples were analyzed in replicate binding cycles. Each panel shows an example data set (N of 1) where the measured data (colored lines) were fit globally to a simple model (black lines). The KD values are the mean ± SD for N independent measurements. See Table 1.
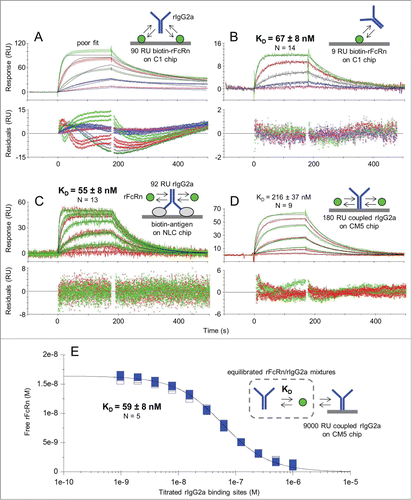
To address whether an IgG's variable domain sequence affects its binding interaction with FcRn, which has been a point of debate in the literature,Citation22,24 we studied a panel of 5 unrelated mAbs of subtype rIgG2a using the assay format shown in . We obtained indistinguishable KD values across the panel, regardless of whether the IgGs were antigen-captured or biotinylated and captured directly on the chip, suggesting that an IgG's variable region does not influence its interaction with FcRn (see ). In contrast, when we flowed rFcRn as analyte over amine-coupled rIgG2a on a Biacore CM5 chip (), the apparent affinity was 4-fold weaker (due mainly to a slower association rate constant, ka), the binding responses were slightly heterogeneous, but could still be fit fairly well to a simple model, and the Rmax values were significantly lower than dictated by the immobilized capacity. These observations suggested that the FcRn-binding activity of rIgG2a was altered upon amine-coupling to a CM5 chip, consistent with the coupling-dependent affinity loss reported when rFcRn was flowed over amine-coupled rIgG2a Fc homodimers on Biacore CM5 chips.Citation12
Table 1. Affinity determination of the rFcRn/rIgG2a interaction at pH 5.8 using 3 different assay formats on SPR platforms. Parameter values represent the mean ± SD of N independent measurements on 5 unrelated mAbs. The range of KD values obtained is also shown in parenthesis. ND = not determined
To bypass the need to immobilize either binding partner, a solution affinity was determined for the rFcRn/rIgG2a interaction using Biacore (). In calculating the concentrations of each binding partner used to prepare the samples for these experiments, we determined their active concentrations (in molecules) using a CFCA assay, which does not rely on a standard compound, and then worked in terms of binding sites by assuming one per rFcRn molecule and 2 per rIgG2a homodimer, e.g., 1 g/l rIgG2a was equivalent to 13.3 μM binding sites in these assays. The titration data fit very well to a simple bimolecular binding equation that assumes a single KD value. The solution affinity was in good agreement with that obtained from low capacity FcRn surfaces () and antigen-captured IgG surfaces (), which shows that when kinetic experiments are performed on SPR biosensors under conditions that minimize immobilization artifacts, they can yield KD values that are comparable to solution values (). Throughout this work, CFCA experiments were used to determine the “active” or “effective” concentration of all analytes and these values were used in calculating their kinetic and affinity constants (see Methods).
When hIgG1 homodimers are analyzed under conditions that dilute out the avidity of a bivalent analyte, their binding kinetics recapitulate those of the corresponding monovalent heterodimers
To test the generality of our findings described above for interactions that are more relevant to the development of therapeutic mAbs, we studied a multi-species panel of FcRn proteins binding to a panel of hIgG1 variants. We selected anti-human ErbB2 (hErbB2) trastuzumab as a model compound and used bispecific antibody technologyCitation29 to produce monovalent hIgG1 heterodimers that contain only one functional FcRn-binding site (see the Antibodies section in the Methods). Our strategy paired a wild-type “R” arm with an “E” arm that contained an additional triple mutation (I253A+H310A+H435A = AAA) known to disable FcRn-bindingCitation30 to yield a high purity AAA:WT hIgG1 “E:R” heterodimer (Fig. S1A). To demonstrate that our results were consistent across FcRn/IgG interactions spanning a range of affinities, we also paired the “E” arm containing the AAA mutations with an “R” arm containing either the N434H or the N434Y mutations that have been shown to bind FcRn with higher affinity than that of hIgG1-WT.Citation3 All of these hIgG1 heterodimers behaved as monovalent analytes when flowed over medium capacity FcRn-coated chips as judged by their homogenous binding responses (Fig. S1B and Table S1), whereas their corresponding homodimers yielded heterogeneous binding responses, as expected for bivalent analytes (data not shown). By transferring the assay from ProteOn's GLM chip to Biacore's C1 chip, we diluted out avidity artifacts and revealed that the hIgG1 homodimers bound to sparsely immobilized FcRn with homogeneous responses (Fig. S2A) that were kinetically indistinguishable from those of their corresponding monovalent heterodimers (Fig. S2B).
Sandwiching experiments show that an IgG homodimer offers 2 independent and equal affinity binding sites for FcRn
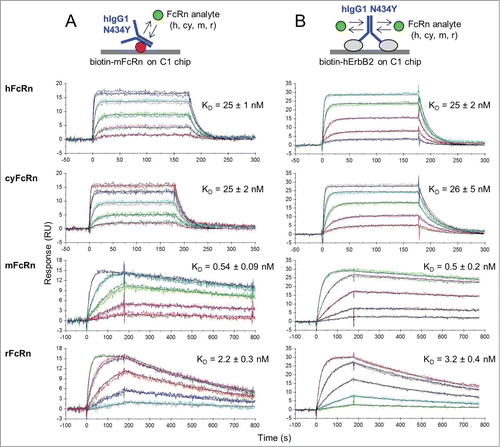
The properties of the second FcRn-binding site on an IgG homodimer were further confirmed to be identical to the first binding site by flowing FcRn as analyte over hIgG1-N434Y homodimer that was first captured via immobilized mouse FcRn (mFcRn). We chose the high affinity hIgG1-N434Y/mFcRn interaction to provide a stable IgG surface and used a Biacore C1 chip to favor IgG capture in a mostly monovalent manner. Others have employed a similar strategy to probe the second functional binding site of growth hormone ligand.Citation31 When a multi-species panel of FcRn analytes was analyzed over hIgG1-N434Y that was tethered via immobilized mFcRn (), their binding kinetics were identical to those obtained on hErbB2-captured hIgG1-N434Y (). Thus, using mFcRn to occupy one site on an IgG homodimer did not perturb the binding kinetics of the IgG's second site, both of which have identical binding kinetics.
Figure 2. Kinetic analysis at pH 5.8 of a multi-species panel of FcRn proteins binding as analytes to trastuzumab hIgG1-N434Y that was first captured via (A) biotinylated mFcRn or (B) biotinylated hErbB2 on Biacore C1 chips to which neutravidin was amine-coupled. Analytes were injected in duplicate binding cycles as a 3-fold dilution series with top at 180, 300, 60, or 100 nM for hFcRn, cyFcRn, mFcRn, and rFcRn respectively. Each overlay plot shows a representative example of the measured data (colored lines) and the global fit to a simple model (black lines) for a typical experiment (N of 1) and the KD values are the mean ± SD of N = 6.
IgG heterodimers and homodimers bind FcRn with identical affinities but differ in binding stoichiometry
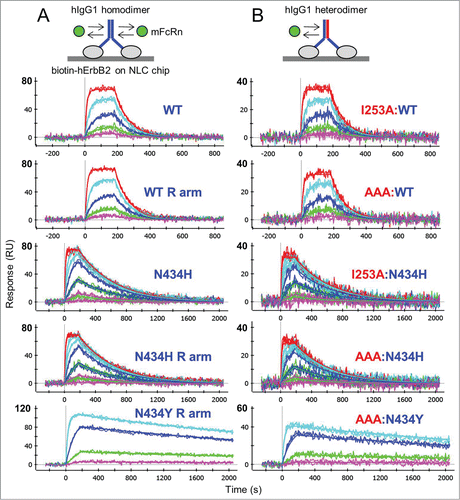
For a direct comparison of monovalent IgG heterodimers (AAA:WT, AAA:N434H, and AAA:N434Y) with their corresponding IgG homodimers (WT, N434H, and N434Y), we captured them at similar levels on chips coated with hErbB2 and flowed FcRn over them. We show that mFcRn bound the hIgG1 homodimers () with kinetics identical to those of the corresponding heterodimers (). However, the homodimer surfaces were saturated at 2-fold higher experimental Rmax values, a measure that is directly proportional to the mass of analyte required for saturation. Table S2 summarizes the global analysis of all interactions tested in this way, which included the above-mentioned constructs in addition to other homodimers (e.g., WT:WT and parental E and R arms) and heterodimers (e.g., I253A:WT and I253A:N434H) prepared by bispecific antibody technology. Consistent with the kinetic analysis of rFcRn binding to antigen-captured rIgG2a (), the observed Rmax values for hErbB2-captured trastuzumab variants reached their theoretical values, assuming that 2 FcRn molecules saturate an IgG homodimer and one FcRn molecule saturates an IgG heterodimer. While others have shown that soluble FcRn binds immobilized Fc homodimers and heterodimers with very similar KD values,Citation12 here we show that IgG homodimers offer twice the number of binding sites as their corresponding IgG heterodimers, as judged by their relative Rmax values. This implies that an IgG homodimer offers 2 equal affinity and independent binding sites for FcRn.
Figure 3. One-shot kinetic analysis at pH 5.8 of mFcRn binding as analyte to (A) trastuzumab homodimers and (B) their corresponding heterodimers that were captured at similar levels via ProteOn NLC or GLC chips coated with biotinylated hErbB2. Mouse FcRn was injected as a 3-fold or 5-fold dilution series with top concentrations of 180 nM (WT and N434H) or 85 nM (N434Y). Each overlay plot shows a representative example of the measured data (noisy lines) and the global fit (smooth lines) for a subset of the constructs tested. Multiple constructs per variants were fit simultaneously to give global apparent mean KD ± SD (N = 4) values of 58 ± 10 nM (WT-hIgG1, WT:WT, WT E arm, WT R arm, I253A:WT and AAA:WT), 5 ± 1 nM (N434H, N434H R arm, I253A:N434H and AAA:N434H), and 0.47 ± 0.07 nM (N434Y, N434Y R arm, AAA:N434Y). For a summary of all interactions tested in this way, see Supplemental Material (Table S2).
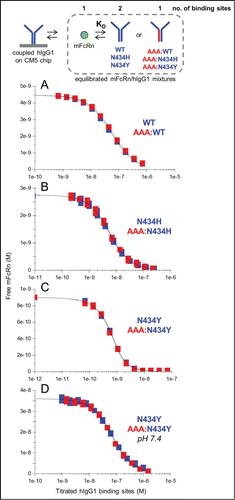
The affinities deduced from the kinetic measurements described above were corroborated by performing a series of solution affinity experiments with a subset of these IgGs. shows examples of the overlapping KD-controlled titration curves obtained when comparing the interactions of mFcRn with hIgG1 homodimers (blue curves) and their corresponding heterodimers (red curves), assuming 2 binding sites per IgG homodimer, one per IgG heterodimer, and one per FcRn. summarizes the affinities determined for the interactions of mFcRn and rFcRn with hIgG1-WT, N434H, and N434Y when studied in 3 complementary assay orientations.
Table 2. Summary of the apparent affinities determined in 3 different assay formats for the interactions of mFcRn and rFcRn with trastuzumab variants. Reported KD values (nM) are the mean (± SD) of N independent measurements. The IgG kinetic data are a subset of those shown in Table S1, where hIgG1 heterodimers were flowed as monovalent analytes over biotinylated FcRn on ProteOn GLM sensor chips. The FcRn kinetic data are a subset of those shown in Table S2, where FcRn was flowed as analyte over hErbB2-captured trastuzumab on ProteOn NLC or GLC sensor chips
Figure 4. Solution affinity determinations of mFcRn binding to trastuzumab hIgG1 variants. (A) WT and AAA:WT at pH 5.8, (B) N434H and AAA:N434H at pH 5.8, (C) N434Y and AAA:N434Y at pH 5.8 and (D) N434Y and AAA:N434Y at pH 7.4. Each overlay plot shows an example of the KD-controlled curves obtained for titrations of a hIgG1 homodimer (blue symbols) and its corresponding heterodimer (red symbols) into a fixed concentration of mFcRn (4.5, 2.7, 0.9, and 36 nM for panels A-D respectively). See .
IgG variable region does not influence affinity for FcRn
It has been suggested that the variable region of an IgG can influence its interaction with FcRn.Citation22 However, in the current work, the affinity of the rFcRn/rIgG2a interaction was independent of the rIgG2a's variable region (). To investigate whether this held true for other FcRn/IgG interactions, we performed a kinetic analysis of both mFcRn and rFcRn on a panel of hIgG1 and hIgG2 molecules, including hIgG2Δa,Citation32 by arraying them onto 36 individual spots of a ProteOn chip. An NLC chip was coated with 6 different antigens and used to capture 11 different recombinant mAbs representative of both hIgG1 and hIgG2 subtypes. A GLC chip was amine-coupled with a larger, more diverse panel of IgGs comprised of 23 unique recombinant mAbs specific for 14 unrelated antigens, 2 polyclonal antibodies, and a hIgG2Δa Fc stump (with a molecular mass of 50 kDa) lacking a variable region. Irrespective of the IgG immobilization method used, there was no correlation between an IgG's variable region and its affinity for FcRn. This is reinforced in Fig. S3 which shows rFcRn binding with similar kinetics to intact hIgG molecules (of subtypes 1, 2, and 2Δa) and to a hIgG2Δa Fc stump; a similar result was observed for mFcRn (data not shown). While amine-coupling provided a universal method for immobilizing a diverse panel of IgGs, it yielded apparent affinities that were slightly weaker than (within 2-fold of) their antigen-captured or solution affinity counterparts.
Immobilization method and chip type influence kinetic analyses of FcRn on IgG surfaces
To investigate the effect of immobilization method on the apparent affinities of FcRn binding to IgG surfaces, we performed a series of kinetic experiments by Biacore using various FcRn/IgG pairs chosen to span a range of KD values.Citation3 Table S3 shows that the immobilization method mattered, since biotinylating an IgG recapitulated the affinity observed via antigen-capture, whereas amine-coupling an IgG resulted in a 2- to 3-fold weaker affinity, consistent with the data acquired by ProteOn. In addition to immobilization chemistry, we observed that chip type influenced affinity measurements on both instruments. Within a given assay format, such as FcRn kinetics on antigen-captured IgGs, the apparent affinities determined on ProteOn's NLC, GLC, and GLM chips and Biacore's C1 chip agreed more closely with solution measurements than those obtained on Biacore's SA, CM4, or CM5 chips, which yielded weaker affinities driven by slower apparent association rates in these chip types, as noted in the literature for other interaction systems.Citation33,34 Taken together, we observed that, amine-coupling an IgG on a Biacore CM4 or CM5 chip perturbed the affinity the most (toward weaker affinity) from that determined in solution.
FcRn/IgG binding exhibits affinity differences across species and IgG subtypes
To generate a data set that was relevant to in vitro cell-based experiments and in vivo studies, we determined the affinities of a multi-species panel of FcRn interacting with a multi-species and multi-subtype panel of IgGs at both 25°C and 37°C. summarizes the results from a series of kinetic experiments where FcRn was flowed over antigen-captured or biotinylated IgGs on ProteOn's NLC sensor chips; this assay format was chosen due to its speed and excellent agreement with solution affinity values for these interacting pairs (). Our data show that, for species-matched FcRn/IgG interactions, hFcRn barely discriminated across hIgG subtypes, whereas both rFcRn and mFcRn exhibited a more significant subtype-dependency since rFcRn bound rIgG2a with a 10-fold higher affinity than rIgG2c, and mFcRn bound mIgG3 with a 10-fold higher affinity than mIgG1. The FcRn/IgG interactions showed only a 2-fold weaker affinity at 37°C compared with 25°C.
Table 3. Affinities for a multi-species panel of FcRn/IgG interactions determined at pH 5.8 at both 25°C and 37°C. These data were generated in a series of one-shot kinetic experiments by flowing FcRn over antigen-captured, anti-Id Fab-captured, or biotinylated IgGs on ProteOn NLC chips. The KD values (nM) represent the mean (± SD) of N independent measurements. The hIgG1 category includes hIgG1 Fc stump (no variable region) and the hIgG2 category includes hIgG2Δa. NB = no binding (or barely binds) at 6 μM. NT = not tested
Longer IgG serum half-life is modulated by 2 independent and equal affinity FcRn binding sites
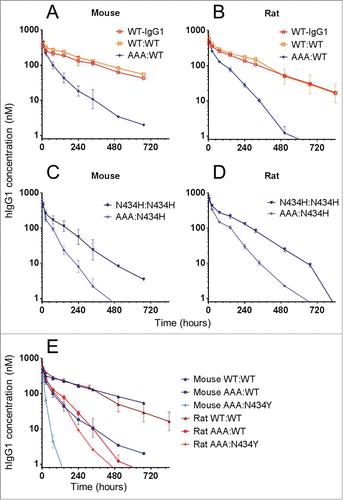
To determine whether our biosensor-based affinity determinations on recombinant FcRn were biologically relevant, we determined the clearance of a panel of trastuzumab variants in mice and rats (). Our in vivo data revealed the importance of avidity in extending an IgG's serum half-life, as monovalent IgG heterodimers AAA:WT () and AAA:N434H () cleared significantly faster than their corresponding homodimers. Since receptor density influences avidity, some animal models, especially transgenic ones that often express non-native FcRn levels, may function as poor surrogates.Citation35 The data also illustrate the influence of affinity, with monovalent IgG heterodimer AAA:N434Y clearing significantly faster in mice than rats (). This is consistent with its 10-fold higher affinity for mFcRn than rFcRn (), resulting in poorer release at neutral pH, and thus less efficient recycling, in mice than rats. These results also revealed that, while hIgG1-WT binds mFcRn and rFcRn with indistinguishable affinities, introduction of a single point mutation, e.g., N434Y, can create significant affinity-discrimination that manifests as different biological outcomes in mice and rats. Therefore, care should be taken to consider both affinity and avidity factors when choosing animal models.
Figure 5. In vivo clearance of a panel of trastuzumab hIgG1 homodimers and heterodimers in mice and rats. Overlay plot of WT-hIgG1, WT:WT and AAA:WT in (A) mice and (B) rats. Overlay plot of N434H:N434H and AAA:N434H in (C) mice and (D) rats. (E) Overlay plot of WT:WT, AAA:WT, and AAA:N434Y in mice and rats. Data points represent the mean ± SD of 3 or 4 animals per group.
Discussion
The success of therapeutic antibodies is based on their exquisite specificity and long half-life. A better understanding of the latter is therefore essential for guiding the rational design of superior therapeutics. We report a refined understanding of the binding mechanism of FcRn/IgG interactions through the use of multiple orthogonal binding assays using SPR biosensors. The work described herein supports the conclusion that there are 2 equal affinity and independent FcRn-binding sites per IgG. We also show that experimental artifacts can result in discrepancies for the FcRn/IgG affinity values, which may explain disparate results previously reported in the literature. The experimental techniques applied herein establish reliable in vitro assays for measuring the apparent KD of an FcRn/IgG interaction, which is essential to any endeavor to find a correlation between an IgG's affinity for FcRn and its in vivo clearance.
Our characterization of a panel of trastuzumab hIgG1 variants produced as both homodimers and heterodimers provides compelling evidence that 2 FcRn molecules saturate an IgG with identical affinities at independent sites, consistent with the symmetrical arrangement observed in the crystal structure of the FcRn/Fc complexes.Citation4,11 First, we demonstrated that the 2 binding sites were independent of one another in a sandwiching experiment, where we found that the binding kinetics of human, cynomolgus monkey, mouse, and rat FcRn analytes toward hIgG1-N434Y homodimer that was tethered via one of its FcRn-binding sites using mFcRn-coated chips () were indistinguishable from those obtained on hIgG1-N434Y homodimer that was captured via the IgG's variable domains to hErbB2-coated chips, where both FcRn-binding sites were exposed (). Thus, occupying one FcRn-binding site of an IgG homodimer did not perturb the kinetics of the other FcRn-binding site. Second, we showed that an IgG homodimer offers 2 equal affinity and independent binding sites for FcRn because trastuzumab homodimers and heterodimers that were captured at very similar levels via hErbB2-coated chips gave identical FcRn-binding kinetics, but the homodimer surfaces required twice the number of FcRn molecules than their corresponding heterodimers to achieve saturation, as judged by their relative Rmax values (). Third, our solution affinity measurements showed that FcRn's binding to an hIgG1 homodimer was well-described by a single KD value that was identical to that of the corresponding monovalent heterodimer, with the homodimer again offering twice the number of binding sites ().
Although there have been conflicting reports regarding the effect of an IgG's variable domain on FcRn binding interactions,Citation22,24 we found no correlation between the KD of an FcRn/IgG interaction and an IgG's variable region. Furthermore, we did not observe any trend between an IgG's isoelectric point (pI) and its affinity for FcRn, when studying hIgG molecules with pI values ranging from 6.31 to 9.11, consistent with a study that proposed that an IgG's pI may affect its serum half-life, but via an FcRn-independent mechanism.Citation36
Our studies demonstrate how an interplay of experimental parameters, such as assay orientation, avidity factors, conjugation/capture method and chip planarity affect the quality of biosensor data and the apparent affinity values determined for FcRn/IgG interactions. Sub-optimal assay design combined with the acceptance of an earlier hypothesis of disparate FcRn-binding sites on an IgG, has led to an inconsistent set of FcRn/IgG affinity values reported in the literature. Differences in pharmacokinetic behaviors of different antibodies have often been attributed to these inconsistent values, resulting in conflicting conclusions. Based on our hypothesis of 2 independent and equal affinity FcRn-binding sites per IgG, we present in a detailed set of FcRn/IgG affinities for the interactions of multiple species that can be used for future measurement comparison.
The difference in the half-life between hIgG1 heterodimers (presenting one functional FcRn-binding site) versus their corresponding hIgG1 homodimers (presenting 2 functional FcRn –binding sites) clearly demonstrate that avidity plays an important role in prolonging the in vivo serum half-life of wild-type IgGs. The strict pH-dependence of the FcRn/IgG interaction presents substantial challenges for engineering a therapeutic IgG with enhanced affinity at acidic pH to promote its binding in the lysosome, where FcRn serves to rescue an IgG from degradation, and naturally weaker binding at neutral pH to enable its release back into circulation. As of yet, it remains elusive to accomplish both these tasks simultaneously because an increase in affinity at acidic pH is usually coupled to an increase at neutral pH.Citation3
In addition to affinity, our work also illustrates the importance of avidity associated with an IgG's interaction with FcRn, which can be used to manipulate the pharmacokinetics of antibody therapeutics. Modeling the pharmacokinetics of hIgGs in human using transgenic animals is complicated by the need to replicate the correct expression levels of hFcRn in humans. In addition, in vitro transcytosis assays are often performed with hFcRn transfected into dog cells,Citation37 which do not reflect FcRn expression levels in humans. Therefore, both affinity and avidity effects should be considered when engineering therapeutic antibodies to have increased half-lives.
Materials and Methods
General biosensor methods
Biacore T200, Biacore 2000, sensors chips (CM5, CM4, C1, and SA), and amine-coupling reagents (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and 1.0 M ethanolamine-HCl pH 8.5) were purchased from GE Healthcare Life Sciences. A ProteOn-XPR36 biosensor and sensor chips (GLC, GLM, and NLC) were purchased from BioRad Inc. and sulfo-NHS (for activating ProteOn chips) was purchased from Thermo Scientific Pierce, Inc. Standard amine-coupling was used to prepare high capacity reaction surfaces when performing calibration-free concentration analysis (CFCA), solution affinity experiments, and coating chips with neutravidin (catalog number PI-31000, Thermo Scientific Pierce, Inc.). All interaction analysis was performed at 25°C (or 37°C, where noted) at a flow rate of 30 μl/min in a running buffer of 10 mM sodium phosphate pH 5.8, 150 mM NaCl supplemented with 0.05% Tween-20 (PBST+ pH 5.8) for Biacore experiments, or 0.01% Tween-20 (PBST pH 5.8) for ProteOn experiments, unless stated otherwise. All surfaces were prepared in PBST+ pH 7.4 (Biacore) or PBST pH 7.4 (ProteOn) running buffers.
Detailed methods are provided in the supplementary material. All kinetic analyses were performed on low surface capacities of a specific binding partner that was immobilized under gentle conditions. Thus, streptavidin- or neutravidin-coated chips were used to capture biotinylated FcRn and IgG to provide reaction surfaces for measuring the binding kinetics of IgG and FcRn analytes, respectively. Alternatively, IgGs were captured via a biotinylated form of their specific antigen. Appropriate reference surfaces were prepared, lacking the specific immobilized partner. Biacore CM5 chips were used when high capacity surfaces were needed (see above) and Biacore C1 chips were used to dilute out the avidity of a bivalent analyte and for sandwiching experiments. Analytes were prepared by buffer-exchanging them into the running buffer using spin columns and their “active” or “effective” concentrations determined under conditions of mass transport limitation using a CFCA method on a Biacore T200, as described elsewhere.Citation38 CFCA experiments confirmed that all analytes used herein were >70% active relative to their nominal or total protein concentration as determined by light absorbance at 280 nm with use of an appropriate extinction coefficient. All kinetic and affinity calculations used active analyte concentrations, rather than nominal ones, as input values. Antibody concentrations are reported in binding sites, assuming 2 per IgG homodimer and one per IgG heterodimer. Analytes were prepared as a 3-fold or 5-fold dilution series and injected in duplicate or triplicate binding cycles using no regeneration or the mildest possible regeneration (PBS pH 7.4 or 100 mM Tris pH 8.0). When using the ProteOn, analytes were injected in a “one-shot kinetic mode” over 6 different binding partners immobilized along parallel ligand channelsCitation39 or in a “kinetic titration mode” over a 36-ligand array.Citation33 All samples were injected in duplicate binding cycles and experiments were repeated on independent chips. Kinetic data were double-referencedCitation25 and fit globally to a simple 1:1 Langmuir binding model with 3 floating parameters (ka, kd and Rmax). Biacore 2000 and ProteOn data were processed and analyzed in BIAevaluation v.4.1.1 and ProteOn Manager software v.3.1.0.6, respectively.
Solution affinity experiments were performed on a Biacore 2000 equipped with CM5 chips onto which a high capacity of an IgG was amine-coupled to compete out the binding of the titrated IgG. Samples were prepared by titrating an IgG as a 12-membered 2-fold dilution series into FcRn fixed at 2 different concentrations, typically 10-fold apart, allowing the samples to reach equilibrium and then injecting them over the chip in duplicate binding cycles. Samples of FcRn alone, as a 2-fold or 3-fold dilution series, were used to provide a standard curve. The binding responses obtained from solution affinity experiments were double-referenced, converted to a “free” FcRn concentration using the standard curve and fit to a “solution affinity” model in the BIAevaluation v.4.1.1 software or were analyzed in Sapidyne's KinExA Pro software v.3.7.9 using the N-curve tool, by inputting the concentration of the fixed binding partner (FcRn) and floating the concentration of the titrated partner (IgG), as described elsewhere.Citation38
FcRn proteins
Purified recombinant FcRn proteins were prepared in-house by transient expression in Expi293 or HEK293F cell lines. Two plasmids were generated per FcRn construct, one encoded the soluble extracellular domain of FcRn and the other encoded a species-matched beta2-microglobulin (β2m). The accession number (and amino acids that were expressed) were: NP_001129491 (24–290) for hFcRn and NP_004039 (21–119) for human β2m; NP_001271480 (24–290) for cynomolgus monkey FcRn (cyFcRn) and NP_001271618 (21–119) for cynomolgus monkey β2m; NP_034319 (22–290) for mFcRn and NP_033865 (21–119) for mouse β2m; and NP_203502 (23–291) for rFcRn and NP_036644 (21–119) for rat β2m. Purification of the FcRn/β2m heterodimer (herein referred to as “FcRn”) was performed by affinity chromatography using a human IgG column, as described elsewhere.Citation40
Antibodies
A panel of unrelated mAbs of subtype rIgG2a were purchased from R&D systems Inc. (catalog numbers MAB2365, MAB006, and MAB3388) and BioLegend Inc. (catalog number 400501). Purified polyclonal human IgG1-κ, IgG2-κ, and IgG3-κ from myeloma plasma were purchased from Sigma-Aldrich Corp. (catalog numbers I5154, I5404, and I5654). Human IgG1 heterodimers were generated by our bispecific antibody technology,Citation29 using trastuzumab as a model compound. Briefly, an “E” arm of trastuzumab carrying D221E, P228E, and L368E mutations was exchanged with an “R” arm of trastuzumab carrying D221R, P228R, and K409R mutations under reducing condition to generate “E:R” heterodimers, which were purified using ion exchange chromatography. Namely, WT, I253A and I253A+H310A+H435A (AAA) variants of the E arm and WT, N434H, and N434Y of the R arm were generated for the various bispecific exchanges.
A panel of recombinant mAbs against bovine herpes virus with the same variable region was produced in-house by PCR-based amplification of IgG heavy chain and light chain variable domain cDNA reverse transcribed from hybridoma CRL-1853 (ATCC) RNA, an anti-bovine herpes virus (anti-BHV) mAb. The IgG heavy and light chain genes were cloned into a CMV-based expression vector, and the anti-BHV mAb was recombinantly produced as the following subtypes; human IgG1 and IgG2Δa, cynomolgus monkey IgG1 and IgG2, rhesus monkey IgG1 and IgG2, mouse IgG1, IgG2a, IgG2b, and IgG3, and rat IgG1, IgG2a, IgG2b, and IgG2c. All recombinant mAbs were produced in-house by transient transfection of HEK293F cells followed by protein A (or protein G for rat IgG2a and rat IgG2c antibodies) purification. Recombinant human IgG2Δa Fc stump (50 kDa homodimer) was expressed in-house in HEK293F cells and purified on a MabSelect SuRe column. Recombinant human IgG1 Fc stump was purchased from R&D systems (catalog number 110-HG).
Antigens
Recombinant purified antigens were either prepared in-house or purchased from Sino Biological, Creative Biomart or R&D systems. To provide a high affinity and specific binding partner for our panel of anti-BHV mAbs, an anti-idiotypic (anti-Id) mouse mAb (10H11, mouse IgG1κ) was raised in-house by standard hybridoma technology using the anti-BHV Fab fragment as immunogen. Purified anti-Id 10H11 Fab was prepared by papain-digestion of the parental mAb.
Biotinylations were performed using EZ-link NHS-LC-LC-biotin (catalog number 21343, Thermo Scientific Pierce, Inc.) using an equimolar ratio of linker:protein for IgGs and FcRn (when immobilizing them for kinetic analyses) and a 1:1 to 5:1 molar ratio of linker:protein for antigens (when using them to coat chips to enable the oriented capture of IgGs for kinetic analyses).
In vivo measurements
Animal studies were conducted in accordance with approved protocols by the Institutional Animal Care and Use Committee at Pfizer Inc. Mouse and rat sera were analyzed by ELISA as described previouslyCitation29 using purified recombinant hErbB2-Fc fusion protein (catalog number 1129-ER, R&D Systems) to coat plates, trastuzumab hIgG1 WT as the standard and biotinylated anti-human κ mouse mAb (catalog number 9230–08, Southern Biotech, Inc.) followed by streptavidin-horseradish peroxidase (catalog number 21130, Thermo Scientific Pierce, Inc.) as the 2-step secondary detection.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
KMAB_I_7_02_Supplemental_material.docx
Download MS Word (1.1 MB)Acknowledgments
We thank Thomas J Van Blarcom and Donna Stone for critical reading of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/10.1038/nri2155
- Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW, Lencer WI, Pierce GF, Blumberg RS. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol 2013; doi:10.3109/07388551.2013.834293; PMID:24156398
- Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol 2009; 182:7663-71; PMID:19494290; http://dx.doi.org/10.4049/jimmunol.0804182
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994; 372:379-83; PMID:7969498; http://dx.doi.org/10.1038/372379a0
- Popov S, Hubbard JG, Kim J, Ober B, Ghetie V, Ward ES. The stoichiometry and affinity of the interaction of murine Fc fragments with the MHC class I-related receptor, FcRn. Mol Immunol 1996; 33:521-30; PMID:8700168; http://dx.doi.org/10.1016/0161-5890(96)00004-1
- Schuck P, Radu CG, Ward ES. Sedimentation equilibrium analysis of recombinant mouse FcRn with murine IgG1. Mol Immunol 1999; 36:1117-25; PMID:10698313; http://dx.doi.org/10.1016/S0161-5890(99)00093-0
- Huber AH, Kelley RF, Gastinel LN, Bjorkman PJ. Crystallization and stoichiometry of binding of a complex between a rat intestinal Fc receptor and Fc. J Mol Biol 1993; 230:1077-83; PMID:8478919; http://dx.doi.org/10.1006/jmbi.1993.1220
- Sánchez LM, Penny DM, Bjorkman PJ. Stoichiometry of the interaction between the major histocompatibility complex-related Fc receptor and its Fc ligand. Biochemistry 1999; 38:9471-76; PMID:10413524; http://dx.doi.org/10.1021/bi9907330
- West AP Jr, Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor. Biochemistry 2000; 39:9698-08; PMID:10933786; http://dx.doi.org/10.1021/bi000749m
- Raghavan M, Wang Y, Bjorkman PJ. Effects of receptor dimerization on the interaction between the class I major histocompatibility complex-related Fc receptor and IgG. Proc Natl Acad Sci USA 1995; 92:11200-204; PMID:7479965; http://dx.doi.org/10.1073/pnas.92.24.11200
- Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem 2014; 289:7812-24; PMID:24469444; http://dx.doi.org/10.1074/jbc.M113.537563
- Martin WL, Bjorkman PJ. Characterization of the 2:1 complex between the class I MHC-related Fc receptor and its Fc ligand in solution. Biochemistry 1999; 38:12639-47; PMID:10504233; http://dx.doi.org/10.1021/bi9913505
- Bitonti AJ, Dumont JA, Low SC, Peters RT, Kropp KE, Palombella VJ, Stattel JM, Lu Y, Tan CA, Song JJ et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc Natl Acad Sci USA 2004; 101:9763-68; PMID:15210944; http://dx.doi.org/10.1073/pnas.0403235101
- Kamei DT, Lao BJ, Ricci MS, Deshpande R, Xu H, Tidor B, Lauffenburger DA. Quantitative methods for developing Fc mutants with extended half-lives. Biotechnol Bioeng 2005; 92:748-60; PMID:16136591; http://dx.doi.org/10.1002/bit.20624
- Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 2006; 43:1462-73; PMID:16139891; http://dx.doi.org/10.1016/j.molimm.2005.07.032
- Kacskovics I, Kis Z, Mayer B, West AP Jr, Tiangco NE, Tilahun M, Cervenak L, Bjorkman PJ, Goldsby RA, Szenci O, et al. FcRn mediates elongated serum half-life of human IgG in cattle. Int Immunol 2006; 18:525-36; PMID:16481343; http://dx.doi.org/10.1093/intimm/dxh393
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos 2007; 35:86-94; PMID:17050651; http://dx.doi.org/10.1124/dmd.106.011734
- Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem 2010; 285:4826-36; PMID:20018855; http://dx.doi.org/10.1074/jbc.M109.081828
- Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil FP, Lowman HB, Fielder PJ, Prabhu S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{alpha} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos 2010; 38:600-05; PMID:20071453; http://dx.doi.org/10.1124/dmd.109.031310
- Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol 2010; 184:1968-76; PMID:20083659; http://dx.doi.org/10.4049/jimmunol.0903296
- Magistrelli G, Malinge P, Anceriz N, Desmurs M, Venet S, Calloud S, Daubeuf B, Kosco-Vilbois M, Fischer N. Robust recombinant FcRn production in mammalian cells enabling oriented immobilization for IgG binding studies. J Immunol Methods 2012; 375:20-9; PMID:21939661; http://dx.doi.org/10.1016/j.jim.2011.09.002
- Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011; 39:1469-77; PMID:21610128; http://dx.doi.org/10.1124/dmd.111.039453
- Andersen JT, Foss S, Kenanova VE, Olafsen T, Leikfoss IS, Roopenian DC, Wu AM, Sandlie I. Anti-carcinoembryonic antigen single-chain variable fragment antibody variants bind mouse and human neonatal Fc receptor with different affinities that reveal distinct cross-species differences in serum half-life. J Biol Chem 2012; 287:22927-37; PMID:22570488; http://dx.doi.org/10.1074/jbc.M112.355131
- Neuber T, Frese K, Jaehrling J, Jäger S, Daubert D, Felderer K, Linnemann M, Höhne A, Kaden S, Kölln J, et al Characterization and screening of IgG binding to the neonatal Fc receptor. mAbs 2014; 6:928-42; PMID:24802048; http://dx.doi.org/10.4161/mabs.28744
- Myszka DG. Improving biosensor analysis. J Mol Recognit 1999; 12:279-84; PMID:10556875; http://dx.doi.org/10.1002/(SICI)1099-1352(199909/10)12:5%3c279::AID-JMR473%3e3.0.CO;2-3
- Jakoi ER, Cambier J, Saslow S. Transepithelial transport of maternal antibody: purification of IgG receptor from newborn rat intestine. J Immunol 1985; 135:3360-64; PMID:2931482
- Hobbs SM, Jackson LE, Peppard JV. Binding of subclasses of rat immunoglobulin G to detergent-isolated Fc receptor from neonatal rat intestine. J Biol Chem 1987; 262:8041-46; PMID:3597360
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 2001; 13:1551-59; PMID:11717196; http://dx.doi.org/10.1093/intimm/13.12.1551
- Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol 2012; 420:204-19; PMID:22543237; http://dx.doi.org/10.1016/j.jmb.2012.04.020
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001; 276:6591-604; PMID:11096108; http://dx.doi.org/10.1074/jbc.M009483200
- Walsh ST, Jevitts LM, Sylvester JE, Kossiakoff AA. Site2 binding energetics of the regulatory step of growth hormone-induced receptor homodimerization. Protein Sci 2003; 12:1960-70; PMID:12930995; http://dx.doi.org/10.1110/ps.03133903
- Armour KL, Clark MR, Hadley AG, Williamson LM. Recombinant human IgG molecules lacking Fcgamma receptor binding and monocyte triggering activities. Eur J Immunol 1999; 29:2613-24; PMID:10458776; http://dx.doi.org/10.1002/(SICI)1521-4141(199908)29:08%3c2613::AID-IMMU2613%3e3.0.CO;2-J
- Abdiche YN, Lindquist KC, Pinkerton A, Pons J, Rajpal A. Expanding the ProteOn XPR36 biosensor into a 36-ligand array expedites protein interaction analysis. Anal Biochem 2011; 411:139-51; PMID:21168382; http://dx.doi.org/10.1016/j.ab.2010.12.020
- Drake AW, Tang ML, Papalia GA, Landes G, Haak-Frendscho M, Klakamp SL. Biacore surface matrix effects on the binding kinetics and affinity of an antigen/antibody complex. Anal Biochem 2012; 429:58-69; PMID:22766435; http://dx.doi.org/10.1016/j.ab.2012.06.024
- Roopenian DC, Christianson GJ, Sproule TJ. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol 2010; 602:93-104; PMID:20012394; http://dx.doi.org/10.1007/978-1-60761-058-8_6
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385-92; PMID:20159773; http://dx.doi.org/10.1093/protein/gzq009
- Tesar DB, Tiangco NE, Bjorkman PJ. Ligand valency affects transcytosis, recycling and intracellular trafficking mediated by the neonatal Fc receptor. Traffic 2006; 7:1127-2; PMID:17004319; http://dx.doi.org/10.1111/j.1600-0854.2006.00457.x
- Bee C, Abdiche YN, Pons J, Rajpal A. Determining the binding affinity of therapeutic monoclonal antibodies towards their native unpurified antigens in human serum. PLoS One 2013; 8(11):e80501; PMID:24223227; http://dx.doi.org/10.1371/journal.pone.0080501
- Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG. See comment in PubMed Commons belowExploring "one-shot" kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal Biochem 2006; 358:281-88; PMID:16962556; http://dx.doi.org/10.1016/j.ab.2006.08.005
- Gastinel LN, Simister NE, Bjorkman PJ. Expression and crystallization of a soluble and functional form of an Fc receptor related to class I histocompatibility molecules. Proc Natl Acad Sci USA 1992; 89:638-42; PMID:1530991; http://dx.doi.org/10.1073/pnas.89.2.638