
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The application of monoclonal antibodies as commercial therapeutics poses substantial demands on stability and properties of an antibody. Therapeutic molecules that exhibit favorable properties increase the success rate in development. However, it is not yet fully understood how the protein sequences of an antibody translates into favorable in vitro molecule properties. In this work, computational design strategies based on heuristic sequence analysis were used to systematically modify an antibody that exhibited a tendency to precipitation in vitro. The resulting series of closely related antibodies showed improved stability as assessed by biophysical methods and long-term stability experiments. As a notable observation, expression levels also improved in comparison with the wild-type candidate. The methods employed to optimize the protein sequences, as well as the biophysical data used to determine the effect on stability under conditions commonly used in the formulation of therapeutic proteins, are described. Together, the experimental and computational data led to consistent conclusions regarding the effect of the introduced mutations. Our approach exemplifies how computational methods can be used to guide antibody optimization for increased stability.
Abbreviations
| mAb | = | monoclonal antibody |
| CMC | = | chemistry, manufacturing and control |
| PK | = | pharmacokinetics |
| DSC | = | differential scanning calorimetry |
| RALS | = | right angle light scattering |
| WT | = | wild-type |
| HC | = | heavy chain |
| LC | = | light chain |
Introduction
Although monoclonal antibodies (mAbs) have gained a substantial share of the pharmaceutical market and the number of mAbs in clinical trials is ever increasing,Citation1 the path from bench science to the final drug product is often hindered by substantial roadblocks arising from unfavorable, but intrinsic, molecular properties. For example, low expression levels, high aggregation rates and challenging formulation conditions can lead to high costs of goods, potentially delayed entry into clinical trials and, consequently, delayed entry into market, or project termination in the worst case.Citation2-4 To avoid such setbacks, risk mitigation strategies increasingly center around "Quality-by-Design" (QbD) approaches, which consider developability-related issues early in the drug discovery phase and involve rationally engineering antibodies toward favorable Chemistry, Manufacturing and Control (CMC) and pharmacokinetics (PK) properties.Citation5-7 Choosing molecules that exhibit low intrinsic immunogenicity and that are robust against the formation of immunogenic degradation products can also reduce safety risks. However, the large number of different antibodies typically considered in early projects, and the small amount of each antibody that is typically available, precludes extensive experimental characterization and determination of CMC properties such as yield, chemical and physical stability. Therefore, attention is increasingly being paid to the development and application of computational methods aiming at developability predictions solely based on amino acid sequence information.
The key prerequisite for a computationally assisted QbD approach is a sound understanding of how primary sequences translate into antibodies with favorable properties, or vice versa, which sequence patterns are likely to compromise successful development. Antibodies are very complex molecules that are susceptible to degradation under many conditions that occur in vitro during development, commercial-scale manufacturing and finally as a therapeutic drug.Citation8 Thus, a favored antibody candidate is robust toward degradation under challenging process conditions such as pH-shifts, interfacial stress, high protein concentrations and temperature changes. Antibody degradation pathways are complex and multifaceted. Chemical modifications can occur through oxidation of solvent-exposed cysteine, methionine and tryptophan residues, cleavage of disulfide bonds, proteolysis, β-elimination, deamidation of asparagine, and isomerization of aspartic acid.Citation9 All of these modifications can compromise efficacy if the respective residues are engaged in target binding. They also can give rise to reduced thermodynamic stability (i.e., conformational stability) or increased immunogenicity of the modified antibody.
A second major degradation pathway is protein aggregation.Citation8 From a macroscopic view, aggregation manifests as particles with high molecular weight, and ultimately precipitation is observed. However, antibody aggregation should be regarded as an umbrella term for a whole family of different degradation pathways that lead to the formation of large soluble particles of different structures that eventually precipitate irreversibly. Several studies have shown that the formation of intermolecular cross-beta-sheets and amyloidogenic substructures is a crucial step for some aggregation pathways.Citation8,10 This mechanism requires a preceding, at least partial, unfolding step of the protein to allow exposure and alignment of the reactive sequence parts. Protein self-association or aggregate growth is then thought to follow a mechanism similar to polymerization, but this process is reversible up to a certain degree. A second mechanism that is thought to limit developability depends on intermolecular interactions compromising the colloidal stability of proteins. Here, in contrast to cross-beta-sheet dependent aggregates, antibodies associate out of their natively folded conformation either through hydrophobic interactions or patches of locally accumulated charged amino acids on their surface.Citation11-13 These interactions can also affect the conformational equilibrium between folded and partially unfolded states such that association out of the folded state in combination with ever-present transient unfolding increases the likelihood for the formation of cross-beta-sheet aggregates. In addition, transiently formed associated proteins that retain their native conformation can give rise to an increase in the apparent particle size resulting in an exponential increase of viscosity at high protein concentrations.Citation14 As viscosity can be a critical parameter in downstream processing, as well as in fill and finish operations and drug delivery, viscosity is a developability attribute.Citation11
In summary, one can say that none of the above described aggregation pathways occurs isolated and exclusively in a solution of a certain antibody. Rather, they are intertwined with many possible paths eventually leading to insoluble aggregates. With the current understanding of antibody degradation and with the experimental and computational tools available, a thorough description or even prediction of the degradation pathway for a given antibody is not possible.Citation15 However, from the knowledge accumulated to date, a number of paradigms have emerged to mitigate the risk of antibody development: 1) the number of reactive sites (oxidation sites, deamidation sites, sites susceptible to proteolysis) should be minimal; 2) the thermodynamic stability should be high such that the fraction of unfolded protein is small; 3) the structure should not contain hydrophobic or charged patches on the surface; and 4) the sequence should not contain cross-beta-sheet aggregation hotspots.
Each of these 4 points is an important surrogate parameter for the prediction of shelf-life. More importantly, each of these properties (except for the thermodynamic stability) can be assessed computationally based on the protein sequence or a homology model of the structure. Deamidation rates depend on the local sequence and the conformational flexibility of substructures, whereas oxidation rates primarily depend on solvent accessibility. Both flexibility and solvent accessibility can be computed from molecular dynamics simulations or computationally less demanding methods that generate estimates of the conformational flexibility.Citation16-18 The calculation of absolute thermostability from a given sequence or structure is not possible, but changes in stability of mutants with respect to a wild-type (WT) reference can be predicted with reasonable accuracy.Citation19-21 Hydrophobic patches and inhomogeneous charge distributions can also be calculated.Citation12,22 A number of different software packages have been developed to predict cross-beta-sheet aggregation-propensities based on the sequence.Citation23-28
There is no single computational method that is able to estimate antibody developability and shelf-life, but a number of established methods, developed for different purposes, can be utilized to assemble important aspects of rate-limiting steps of antibody degradation pathways and provide valuable estimations for antibody developability. Computational methods pinpoint potential liabilities to distinct sequence patterns, paving the way for rational engineering toward improved developability.
Here, we describe a systematic application of the above mentioned paradigms to an antibody with nanomolar affinity to its target that was generated in a research project. The antibody showed particularly poor CMC properties, which precluded further development. Based solely on the sequence of the variable domains, different computational techniques were applied to generate hypotheses about the most likely origins of the undesired properties. Through a small number of targeted mutations, these hypotheses were tested experimentally, resulting in the production of antibodies that completely retained biological function, but also had substantially improved CMC properties.
Results
Generated using classical hybridoma technology, the antibody used in this study, mAb1, targets the extracellular domain of the human oxidized low-density lipoprotein receptor 1 (Ox-LDL receptor 1), also known as lectin-type oxidized LDL receptor 1 (LOX-1). MAb1 was expressed as a chimeric antibody comprising mouse variable and human constant regions. It binds with nanomolar affinity to LOX-1 as determined by a kinetic exclusion assay (KinExa). However, mAb1 showed extremely unfavorable properties, which eliminated it from further profiling and development. The antibody was constructed in 2 Fc versions (Fig. 1): a G1m17 allotype EEM version (named mAb1_WT) and a hinge LL->AA version (named mAb1*_WT).Citation29 Expression levels of both Fc versions in a stably transfected recombinant Chinese hamster ovary (CHO) cell line was extremely low, with a titer below 1 mg/l. Hence, the poor expression was independent of the constant region. For mAb1*_WT, it was possible to obtain a few mg of purified protein for biophysical characterization. Even so, the stability of this mAb was particularly poor and visible precipitation of a sub mg/ml solution was observed after a few hours at room temperature. Compendial quality and stability assays were therefore not performed.
Design of mutants
The integrated computational antibody analysis platform developed at Boehringer Ingelheim, which comprises sequence and structure-based analysis tools, was applied to mAb1. Since only the Fv region of the sequence/structure is analyzed, the results obtained for mAb1_WT and mAb1*_WT, which differ only in the constant region, are identical in these analyses.
The analyses revealed no chemical degradation sites like deamidation or aspartate isomerization sites. Statistical analysis of the complementarity-determining region hydrophobicity and charge furthermore showed that these properties are close to the average of the distributions found in known antibody sequences. This also translates into the 3-dimensional structure, where no hydrophobic patches or charge spots could be identified. Likewise, the search for aggregation-prone sequence stretches using cross beta-sheet aggregation prediction with TANGO revealed no highly aggregation-prone regions.Citation23
Subsequently, we applied a statistical sequence analysis method described in the literature.Citation30 In brief, the antibody sequence was evaluated by scoring functions developed from the entire pool of antibody sequences available in public databases. This allows for a statistical assessment of each individual position in the sequence. For each position in the framework, the algorithm computes a statistical score based on intrinsic and conditional probabilities that reflects the likelihood to find a particular amino acid at a certain position in the context of the entire sequence. The results of these calculations are shown in . Low scores, visualized by blue bars, indicate that the amino acid in this position and in the context of the entire sequence is very common. High scores, visualized by red bars, denote an unusual amino acid in this position. Here the sequence of the mAb1 variable region reveals several high-scoring positions in both the heavy and light chains. Closer examination of the high-scoring positions in the light chain (LC) reveals deviations from the underlying germline sequence at positions that are usually quite conserved: Ile107 (Kabat numbering) located at the interface to the constant light domain CL is a conserved lysine (96% conservation in murine kappa-sequence). Phe75 points into the hydrophobic core of the VL domain and is an isoleucine in 99% of all murine kappa chains. Gln43 in frame 2, located at the interface between heavy and light chain is also very uncommon (71% Ser, 12% Pro and 10% Thr dominate this position). Furthermore, Gly18 and Ile20 were identified as unusual in the LC of mAb1.
Figure 1. Schematic of the antibody constructs used in this study. The sequences of the heavy chain and light chain variable regions for wild-type and variants is given on the upper part of the figure. The lower part illustrates the combination of the corresponding heavy and light chain variants in the antibodies. The wild-type protein was constructed bearing 2 different Fc parts; heavy chain Fc of G1m17 allotype EEM (white boxes) or a heavy chain hinge LL->AA version (striped boxes).Citation29 The variants with engineered variable regions were constructed as G1m17 allotype EEM Fc version. Engineered positions in the variable regions are highlighted by arrows.
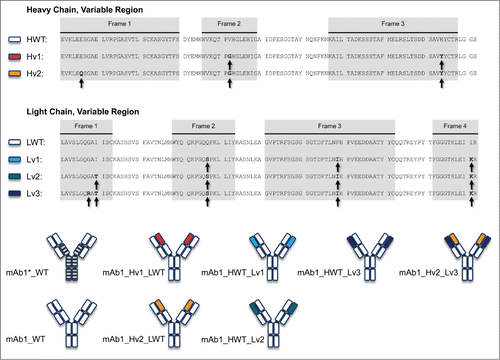
Figure 2. Statistical Sequence Analysis of the heavy and light chain variable regions of mAb1. Negative sequence scores (blue bars) indicate that the amino acid found in this positions is commonly observed in the context of the rest of the sequence. Positive values (red bars) indicate that this sequence pattern is unusual. Both chains contain framework residues with high statistical scores which were chosen as sites for engineering.
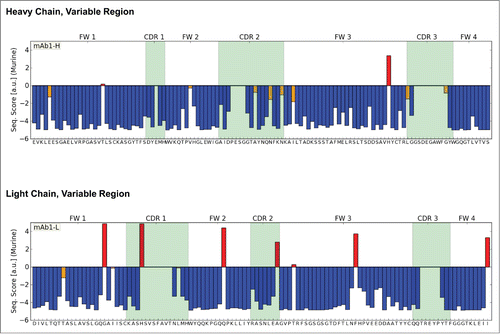
Examination of the heavy chain (HC) of mAb1 also reveals positions with high statistical score, e.g., His90. Although this variant occurs in several murine germlines, it is extremely rare in the phenotype of murine HCs. At this position, a tyrosine is found in more than 96% of all sequences. A second position with noticeable statistical score was Val42, located in frame 2. This position is usually occupied by glycine (74%) or glutamic acid (19%) in murine HCs. As a third position, we identified position 5 in frame 1, which is occupied by glutamic acid in mAb1 and where mutation to glutamine was recommended by the algorithm. However, by mistake, position 6 (also glutamic acid) instead of 5 was mutated to glutamine in one variant of the heavy chain. Since both glutamic acid and glutamine occur with comparable frequency at this position in murine HCs, we did not expect this mutation to negatively affect CMC properties.
In order to scrutinize the effect of each mutation as closely as possible, and to study the effect of a larger set of mutations, we designed constructs in which one of the 2 chains remained the wild-type while positions in the second chain were mutated gradually to the computationally suggested amino acid. As we identified 5 positions in the LC and 3 in the HC that were of interest, exploration of all combinations and large-scale production of all possible variants would have been time-consuming and expensive. We thus chose combinations that we expected to result in distinct effects on CMC properties. Following this paradigm, we designed 2 variants of the HC and 3 of the LC and expressed 6 different combinations of heavy and light chains: 2 variants with modified HCs (2 and 3 mutations) and WT LC; 3 variants of LCs (3, 4 and 5 mutations) with WT HC; and 1 variant with both chains modified (3 mutations in the HC, 5 in the LC). The sequences for each chain are shown in . Mutations were chosen based on the score and the degree of conservation at the respective sequence position. Therefore, we decided to start with at least 3 mutations in the LC at positions that show a high degree of conservation.
The variants were constructed bearing the G1m17 EEM Fc part. All designed variants expressed with substantially higher titers (50–150 mg/l) compared to the WT versions mAb1_WT and mAb1*_WT (titer <<1 mg/l for both; ), and could be purified and subjected to a diverse array of analytical methods.
Table 1. Antibody expression levels as noted during expression trials or recombinant production of the protein material used in this study
Biophysical characterization
To gain a deeper understanding of the unfavorable CMC properties of the WT protein (e.g., remarkably low titer and precipitation tendency) on a molecular level, biophysical characterization of mAb1*_WT was performed. shows the differential scanning calorimetry (DSC) thermogram for the protein unfolding. Upon temperature ramping, the first endothermic transition occurs with a TM of 68 °C. In a complementary experiment, the protein unfolding was also monitored via the signal change in tryptophan fluorescence.Citation31 In this experiment, a transition midpoint of 61.5°C was identified. The F350/F330 ratio of tryptophan fluorescence at 23 °C was 0.75.
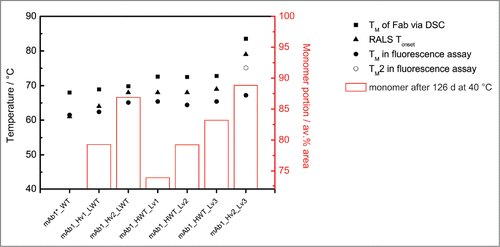
Figure 3. (A) DSC thermograms and (B) RALS curves for wild-type antibody mAb1*_WT and variants. (A) The major endothermic transition can be assigned to the unfolding of the Fab region of the antibodies.Citation32,33 The position of this peak is listed in : Notice a drastic shift of this peak when comparing the wild-type protein with the variant with engineered heavy and light chain mAb1_Hv2_Lv3. (B) Upon temperature ramping the light scattering of the sample is recorded, increasing scattering is commonly attributed to the formation of aggregates and protein precipitate. The corresponding onset temperatures are listed in . While the wild-type starts aggregating at 61 °C, the onset for the variant with engineered heavy and light chain mAb1_Hv2_Lv3 is at 79 °C.
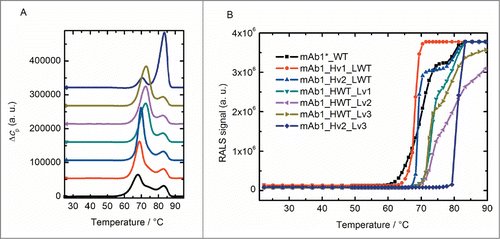
The same biophysical characterization was performed for the antibody variants. also summarizes the DSC thermograms for these proteins. For the mutants with an engineered HC, mAb1_Hv1_LWT and mAb1_Hv2_LWT, the first endothermic transitions take place with a TM of 68.9 °C and 69.8 °C, respectively. The mutants with an engineered LC, mAb1_HWT_Lv1, mAb1_HWT_Lv2 and mAb1_HWT_Lv3, exhibit a first unfolding event with a TM of 72.6 °C, 72.5 °C and 72.8 °C, respectively. In the mutant with engineered HC and LC (mAb1_Hv2_Lv3), the first endotherm is observed at 70.5 °C. In contrast to all other variants, this first event exhibits a markedly lower calorimetric enthalpy (ΔHcal, the peak area under the Δcp vs. T curve). The major endothermic transition is observed at 83.5 °C. As previously described, the DSC peak with the largest peak area can be assigned to the unfolding of the Fab part of antibodies.Citation32,33 Therefore, we concluded from the data shown in that the TM for the antibody Fab unfolding increases about 16 K after removal of predicted liabilities in HC plus LC (from 68 °C for WT to 83.5 °C for mAb1_Hv2_Lv3).
When unfolding was monitored via the change in tryptophan fluorescence, the same trend was observed. The corresponding unfolding transition temperatures for all mutants are summarized in . For mAb1_Hv2_Lv3, 2 unfolding events could be resolved. Due the nature of the experiment, we cannot assign certain structural elements to these 2 events. The DSC data, however, indicate that the second transition (TM of 75.1 °C) reflects the unfolding of the Fab part of the protein. In summary, the thermal (i.e., conformational) stability of mAb1 as monitored by 2 independent assays is increased in the engineered mutants. As discussed below, the engineering of both HC and LC actually provokes an increase in stability that may reflect a cooperativity effect.
Table 2. Biophysical characteristics of mAb1 wild-type and variants
The F350/F330 ratios of tryptophan fluorescence for all mAb1 variants at 23 °C are also shown in . In general, a higher ratio is indicative of the tryptophan residues becoming exposed to a more polar environment.Citation34,35 Based on our experience, full-length mAbs exhibit F350/F330 values from ∼0.6 to ∼1. For mAb1, the ratio ranges from 0.75 for mAb1*_WT to 0.69 for mAb1_Hv2_Lv3, and thus falls within a narrow range compared to the variability between various full-length mAbs. This observation indicates that the local environment of the tryptophan residues, and therefore the overall 3-dimensional fold of all mAb1 variants, are similar. The slight decrease in F350/F330 ratio with increasing degree of engineering might be due to a more compact structure in which some tryptophan residues have less access to the polar solvent. It should be noted that no tryptophan residues have been replaced or introduced during the design of the variants.
In a next step, we tested for heat-induced aggregation of the WT protein and the engineered variants. summarizes the change in the light scattering signal (at 90 °, right angle light scattering, RALS) upon heating for the corresponding protein solutions. The onset temperature for the steep increase in light scattering is included in . While mAb1*_WT starts to aggregate at 61 °C, successive engineering of the protein increases its Tonset value up to 79 °C for mAb1_Hv2_Lv3 (ΔT = 19 K). Comparing these data with the DSC data, we find the RALS Tonset correlates with the Fab unfolding temperature. This observation indicates the unfolded Fab is the part of the antibody that triggers aggregation onset here.
Long-term stability
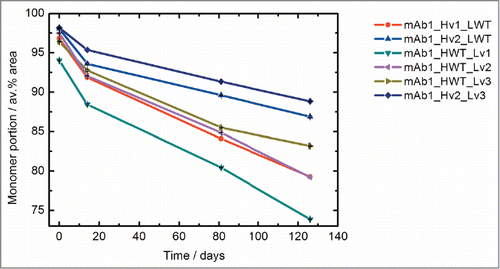
The stability of the engineered variants was compared under conditions that are relevant to the biopharmaceutical development process. For that, the aggregation tendency of the 10 mg/ml protein solutions at accelerated conditions (40 °C) was monitored via size-exclusion chromatography (SEC) analysis. depicts the monomer content of the samples over time. After 40 °C storage for 126 d the monomer content decreased for all variants. For mAb1_Hv2_Lv3, the monomer content decreased from the initial value of 98% to 89% after storage. The mutants with the lowest degree of engineering showed the largest absolute monomer loss (for mAb1_HWT_Lv1 from 94% to 74% and for mAb1_Hv1_LWT from 97% to 79% monomer). As the protein preparations contained different amounts of impurities (e.g., aggregates) already at the beginning of the experiment, we hesitate to derive an absolute stability ranking of the antibody variants from this experiment. Nevertheless, the data illustrate that stepwise sequence engineering improves the 40 °C stability of mAb1. The variant comprising both engineered HC and LC retains the highest monomer content over the course of the experiment.
Figure 4. Long-term stability of the mAb1 variants. The monomer content of the samples was assessed via size exclusion chromatography at different timepoints after storage at 40 °C. Error bars (smaller than the symbols) indicate the range for the duplicate SEC analytical results. The time-dependent decrease of monomer content is smallest for the variant with engineered heavy and light chain mAb1_Hv2_Lv3.
Antigen binding
To test whether the engineering affects the antigen binding capabilities of the antibody, we performed affinity measurements with WT protein and all variants. As listed in , all variants bind the antigen with a KD of around 1 nM, demonstrating the engineering preserves this key function of the antibody.
Table 3. Antigen binding affinities (dissociation constants KD) of mAb1 wild-type and variants as determined in surface plasmon resonance experiments
Protein stability calculations
Structure determination of mAb1_HWT_Lv3 enabled us to employ structure-based computational methods to study the effects of the introduced mutations on an atomistic level. Based on the locations of the respective amino acids, 2 mechanisms were identified by which the stability of the structure could be compromised. Those amino acids that point into the core of the immunoglobulin domains could destabilize its fold, whereas others located at the interface between heavy and light chain could perturb the stability of the interface. In order to address all possible mechanisms, we constructed thermodynamic cycles from which the relative free energy differences were computed using molecular dynamics-based alchemical free energy calculations that allow for an interpretation of the effect of every mutation on the thermodynamic stability. Results are shown in . denotes the change in the free energy of folding caused by a single mutation in the Fab, whereas
is the free energy change in the individual heavy or light chain in the absence of the second chain.
is the change in binding free energy between heavy and light chain upon mutation. The computational approach allows determination of the effects of mutations on the stability of individual domains and the interaction between them, which cannot be done in an experiment. Negative values for the different
denote that the stability of the individual chain (or the interaction between the 2 chains) has increased as a consequence of the mutation. Except for the mutation I107K in the LC, all introduced mutations are predicted to increase the stability of the individual chain, the Fab, or both. In particular H:H90Y and L:F75I give rise to substantial stability gains of several kcal/mol compared to WT. Interestingly, the mutation E6Q, which was not introduced deliberately, also contributes favorably to protein stability, in line with our findings in the DSC experiments. The largest increase for
is predicted for L:Q43S. The amino acid is located at the interface between heavy and light chain. While the effect on the stability of the isolated LC is marginal, the mutation causes a substantial stabilization of the interaction between heavy and light chain.
Table 4. Results of protein stability calculations
Discussion
As our data show, a limited number of targeted mutations can be sufficient to fundamentally transform the stability and developability of an antibody with highly unfavorable CMC properties. Moreover, the antibody variants designed for this study retained target-binding abilities, clearly showing that those parts of the sequence that caused the undesired properties did not evolve as functional constraints during affinity maturation. They are rather the consequence of random mutations in in vivo evolution. Obviously, the properties of the WT antibody were not detrimental in vivo in mice, but large-scale production of such an antibody and its use as a drug would be impossible. A thorough understanding of antibody structure and function, as well as heuristic knowledge of natural sequence variability, is therefore the basis for rational sequence optimization designed to improve manufacturability-related properties of antibodies.
Since degradation pathways of proteins, and antibodies in particular, are manifold and often interdependent, computational methods can identify potential liabilities or reduce insufficient stability to distinct sequence patterns. The result is a rational guide for sequence optimization. As discussed here, we employed a variety of computational methods to identify those sequence patterns that contribute to the insufficient stability of mAb1. While the search for aggregation-prone motifs, charged and hydrophobic patches did not yield discoveries, statistical analysis of the sequences pointed toward distinct amino acids in the framework. Based on these observations, several variants of mAb1 with an increasing number of mutations were designed and characterized experimentally with a variety of biophysical methods. In addition, because we could not characterize all possible combinations of the suggested mutations experimentally, we used alchemical free energy simulations to study the effect of each individual mutation on the thermodynamic stability of the protein. The results of these simulations strongly suggested a stabilizing effect on the individual immunoglobulin domains for most mutations, and a minor effect on the interaction between heavy and light chain. Since we identified these critical positions using a statistical comparison of the mAb1 sequences with a large pool of naturally matured antibodies, our findings underpin the notion that framework mutations that occur rarely in the phenotypic pool of antibodies are likely to compromise the thermodynamic stability of the protein.
The simulation predicts that most introduced mutations increase thermodynamic stability. These results are in line with those obtained from the DSC and RALS measurements. Both experiments demonstrate a continuous increase in stability with increasing number of mutations. The DSC data show only a marginal (but significant and reproducible) increase of the Fab unfolding temperature for mAbs in which only one of the 2 chains has been engineered. But, for the variant with optimized amino acids in both chains (mAb1_Hv2_Lv3), the unfolding temperature of the Fab increases 16 K compared to mAb1_WT. The RALS data yield a similar picture. We see a steady but small increase of the onset of the denaturation for all variants except for the fully engineered one. The respective Tonset is shifted to 18 K. From both experiments we also find that engineering the LC has a larger effect on stability, which is in agreement with the calculated changes of the thermostability for each mutation.
While the results of the biophysical experiments and simulations are consistent, the effect on expression levels of the antibodies seem to suggest an entirely different picture. All engineered variants of mAb1 expressed with titers above 50 mg/l, whereas with WT mAb1 titers were below 1 mg/l. The dramatic increase of the expression levels for the variants harboring only one engineered chain therefore contrasts with the results from DSC and RALS, which show a small stability increase for these variants. However, the calculations suggest that the stability of the individual immunoglobulin domains is improved with the introduced mutations. The generated mAb1 variants, except for the fully engineered mAb1_Hv2_Lv3, likely consist of one stabilized chain while the second WT chain exhibits substantially lower stability. In experiments involving heating, it is plausible that the stability of each individual chain is the limiting factor, indicating that we observed kinetic rather than equilibrium thermodynamic effects. As soon as one domain unfolds, the stabilizing effect of the complex formation vanishes, and, as a consequence, the second chain starts to unfold as well. For protein folding in cells, and thus for expression, however, the presence of one stably folded domain to serve as nucleation point for a less stable chain could be sufficient to substantially increase the amount of successfully folded antibody.Citation36 In fact, studies have revealed that CH1 requires the interaction with the folded constant LC region controlling the assembly of the full-length antibody in vivo.Citation37 In addition, pairing of the variable domains increases the stability of the variable LC domain due to interface contacts.Citation38 Ultimately, the folding of the multi-domain Fab is an interdependent processCitation39 and the above interpretation is consistent with the observations from the biophysical characterization of the antibody set we studied.
From a drug developer's perspective, the most interesting question is whether the step-wise increased protein stability, as assessed by biophysical experiments, translate into step-wise improved long-term stability and thus into improved shelf-life. The 40 °C long-term stability study we conducted with the engineered variants clearly show substantial improvements in long-term stability as judged by monomer content. The fully-engineered variant was also the most stable over the entire period of 4 months. Hence, the thermodynamic stability of the variable domains certainly is an important factor for long-term stability. But it is also evident from our data (see ) that thermodynamic stability and shelf-life cannot be readily correlated in every case, as has been discussed previously.Citation40,41 Although thermodynamic (conformational) stability contributes to the storage stability of proteins,Citation41,42 other factors also play a considerable role. The degradation over an extended time is more complex as slow, covalent degradation processes become more important, and the monomer loss is driven by both aggregation and fragmentation processes. Moreover, the rate-limiting step for degradation is not necessarily constant over a range of temperatures and the actual solution conditions will determine the contribution of conformational stability to the complex aggregation reaction scheme.Citation8 The observed degradation rate constant or endpoint read-out in long-term stability experiments can be different depending on the actual assay chosen.Citation43 However, as shown in , there is an obvious trend in the current data. For the series of antibodies where only one chain is engineered, we see that long-term stability, as judged by SEC-monomer content after 126 d, increases with the number of mutations introduced. This effect is the same for both arms of the antibody variants. No matter which chain is stabilized, the long-term stability of the antibody increases, and, if the 2 engineered chains are combined, there is an additional small gain in stability as judged by the content of protein monomer. As observed for expression, long-term stability in this case is dominated by the most stable compartment of the antibody molecule, in contrast to the results of experiments involving heating, which are dominated by the least stable part.
Figure 5. Wet-lab stability data for the mAb1 variants. The melting temperatures TM as observed in DSC (TM of Fab unfolding, filled square) or tryptophan fluorescence experiments (filled circle and if 2 transitions are observed open circle) as well as the light scattering onset temperature Tonset in RALS experiments (triangle) are plotted in reference to the left y-axis. The bars denote the relative amount of monomer after 40 °C storage for 126 d as assessed via SEC and refer to the right (secondary) y-axis. For the wild-type protein 40 °C stability data are not available due to material constraints.
In conclusion, our work systematically describes the computational design, as well as the experimental and computational characterization, of a series of antibodies. Starting from the in vivo maturated antibody mAb1, which exhibited unfavorable CMC properties, we used heuristic sequence analysis to identify the most likely sequence patterns responsible for the undesired behavior and designed variants that consistently showed improved stability. Free energy calculations suggest that the introduced mutations stabilize the individual protein domains and these findings are in strong agreement with biophysical measurements. Our results provide a reasonable rationale for the substantially improved expression and long-term stability of the designed variants. We also showed that CMC properties of antibodies can be dramatically improved with a limited number of targeted mutations. Therefore, the consideration of developability-related properties early in drug development, before large quantities of a single candidate are produced for advanced pharmacological profiling, is critical. Computational methods, in particular rapid sequence analyses, can play a key role in this "Quality-by-Design"-approach and provide valuable hints for sequence optimization.
Materials and Methods
Protein production and purification
Variable regions of mAb candidates were synthesized and inserts cloned into expression vectors, already containing constant regions of an IgG1 isotype. HC and LC were cloned into individual expression vectors. HC and LC expression vectors were co-transfected into CHO DG44 cells by electroporation and cells were cultured subsequently for 48–72h in CD-OptiCHO media (Life Technologies) without selection pressure. For the generation of stable pools, cultivation of cells was performed in CD-OptiCHO + 500 μg/ml G418 for ∼14 d. Productivity of cell pools was determined by cultivating the pools in shake flasks in batch culture for 6 days, followed by titer measurement using a ForteBio Octet with ProA biosensors (Pall, ForteBio). MAbs were purified by Protein A and analyzed by A280 and SDS-PAGE. Top pools were subcloned by limited dilution in 5x 96-well plates, each. During scale up, the top 6 clones were identified by titer measurement in 96- and 24-well plate culture. Subsequently, the productivity of these clones was determined in 30 ml shake flask culture (as described below).
For protein production, the top clone of each mAb was cultivated in a 10-day fed-batch culture in CD-OptiCHO with a 10% bolus feed on day 3 and day 6 using CD EfficientFeed C (Life Technologies) (4–50 L in shake flasks or WAVE bags, depending on scale).
MAb1 and variants thereof were purified from cell culture supernatant via protein A affinity chromatography (MabSelect or rProtein A Sepharose, GE Healthcare). The purified mAbs were analyzed by reducing and non-reducing SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Quantification of mAb aggregation/fragmentation was performed by SEC.
Differential scanning calorimetry
DSC experiments in 25 mM sodium citrate, 125 mM NaCl, pH 6 were performed on a VP-capillary-DSC calorimeter (MicroCal™, Inc., Northampton, MA, USA) ramping from 13 to 95 °C at a scan rate of 1 K/min. Data were processed employing the MicroCal™, DSC add-on routine running in Origin data analysis software (OriginLab Corporation). The signal of a buffer reference measured under identical conditions was subtracted from the signal of the protein samples, the data were normalized to the molar protein concentration, and an interpolated baseline was subtracted. Transition midpoints (TM values) were determined from the maxima of the heat capacity curves.
Tryptophan fluorescence
The intrinsic tryptophan fluorescence of the mAbs was assayed as previously described.Citation31 Briefly, emission spectra of the 0.1 mg/ml antibody solutions (25 mM sodium citrate, 125 mM NaCl, pH 6) in quartz cuvettes (1.2 ml sample volume) were recorded upon excitation at λexc = 295 nm. Temperature was controlled by placing the cuvettes in a self-manufactured, peltier-controlled sample holder and signals recorded from 23 to 90 °C. All experiments were performed at least in duplicate. For data evaluation, the ratio of the fluorescence signal at λexc = 330 nm and at λexc = 350 nm (F330/F350) was plotted versus temperature and the curve was smoothed using a 7-point Savitzky-Golay algorithm. Transition temperatures were determined from the minima of the curves' first derivatives.
Right Angle Light Scattering (RALS)
The RALS signal change upon heating was recorded as previously described.Citation44 Briefly, the light scattering of 0.1 mg/ml antibody samples in 25 mM sodium citrate, 125 mM NaCl, pH 6 was monitored during thermal ramping employing a fluorimeter setup (λexc = 320 nm, λem = 320 nm) and quartz cuvettes (sample volume 800 μl). Temperature was controlled by placing the cuvettes in a self-manufactured, peltier-controlled sample holder, and light scattering signals were recorded during ramping from 23 to 90 °C. All experiments were performed at least in duplicate.
Long-term stability
Antibody samples at 10 mg/ml in 25 mM sodium citrate, 125 mM NaCl, pH 6 were incubated at 40 °C for the indicated time span. The unstressed samples (t = 0) were analyzed immediately after preparation. All 40 °C incubated samples were stored at – 70 °C and analyzed together at the end of the study.
Soluble protein aggregates were determined via HP-SEC employing a TSKgel G3000SW column (Tosoh Bioscience LLC)) and 50 mM Tris/HCl, 150 mM NaCl, pH 7.5 running buffer. All analyses were performed in duplicate.
Target binding/affinity measurements
Antigen binding was analyzed by surface plasmon resonance on a Biacore T200 instrument (GE Healthcare). The antibodies were immobilized on a CM5 sensor chip employing the anti-human Fc capture Kit (GE Healthcare) and the assay was run at 25 °C. Data were evaluated employing a 1:1 binding model.
Protein crystallization and data collection
Crystals were produced with the PACT-Screen (Jena Bioscience). The best crystals grew under a condition containing 200 mM Na citrate, 20% PEG3350, 100 mM BIS-TRIS propane pH 6.5, 20 °C after 1–3 d. Crystals were cryo-protected with 30% glycerol and flash-cooled in liquid nitrogen. The crystallographic experiments were performed on the X06SA beam line at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland. Data were processed with MOSFLM,Citation45 XDS,Citation46 SCALA and POINTLESSCitation47 within autoPROC.Citation48
Structure solution and refinement
The structure was determined by molecular replacement with PHASERCitation49 using pdb entry 2OSL as start model and refined with autoBUSTER (Global Phasing Ltd.). Model building was done with COOT.Citation50 The quality of the final model was assessed with MOLPROBITY.Citation51
Protein stability free energy calculations
Free energy differences for point mutants were computed using the approach described by Seeliger and de Groot.Citation20 To assess the difference in stability between a protein and point mutant thereof, the mutation is carried out alchemically in the folded state and in the unfolded state, which is approximated by a capped tripeptide. With three simulations for each mutation (the Fab, the domain and the tripeptide), the effect on the free-energy of folding and the change of the free energy of binding between heavy and light chain can be calculated. For detailed information see supporting material.
Disclosure of Potential Conflicts of Interest
The authors were employees of Boehringer Ingelheim Pharma GmbH & Co. KG during the course of the project.
Supplemental_Material.zip
Download Zip (9 MB)Acknowledgment
The authors would like to thank Andrea Eiperle, Sabrina Sayle, Christian Heinz and Nikolai Roosz for excellent technical assistance, Anita Bloching for excellent support in Fab generation and crystallization, Ingo Presser for continuous support.
Supplementary Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Reichert JM. Antibodies to watch in 2014. mAbs 2014; 6:5-14; PMID:24284914; http://dx.doi.org/10.4161/mabs.27333
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 2004; 3:711-6; PMID:15286737; http://dx.doi.org/10.1038/nrd1470
- Cromwell MEM, Hilario E, Jacobson F. Protein aggregation and bioprocessing. AAPS J 2006; 8:E572-E579; PMID:17025275; http://dx.doi.org/10.1208/aapsj080366
- Farid SS. Process economics of industrial monoclonal antibody manufacture. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 848: 8-18; PMID:16899415
- Wu SJ, Luo J, O'Neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel 2010; 23: 643-51; PMID:20543007; http://dx.doi.org/10.1093/protein/gzq037
- Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, Berenguer M, Poujol D, Stehle J, Stark Y, et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs 2013; 5: 445-70; PMID:23571156
- Neuber T, Frese K, Jaehrling J, Jäger S, Daubert D, Felderer K, Linnemann M, Höhne A, Kaden S, Kölln J, et al. Characterization and screening of IgG binding to the neonatal Fc receptor. mAbs 2014; 6: 928-42; PMID:24802048; http://dx.doi.org/10.4161/mabs.28744
- Wang W, Nema S, Teagarden D. Protein aggregation–pathways and influencing factors. Int J Pharm 2010; 390: 89-99; PMID:20188160; http://dx.doi.org/10.1016/j.ijpharm.2010.02.025
- Topp EM, Zhang L, Zhao H, Payne RW, Evans GJ, Manning MC. Chemical instability in peptide and protein pharmaceuticals, formulation and process development strategies for manufacturing biopharmaceuticals. Taylor & Francis.; 2010; 41-67.
- Weiss WF, Young TM, Roberts CJ. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J Pharm Sci 2009; 98: 1246-77; PMID:18683878; http://dx.doi.org/10.1002/jps.21521
- Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci 2004; 93: 1390-402; PMID:15124199; http://dx.doi.org/10.1002/jps.20079
- Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci USA 2009; 106: 11937-42; PMID:19571001; http://dx.doi.org/10.1073/pnas.0904191106
- Chaudhri A, Zarraga IE, Kamerzell TJ, Brandt JP, Patapoff TW, Shire SJ, Voth GA. Coarse-grained modeling of the self-association of therapeutic monoclonal antibodies. J Phys Chem B 2012; 116(28):8045-57; PMID:22694284
- Yearley EJ, Godfrin PD, Perevozchikova T, Zhang H, Falus P, Porcar L, Nagao M, Curtis JE, Gawande P, Taing R, et al. Observation of small cluster formation in concentrated monoclonal antibody solutions and its implications to solution viscosity. Biophys J 2014; 106: 1763-70; PMID:24739175; http://dx.doi.org/10.1016/j.bpj.2014.02.036
- Roberts CJ. Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol 2014; 32: 372-80; PMID:24908382; http://dx.doi.org/10.1016/j.tibtech.2014.05.005
- Zheng W, Doniach S. A comparative study of motor-protein motions by using a simple elastic-network model. Proc Natl Acad Sci U S A 2003; 100: 13253-8; PMID:14585932
- Suhre K, Sanejouand YH. ElNémo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res 2004; 32: W610-W614; PMID:15215461; http://dx.doi.org/10.1093/nar/gkh368
- Seeliger D, Haas J, de Groot BL. Geometry-based sampling of conformational transitions in proteins. Structure 2007; 15: 1482-92; PMID:17997973; http://dx.doi.org/10.1016/j.str.2007.09.017
- Guerois R, Nielsen JE, Serrano L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J Mol Biol 2002; 320: 369-87; PMID:12079393; http://dx.doi.org/10.1016/S0022-2836(02)00442-4
- Seeliger D, de Groot BL. Protein thermostability calculations using alchemical free energy simulations. Biophys J 2010; 98: 2309-16; PMID:20483340; http://dx.doi.org/10.1016/j.bpj.2010.01.051
- Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011; 79: 830-8; http://dx.doi.org/10.1002/prot.22921
- Jones S, Thornton JM. Prediction of protein-protein interaction sites using patch analysis. J Mol Biol 1997; 272: 133-43; PMID:9299343; http://dx.doi.org/10.1006/jmbi.1997.1233
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat.Biotechnol. 2004; 22: 1302-6.
- Tartaglia GG, Cavalli A, Pellarin R, Caflisch A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci 2005; 14: 2723-34; PMID:16195556; http://dx.doi.org/10.1110/ps.051471205
- Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: A server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics 2007; 8:65.
- Trovato A, Seno F, Tosatto SCE. The PASTA server for protein aggregation prediction. Protein Engineering, Design and Selection 2007; 20: 521-3; http://dx.doi.org/10.1093/protein/gzm042
- Tartaglia GG, Pawar AP, Campioni S, Dobson CM, Chiti F, Vendruscolo M. Prediction of aggregation-prone regions in structured proteins. J Mol Biol 2008; 380: 425-36; PMID:18514226; http://dx.doi.org/10.1016/j.jmb.2008.05.013
- Frousios KK, Iconomidou VA, Karletidi CM, Hamodrakas SJ. Amyloidogenic determinants are usually not buried. BMC Struct Biol 2009; 9:44; PMID:19589171; http://dx.doi.org/10.1186/1472-6807-9-44
- Wines BD, Powell MS, Parren PWHI, Barnes N, Hogarth PM. The IgG Fc contains distinct Fc receptor (FcR) binding sites: The leukocyte receptors FcgRI and FcgRIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J Immunol 2000; 164: 5313-8; http://dx.doi.org/10.4049/jimmunol.164.10.5313
- Seeliger D. Development of scoring functions for antibody sequence assessment and optimization. PLoS One 2013; 8:e76909
- Garidel P, Hegyi M, Bassarab S, Weichel M. A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy. Biotechnol J 2008; 3: 1201-11; PMID:18702089; http://dx.doi.org/10.1002/biot.200800091
- Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochemical Biophys Rese Commun 2007; 355: 751-7; PMID:17321501; http://dx.doi.org/10.1016/j.bbrc.2007.02.042
- Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci 2008; 97: 1414-26; PMID:17721938; http://dx.doi.org/10.1002/jps.21104
- Burstein EA, Vedenkina NS, Ivkova MN. Fluorescence and the location of tryptophan residues in protein molecules. Photochem Photobiol 1973; 18: 263-79; PMID:4583619; http://dx.doi.org/10.1111/j.1751-1097.1973.tb06422.x
- Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J 2001; 80: 2093-109; PMID:11325713; http://dx.doi.org/10.1016/S0006-3495(01)76183-8
- Brandts JF, Hu CQ, Lin LN, Mos MT. A simple model for proteins with interacting domains. Applications to scanning calorimetry data. Biochemistry 1989; 28: 8588-96; PMID:2690944; http://dx.doi.org/10.1021/bi00447a048
- Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, Buchner J. An Unfolded CH1 Domain Controls the Assembly and Secretion of IgG Antibodies. Mol Cell 2009; 34: 569-79; PMID:19524537; http://dx.doi.org/10.1016/j.molcel.2009.04.028
- Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol 2005; 347: 773-89; PMID:15769469; http://dx.doi.org/10.1016/j.jmb.2005.01.053
- Rowe ES. Dissociation and denaturation equilibria and kinetics of a homogeneous human immunoglobulin fab fragment. Biochemistry 1976; 15: 905-16; PMID:813765
- Brummitt RK, Nesta DP, Roberts CJ. Predicting accelerated aggregation rates for monoclonal antibody formulations, and challenges for low-temperature predictions. J Pharm Sci 2011; 100(10):4234-43.
- Youssef AMK, Winter G. A critical evaluation of microcalorimetry as a predictive tool for long term stability of liquid protein formulations: Granulocyte Colony Stimulating Factor (GCSF). Eur J Pharm Biopharm 2013; 84: 145-55; PMID:23333898
- Costanzo JA, O'Brien CJ, Tiller K, Tamargo E, Robinson AS, Roberts CJ, Fernandez EJ. Conformational stability as a design target to control protein aggregation. Protein Eng Des Sel 2014; 27(5):157-67; PMID:24722670
- Maddux NR, Iyer V, Cheng W, Youssef AMK, Joshi SB, Volkin DB, Ralston JP, Winter G, Middaugh CR. High throughput prediction of the long-term stability of pharmaceutical macromolecules from short-term multi-instrument spectroscopic data. J Pharm Sci 2014; 103: 828-39; PMID:24421157; http://dx.doi.org/10.1002/jps.23849
- Garidel P. Right angle light scattering protein thermostability screening: From research to development. 2012; 24: 13-8
- Leslie A, Powell H. Processing diffraction data with mosflm, In: Read R, Sussman J, eds., Evolving methods for macromolecular crystallography. Taylor & Francis Netherlands; 2007; 41-51
- Kabsch W. XDS. Acta Crystallogr D Biol.Crystallogr 2010; 66: 125-32; http://dx.doi.org/10.1107/S0907444909047337
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 2006; 62: 72-82; http://dx.doi.org/10.1107/S0907444905036693
- Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr.D Biol.Crystallogr 2011; 67: 293-302; http://dx.doi.org/10.1107/S0907444911007773
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40: 658-74; PMID:19461840; http://dx.doi.org/10.1107/S0021889807021206
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66: 486-501; http://dx.doi.org/10.1107/S0907444910007493
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010; 66: 12-21; http://dx.doi.org/10.1107/S0907444909042073