
Abstract
Structural characterization of proteins and their antigen complexes is essential to the development of new biologic-based medicines. Amino acid-specific covalent labeling (CL) is well suited to probe such structures, especially for cases that are difficult to examine by alternative means due to size, complexity, or instability. We present here a detailed account of carboxyl group labeling (with glycine ethyl ester (GEE) tagging) applied to a glycosylated monoclonal antibody therapeutic (mAb). The experiments were optimized to preserve the structural integrity of the mAb, and experimental conditions were varied and replicated to establish the reproducibility of the technique. Homology-based models were generated and used to compare the solvent accessibility of the labeled residues, which include aspartic acid (D), glutamic acid (E), and the C-terminus (i.e., the target probes), with the experimental data in order to understand the accuracy of the approach. Data from the mAb were compared to reactivity measures of several model peptides to explain observed variations in reactivity. Attenuation of reactivity in otherwise solvent accessible probes is documented as arising from the effects of positive charge or bond formation between adjacent amine and carboxyl groups, the latter accompanied by observed water loss. A comparison of results with previously published data by Deperalta et al using hydroxyl radical footprinting showed that 55% (32/58) of target residues were GEE labeled in this study whereas the previous study reported 21% of the targets were labeled. Although the number of target residues in GEE labeling is fewer, the two approaches provide complementary information. The results highlight advantages of this approach, such as the ease of use at the bench top, the linearity of the dose response plots at high levels of labeling, reproducibility of replicate experiments (<2% variation in modification extent), the similar reactivity of the three target probes, and significant correlation of reactivity and solvent accessible surface area.
Keywords:
- acetonitrile
- circular dichroism
- covalent labeling
- dose response
- 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- extracted ion chromatogram
- glycine ethyl ester
- heavy chain
- hydrogen-deuterium exchange
- hydroxyl radical footprinting
- immunoglobulin gamma
- ion trap
- light chain
- lysyl endopeptidase
- monoclonal antibody
- mass spectrometry
- rate constant
- solvent accessible surface area
- size-exclusion chromatography
Abbreviations
| ACN | = | acetonitrile |
| CD | = | circular dichroism |
| CL | = | covalent labeling |
| DR | = | dose response |
| EDC | = | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EIC | = | extracted ion chromatogram |
| GEE | = | glycine ethyl ester |
| HC | = | heavy chain |
| HDX | = | hydrogen-deuterium exchange |
| HRF | = | hydroxyl radical footprinting |
| IgG | = | immunoglobulin gamma |
| IT | = | ion trap |
| LC | = | light chain |
| Lys-C | = | lysyl endopeptidase |
| mAb | = | monoclonal antibody |
| MS | = | mass spectrometry |
| RC | = | rate constant |
| SASA | = | solvent accessible surface area |
| SEC | = | size-exclusion chromatography |
Introduction
Biopharmaceutical proteins (biologics) comprise one of the most important and fastest growing classes of drugs in the commercial marketplace today. The function and efficacy of these products are determined by the structure of the protein and its ability to interact with relevant molecules in the body, as well as its kinetics and stability in patients.Citation1,2 In particular, monoclonal antibodies (mAb) are used both in fundamental research and in clinical settings, where they are highly specific therapeutic agents for treating various forms of cancer and immune-mediated diseases.Citation3,4 Currently, recombinant immunoglobulin gamma (IgG) mAbs are the most prevalent protein format in the biopharmaceutical development pipeline,Citation5 which provides strong motivation for the exploration of analytical methods and tools for mAb characterization. Chromatography and mass spectrometry (MS)-based techniques for characterizing primary structure of mAbs have been well established and applied to study common post-translational modifications, such as phosphorylation, oxidation, or glycosylation, as well as sequence variants.Citation6,7
Understanding the secondary and tertiary structures of therapeutic mAbs is also important because their activity and stability are considerably influenced by their conformation. In addition, characterization of such proteins and their antigen complexes is essential to define their epitopes, which represent fundamental elements of intellectual property protection. Therapeutic mAbs tend to form aggregates during the manufacturing process, including during cell culture, purification, and formulation. Changes in the protocols of manufacturing, formulation, or storage conditions may affect the secondary or tertiary structure, thus necessitating suitable techniques for performing analysis to assure proper conformation.Citation8,9 In addition, as the patents for innovator drugs expire, manufacturing of biosimilar products will increase, along with demand for assessment methods and standards to assure stability and potency. Currently, secondary and tertiary structural features of biologic “similarity” are poorly defined.
Methods to describe structures of proteins and their higher order complexes continue to evolve, and various advances have increased speed, efficiency, and resolution of structure determination.Citation10 Most proteins behave according to the function-dictated-by-structure principle, and their proper conformation is central to their role in processes such as enzyme catalysis, cell signaling, or ligand binding.Citation11-13 Conventional biophysical methods used for assessing secondary and tertiary (or quaternary) structure or their changes include circular dichroism spectroscopy, analytical ultracentrifugation, Fourier Transform Infrared spectroscopy, isothermal titration calorimetry, surface plasmon resonance, and fluorescence spectroscopy.Citation14-19 These methods are very useful for monitoring the global state and population average of the biomolecule's higher order structure; however, they are not necessarily suited to detect small, but potentially significant, changes because the differential signal may be lost among the average readout. Nuclear magnetic resonance spectroscopy and X-ray crystallography offer sophisticated ways to probe small but significant structural changes.Citation20,21 Several crystal structures of intact IgG1 antibodies have been reported previously.Citation22,Citation23,Citation24 All these structures demonstrate unique conformations and provide snapshots of otherwise flexible IgG1 antibodies. However, crystallization of large or glycosylated proteins at mg/ml protein concentration remains a critical, but challenging step in X-ray structure determination. Also, the conditions for crystal structure determination may deviate from the relevant formulation of the drug in solution that may influence conformation or dynamics. In addition, X-ray crystallography-based methods cannot probe potentially important dynamics of proteins.
Covalent labeling (CL) techniques coupled with MS have made important contributions to the understanding of secondary and tertiary protein conformations and their dynamics, offering detailed peptide or side chain level information with respect to structure.Citation6, 25 Reversible CL of protein backbone using hydrogen-deuterium exchange (HDX) MS has been increasingly used to characterize biopharmaceuticals such as mAbs.Citation26,Citation27,28 The advantages of HDX include its non-invasive nature and the potential of providing structural information across the length of the protein sequence. In particular, HDX is a very powerful technique for assessing secondary structure of proteins. The main disadvantage of HDX is the transient and labile nature of the labeling probe, and the results can be affected if experimental conditions such as pH and temperature are not tightly controlled. Irreversible CL coupled to MS, a branch of biophysics in the class of “footprinting” technologies, provides information about tertiary structure of proteins. It is typically used to target surface accessible amino acid side chains by attaching permanent, stable modifications that can be detected and quantitated by MS.Citation25,29,Citation30 The advantage of irreversible CL approaches is that a quantitative measure of the solvent accessibility of the target probe residues across different biological states may be provided.Citation31 However, experimental conditions must be carefully controlled in such experiments so that the resulting modifications and reaction conditions themselves do not perturb the native protein structure. A variety of labeling methods such as OH radicals, carbene labeling, carbodiimide, and diethylpyrocarbonate labeling reagents are routinely used in structural MS-based footprinting. The chemistry of these reagents varies such that a wide range of amino acid side chains can be probed, providing structure assessment of proteins with single side chain resolution.Citation32-36 CL-based footprinting methods can provide quantitative assessments of solvent accessibility of the probes, and there is interest in understanding the limits of reproducibility of these assessments in order to understand their potential for reproducible structure assessment for a specific therapeutically active conformation.Citation37-40
Hydroxyl radical-based footprinting (HRF) techniques target a large number of probes, offering excellent coverage of the overall sequence to map protein surfaces. However, these labeling techniques can be complex, require access to specialized facilities (laser, synchrotron, or electron beam sources), and the interpretation of the resulting data is challenging. In this report, we explore the use of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), in the presence of glycine ethyl ester (GEE), to modify solvent accessible C-terminal, glutamate (E) and aspartate (D) residues of a therapeutic mAb to assess the limits of the methodology in probing structure using MS. The GEE labeling method is convenient because it can be performed on a bench top. It has previously been used to probe the structure of a number of proteins including the membrane-attached antenna protein for mapping a protein-protein interface, and to study phosphorylation-induced conformation changes of a membrane associated kinase.Citation41, 42 For this work, we evaluated the reproducibility of protein labeling kinetics at the specific side chain residues with this reagent using a therapeutic mAb. Specifically, the mAb was exposed to carbodiimide in the presence of GEE label for varying amounts of time from 0 to 10 minutes in triplicate and at various concentrations, providing a detailed quantitative characterization of side chain reactivity. Also, model compounds were synthesized to compare their reactivity to those of protein-based probes.
Comparative modeling of proteins refers to generating a structural model of the target protein by comparing its amino acid sequence to the known three-dimensional structure of related homologous proteins.Citation43 The use of footprinting data in conjunction with computational modeling approaches is a powerful method for cross validating results from the two methods. Using a molecular model of the mAb generated by homology modeling, we explored the relationship of reagent reactivity with the predicted solvent accessibility of individual residues. The crystal structure of a Fab fragment almost identical to this sequence has been solved at 2.4 Å in complex with human vascular endothelial growth factor (PDB ID: 1BJ1; ref. 44), but a complete structural model of the intact IgG1 mAb was required for this comparison. This therapeutic mAb was also recently characterized using HRF,Citation45 which gave us an opportunity to compare the results of the two methods side by side. Overall the CL approach using carbodiimide in the presence of GEE tag provided a detailed, precise, and accurate assessment of the structure of the mAb based on an easy to use protocol and automated data analysis (including identification of the modification sites and construction of dose response curves).
Results
Structural integrity
It is important to ensure the structural integrity of the macromolecule in footprinting experiments. Covalent labeling can potentially introduce structural changes by disrupting non-covalent interactions of the protein due to over-labeling. This can result in misleading conclusions about the biomolecular structure. Circular dichroism (CD), fluorescence spectroscopy, or activity assays can be utilized to check the structural integrity following labeling. A foolproof approach is to limit the number of modifications to, on average, one per biomolecule,Citation29,46-48 although this is likely an over-conservative standard. Another strategy is to use reaction kinetics for the individual modification sites at the peptide level.Citation49-53 Deviations from linearity of the dose response plot of a peptide suggest changes in the reaction dynamics arising from alterations of the microenvironment around the modified residue(s). Changes in such reactivity provide direct evidence that the labeled protein has an altered structure compared to its unlabeled isoform.
In the case of footprinting of carboxyl residues, protein samples at physiological pH conditions are modified by carbodiimides such as EDC, creating inherently unstable intermediate products (amine reactive O-acylisourea intermediate) shown in .Citation35 The nucleophilic primary amines of GEE react with the reactive intermediates, forming stable end products with mass shifts of +85.0528 Da or +57.0215 Da after hydrolysis. To illustrate the overall extent of modification on the intact mAb, we provide data on the light chain (LC) of the mAb using intact MS (data from the heavy chain (HC) was too noisy for this analysis). shows an expanded view of the MS data for the LC of the unlabeled mAb with a charge state of +19 seen as the most abundant isoform appearing at m/z = 1235.04, providing a calculated molecular weight of the LC of 23446.53 Da. The experimental isotopic distribution was aligned against the theoretical isotopic distribution using the Matched Filter approach in order to calculate the experimental molecular weight.Citation54 The corresponding theoretical value obtained from the mAb sequence is calculated as 23450.47 Daltons. The difference of 3.94 Daltons among the observed and theoretical values can be explained by the presence of two incompletely reduced intra-chain disulfide bonds known to be present in the LC.Citation55 As indicated in the Methods section, the mAb samples were reduced to remove the disulfide bonds, hence separating the LCs from the HCs. However, the two intra-chain disulfide bonds in the LCs were not completely reduced as suggested by the data. After accounting for the difference in the masses due to the two disulfide linkages, the experimental (23446.53 Da) and theoretical values (23446.44 Da) differ by 0.09 Daltons, and match within 4 ppm. shows the same region of the spectrum from the GEE labeled (conditions used listed in Column 3, ) form of the mAb. A new isotopic distribution signal appears in the bottom panel at an m/z = 1239.51 (z = +19), but it is absent in the unlabeled sample. This shows a mass addition of 85.04 Da, which matches closely to the expected shift of 85.05 Da (C4H7NO) to the mAb corresponding to the mass of the GEE labeled form of the mAb. Although not observed in this case, another commonly observed mass shift is 57.02 Da (C3H3NO), which arises from the hydrolyzed form of the amide end product.Citation35 The isotopic distributions were extracted by averaging the signal across the entire chromatogram. Since the most abundant isotopes provide the highest signal to noise ratio, the ratios of their corresponding ion intensities from the extracted isotopic distributions were used to calculate the percentage of labeling. The relative intensity of the most abundant isotopes from the extracted signal in suggests that about 11% of the total LCs received one GEE label. Any evidence for the doubly labeled species was below the detection limit of this instrument. These conditions limited our modifications to less than one per molecule, on average. Another notable difference is the increase in signal of isotopic distribution centered around m/z = 1236.52 in the labeled form in . This corresponds to a shift of 28 Daltons from the unlabeled form, and can be explained by the artifactual formylation introduced during the reaction with formic acid.Citation56 Formic acid was used during both the sample prep and labeling process to quench the labeling reaction. The increased exposure to formic acid in the labeled form can explain the increase in the signal.
Figure 1. (A) Reaction mechanism of carboxyl footprinting. Mass spectrum of (B) light chain of unlabeled mAb with a charge state of +19. (C) Labeled form showing the incorporation of GEE label with a mass shift of +85; about 11% of the light chain gets labeled.
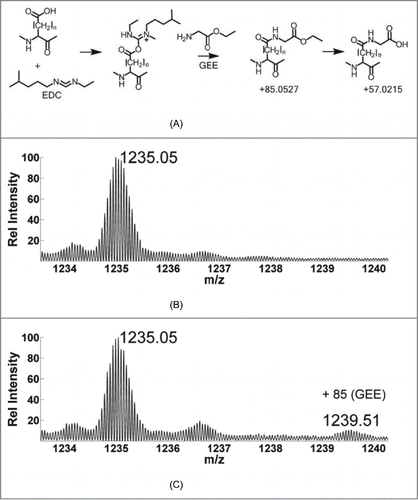
Table 1. Summary of the concentrations of EDC relative to the protein concentration under different conditions of labeling
Reaction conditions
The experimental conditions used in a recently published report by Zhang et alCitation57 and those used in this work are shown in . Although the relative molar concentrations of EDC and the protein are comparable (columns 2 and 3), the number of molecules of EDC per D+E residues of the mAb are reduced to less than half compared to the much smaller biomolecule calmodulin used in the previous work. This leads us to believe that although our approach ensures that there is only one label per molecule, this is a conservative dosage of the reagents compared to the large size of the mAb. In addition, the flexibility of the calmodulin versus the relatively compact and stable conformation of mAbs may lead to different labeling profiles of the two biomolecules. Among other experimental characteristics (e.g., matrix, temperature, time scale), the dosage of the reagents may be adjusted relative to the size of the biomolecule for optimal labeling so that sufficient labeling occurs to provide adequate MS signal while maintaining modest overall labeling extent.
Data analysis
Both the unlabeled and labeled samples were subjected to a dual digestion by Lys-C and trypsin followed by LC-MS/MS analysis. The resulting MS data were interpreted using ProtMapMS.Citation37 As expected, the +85.05 Da and its hydrolysis product +57.02 Da mass shifts represented the primary modified products. The use of iodoacetic acid (+58 mass shift) is preferred over iodoacetamide (+57 mass shift) as the alkylating reagent to avoid confusion between the labeled hydrolysis product and the latter mass adduct. Dose response (DR) plots were constructed and the corresponding rate constants were calculated. The DR curve is examined to determine the adherence to (pseudo) first order reaction kinetics.Citation30 The assignments of labeled species were verified by manual investigation of the tandem MS data, as well as correlations with the retention times of unlabeled peptides.
Summary of dose response plots
A summary of the labeling kinetics data for both the HC and LC tryptic peptides of the mAb is shown in . The overall sequence coverage obtained for the heavy and light chains by Lys-C/tryptic peptide mapping was 93.6% and 91.6%, respectively. The missing regions were composed primarily of peptides that were shorter than 4 residues in length, which can be inherently difficult to detect due to their weak retention properties on the LC column. The first column in denotes the peptides by sequence numbering; the second column represents the rate constant from the carboxyl-group footprinting experiments. The third and fourth columns show the labeled and unlabeled residues, respectively, together with the solvent accessible surface area (SASA) calculated from the homology model. A total of 58 D/E (19 on LC, 39 on HC) residues were present on the mAb, of which 32 were labeled under low EDC conditions (, column 3). This included 9 residues on the LC, and 23 residues on the HC across 19 different peptides.
Table 2. Summary of labeling results for mAb monomer. First column includes peptide sequencing numbering, second column includes GEE labeling rate constants and the associated error bars, third column indicates labeled probes as detected by MS2 together with their Solvent Accessible Surface Area (SASA) values, and the fourth column shows the unlabeled probes from the same peptide with their SASA values (*: Site of modification could not be precisely localized
Reproducibility
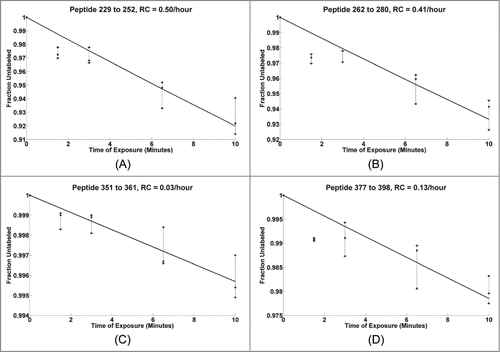
In order to examine the reproducibility of the experiments and to assess the linearity of labeling, the DR plots of the constituent peptides from three independent experiments were compared. shows four examples of DR plots for peptides from different regions (229-252, 262-280, 351-361, and 377-398 along with the best fit to the data) for the HC. The experimental conditions in this case used 633-fold excess of EDC compared to that of the mAb (, column 3), or over 10x the number of potential target probes (58). The mAb sample was exposed to the labeling reaction for 0, 1.5, 3, 6.5, and 10 minutes, as seen on the X-axis. The Y-axis shows the percentage of the peptide that remained unlabeled as a function of the labeling reaction time. The three dotted lines indicate results from three different experiments with identical labeling conditions, while the solid black line shows the best theoretical fit to the first-order exponential for the overall experimental curves. The resulting rate constants (RC) range from 0.03/hour to 0.5/hour as seen in the titles of individual plots. Each of the values on the dose response plots fall within 2% of the corresponding theoretical fit value. The median deviation between the experimental and the theoretical fit values is less than 1%. The HC peptides 229-252 and 262-280 are observed to be about 8% labeled (92% unlabeled, as seen on the Y-axis) when the reaction is carried out for 10 minutes. Typically, the same fraction of labeling is observed in a matter of 20 milliseconds in a typical synchrotron-based HRF experiment, making the GEE labeling 30,000 times slower than its synchrotron-based counterpart.Citation37 A recent study demonstrated that the results from GEE labeling are not compromised by modification-induced protein unfolding.Citation57 The size of the label is relatively small, the label is on the surface, and only about 10% (2 out of total 20) residues in the overall sequence are affected. These factors are likely to ensure minimal perturbation to the protein conformation, allowing for the native structure to be preserved.
Figure 2. Dose response plots for select mAb HC peptides from triplicate runs shown with dashed lines, the solid line shows the best fit for the three replicates.
Linearity under high dose conditions
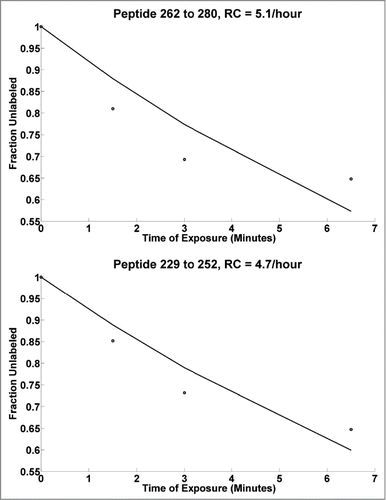
In the case of traditional HRF experiments, the sulfur-containing residues (Met and Cys) are susceptible to secondary oxidation after hydroxyl radical exposure. If the dosage is not carefully controlled, deviation of the reaction from its first order for peptides containing these residues can occur, which interferes with the analysis. However, peptides not containing these residues are minimally affected, unless the oxidation in one site alters structure at another. In order to study the potential for such effects in the case of carboxyl group footprinting, the concentration of EDC was increased 10-fold, while keeping all other variables the same (, column 4). illustrates the observed changes in the DR plots where a 10 fold greater molar excess of EDC (as indicated in , column 4) was added relative to the mAb compared to . Since EDC is responsible for activating the carboxyl groups to be attacked by the nucleophilic GEE, the excess EDC is expected to increase the extent of labeled products. The dotted line represents the experimental data, while the solid black line represents the best theoretical fit to the first-order exponential equation. shows that nearly 35% of the 262-280 (top) and 229-252 (bottom) peptides are labeled at a reaction time of 6.5 minutes. Although the rate constants increase by 10 fold compared to those seen in , the dose response plots remain linear, with the experimental values fitting within 6% of their theoretical fit values. However, some saturation of the labeling is apparent at 6.5 minutes, where the experimental DR curve starts to deviate upwards from the theoretical fit line. Overall, the data suggests that the higher order structure of the mAb is preserved under such a high modification regime. This may in part be due to the large and stable nature of the mAb, allowing it to be tolerant to modifications with minimal effect on the higher order structure. Note that the optimal dosage levels need to be determined on a case-by-case basis because they depend upon the characteristics of the protein system under consideration. In general, tolerance to a high level of modifications can be advantageous over HRF, where such high modification conditions may pose a challenge, and the secondary oxidation reactions may interfere with the correct quantitative characterization.Citation39 It should be noted that ultra-fast HRF techniques, such as those using nanosecond photolysis of peroxide, likely overcome some of these limitations as well.Citation58, 59
Figure 3. Dose response plots for mAb HC peptides at 10 times higher concentration of EDC, with black line showing the best theoretical fit to the first order reaction. Note: Typically, the DR curves are carefully observed for any saturation behavior (non-linearity) in each individual peptide DR curve. Such non-linearity indicates that the high dose is potentially altering structure. Thus, DR plots are truncated at high dose levels if evidence of such saturation is occurring. In general the lowest dose points are the most accurate and representative of the intact biological state.
Homology modeling
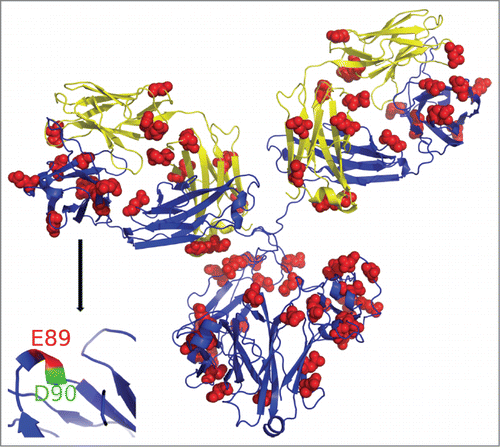
A molecular model of the mAb was generated using homology modeling from the existing templates with high sequence similarity, allowing us to explore the relationship between reactivity of the individual probes against their predicted solvent accessibility profile. shows the homology model of the mAb constructed using Swiss-Model workspace based on available templates of intact hetero-tetrameric IgG1 mAb (PDB ID:1IGY) and Fab fragment (PDB ID:1BJ1).Citation22, 44 The two LCs are shown in yellow, while the HCs are shown in blue. The GEE labeled D/E residues are marked as red spheres, and are roughly consistent with their solvent accessibility predicted by homology model. The sequence identity for Fab LC 1-213 and HC 1-224 are 100%. The hinge and the Fc (225-448) are based on 1IGY and the sequence identity is 60%. As Fab domains of the structural model are based on the existing crystal structure, we expect this part of the model to be quite accurate. Some deviations can be expected in the loop regions of HC 221-227, which were missing in the HC of the template 1BJ1. The hinge regions connecting Fab and Fc are flexible and the model represents a snapshot of an overall flexible antibody structure. The inset in (bottom, left) shows a close up view of a particularly interesting coil region of the HC from peptide 88-98. In this case, although the two probes (E89 and D90) are adjacent to each other, only one of them (E89) becomes labeled. This is consistent with the higher solvent accessibility of E89 residue (shown in red), while D90 (green) has limited solvent accessibility and was not detected as labeled. This illustrates the power of CL-based protein footprinting to be able to provide results at the individual amino acid level. Carboxyl labeling provides a much simpler MS-profile than HRF, making it easier to separate and quantitate the contributions of individual residues within a peptide due to chromatographic separation of isobaric species.
Figure 4. Homology model of mAb structure. GEE labeled residues marked in red spheres. Inset: HC region representing peptide 88-98, where E89 is highly solvent accessible (shown in red) and is modified, while D90 (blue) has limited solvent accessibility and was not detected as labeled.
Comparison of experimental labeling data and homology modeling results
To provide an unbiased comparison of the solvent accessibility values predicted from the homology modeling with the data derived from this study, the rate data for all peptides in are plotted vs. the sum of the solvent accessible surface areas of the D and E residues within the peptides in . The data shows a positive and significant correlation with an overall Pearson's correlation coefficient (R2) of 0.54.Citation60 This provides support for the idea that the SASA of the target residues within the peptide can be predicted from the rate data. The solvent accessible surface area for unhindered D and E side chains is 106 and 138 Å2, respectively.Citation61 Based on these theoretical values, it can be seen from the data that primarily highly solvent accessible peptides (those with most of their surface area exposed) were labeled with GEE. For example, the two highest rate constants are associated with HC peptides 229-252 and 262-280. These two peptides contain one or more fully solvent accessible D/E residues, with peptide 262-280 containing four probes. However, peptide 229-252 is an outlier since it has a similar rate to 262-280, although it has only one highly accessible residue. Peptides with net solvent accessible surface area < 50 Å2 (i.e., <47% of their total accessible area) for their D/E residues (HC 68-76, 99-127, 255-261, 308-323, LC 191-207) did not get labeled. Peptides containing probes with SASA between 50-120 Å2 (HC 44-65, 88-98, 154-211, 281-294, 299-307, 333-340, LC 62-103, 109-126, 170-183) were labeled with intermediate reaction kinetics. Overall, this suggests a threshold under these labeling conditions, where peptides with net solvent accessible surface area under a certain value do not get labeled or their labeling is below the limit of detection; more vigorous labeling would be expected to shift the curve such that modification of these residues would be detected.
Figure 5. Dose response rate constants of all peptides as a function of the total solvent accessibility area of the constituent D/E residues.
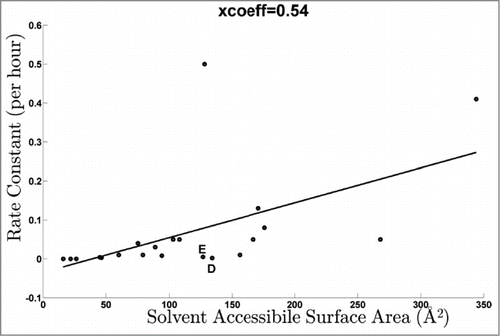
Besides peptide 229-252 and this “threshold” effect, there are other deviations that suggest potential complexities in the data. For example, there was no labeling observed for solvent accessible residues D28 (LC 19-42) and E213 (LC 208-214). This could suggest possible differences in the solution structure of the mAb from its crystal form or other sequence or structure-based factors that attenuate the labeling chemistry. Another specific example of a striking difference in labeling is that the highly solvent accessible E1 on the HC N-term peptide was not observed to be labeled while E6, which is predicted as much less solvent accessible, was found to be labeled. Similarly, the N-terminal D1 of the LC was minimally labeled. These 2 residues (D, E) are indicated in and depart from the overall trend.
Reactivity and attenuations in GEE labeling
Based on the potential discrepancies in the data described above, we explored potential chemical and structural factors underlying the variations in carbodimide chemistry within proteins. Specifically, we hypothesized that the location of probes on the N-terminus may be interfering with the chemistry of the labeling. In order to test this hypothesis, and to determine the relative labeling efficacies of D, E and the C-terminal carboxyl group, we synthesized three peptides, including two that contain D/E as their N-terminal residues, and exposed them to the GEE labeling for 10 minutes using the conditions specified in , column 3. MS analysis of the three synthetic peptides (G-peptide: GIDTPQIESR, E-peptide: EVQPVESGGR, D-peptide: DIQMTQSPSR) was performed and the following results (summary provided in ) were obtained.
Table 3. Summary of labeling from peptide experiments (% of overall modified species)
Results from G-peptide
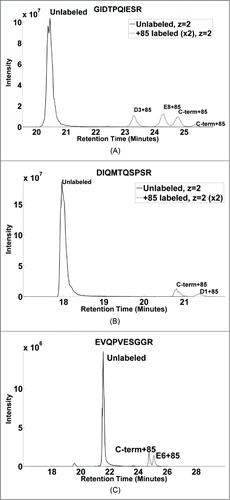
It is important to compare intrinsic reactivity differences of the individual probes to interpret SASA values from the observed rate constants. “G-peptide” (GIDTPQIESR) containing both D/E residues was labeled and analyzed using LC-MS. shows the selected ion chromatograms corresponding to the doubly charged, unlabeled and +85 labeled forms. Three distinct isoforms can be observed corresponding primarily to the modifications D3+85, E8+85, and C-term+85 eluting at 23.4, 24.3, and 24.8 minutes, respectively, allowing us to calculate the contribution of each of the isoforms individually from the peak area of the corresponding selected ion chromatogram. The three isoforms are comparable in their overall intensities with a contribution of 33%, 37%, and 30% from the labeling of D3, E8, and C-terminus, respectively. This indicates that minimal inherent bias exists in the labeling reaction due to the different intrinsic reactivity of the three probes, at least in the context of the sequence of this specific peptide. Thus, the assumption inherent in that the SASA values for D and E can be summed together with equal weighting is supported by this data. Although the major component of the three isoforms could be chromatographically separated, a minor form of each of the modifications was found to elute about 30 seconds following the major component. For example, a small fraction at the beginning of the peak at 24.3 minutes was found to contain D3+85, and a minor portion of the peak at 24.8 minutes could be attributed to E8+85, as evidenced by the MS2 scans containing a mixture of modifications. Similarly, the smallest EIC peak at 25.5 minutes represents the minor modification form of C-term+85. Currently, we cannot attribute any structural differences to these isobaric but non co-eluting species.
Figure 6. Single ion chromatograms of unlabeled and +85 labeled form of synthetic peptides. (A) GIDTPQIESR peptide showing modified isoforms of D3 and E8 at 23.3 and 24.3 minutes, with C-term at 24.8 and 25.4 minutes. (B) DIQMTQSPSR peptide showing two isoforms representing labeled C-terminus and D1+85 eluting at 20.8 and 21.4 minutes, respectively. (C) EVQPVESGGR peptide showing the unlabeled and labeled forms of the cyclized peptide showing a water loss. Two similar sized distinct peaks at 24.75 and 24.06 minutes represent +85 modification at C-terminus and E6, respectively.
Results from D-peptide
In the case of “D-peptide” (DIQMTQSPSR), two modification sites corresponding to signals from D1+85 and C-term+85 forms of the peptide could be separated chromatographically. The EIC of the doubly charged forms of the peptide is shown in , where the peak at 20.8 minutes corresponds to C-term+85, while the peak at 21.3 minutes shows the D1+85 modification, as verified by the MS2 spectra. Area under the curve calculations indicate that the signal from D1+85 (21.3 minutes) is about 30% of the overall modification while the labeling of the C-terminus is 70% of the total. Assuming that the intrinsic reactivity of the two probes is expected to be similar (as suggested by the G-peptide), there seems to be a suppression effect when D is in an N-terminal location.
Results from E-peptide
The sequence of the “E-peptide” (EVQPVESGGR) allowed us to study the effect of the N-terminal position on the relative labeling efficiency of the E residue. Interestingly, the EIC signal of the doubly charged unlabeled peptide was observed to be relatively weak (Figure S1A). Closer examination revealed that about 95% of the unlabeled peptide was associated with a mass shift of -18 Da. We suspect this may arise from a known cyclization phenomenon that can occur when the terminal amine group of E forms an intra-molecular bond with the carboxylate resulting in a meta-stable, five membered ring.Citation62-64 The formation of the 5-membered ring species would be accompanied by loss of a water molecule (-18 Da) for the corresponding peptide. shows the EIC of the doubly charged cyclized forms of the peptide. The predominant unlabeled form of the cyclized peptide elutes at 21.5 minutes. The signal contributions from two of the isoforms (E6, and C-term+85) could be chromatographically separated in this case, and the peaks at 24.75 and 25.06 minutes represent the C-term and E6 labeled isoforms. The area under the curve calculations showed the ratio of E6, and C-term+85 modifications to be nearly 1:1. Although E6 and the C-terminus were labeled with equal efficiency (as in the G-peptide above), no labeling of E1 was observed.
The results from the synthetic peptides are summarized in . The data supports our hypothesis that D and E residues tend to be less susceptible to GEE labeling (the effect being more pronounced for E) when present on the N-terminus of the protein/peptide. One possible source of suppression is the positive charge on the N-terminus. In this scenario the positive charge suppresses the electronegativity of the carboxylate, reducing its reactivity with carbodiimide. However, the suppression effect of cyclization (cataloged by water loss) appears to be dominant. The D-peptide and G-peptides show <1% water loss (Fig. S1B, C), while E-peptide shows 95% water loss (Figure S1A). Also, MS2 evidence localizes the water loss to the N-terminus in the cases of N-terminal E residues, and near T in the G-peptide. To see if N-terminal E-localized cyclization caused water loss in our antibody, we examined the observed ratio of species that had lost/retained water for the peptides in question. Notably, for the case of N-terminal D (LC) and E (HC) in the antibody, E has 12% water loss, nearly 15x that of D (data not shown). Literature reports also have documented water loss in N-terminal E residues in proteins, including antibodies.Citation62-64 Thus, the MS data supports the hypothesis that a 5-membered ring can form to interfere with N-terminal GEE labeling. Although a D-residue in a corresponding position cannot form such a ring, some suppression is still observed suggesting that both ring formation and the N-terminal positive charge can suppress reactivity, but that the latter effect is less than the former. Overall, most of the data can be explained through an examination of SASA, charge, or bonding effects. Exceptions, such as the high reactivity of E239, remain to be explained.
To further explore this potential correlation, we examined the data from a footprinting study by Zhang et alCitation42 in order to examine the relationship between the solvent accessibility area and the relative reactivity of the constituent probes. In that study, carboxyl group footprinting was used to characterize the conformational changes introduced in Her4 (a transmembrane receptor tyrosine kinase) during the dimerization and the phosphorylation process. For the purpose of our study, we derived solvent accessibility areas from the known crystal structure of Her4 monomer (PDB ID: 3BCE), and plotted them against the relative extent of modification of the N-lobe of Her4 kinase domain as calculated in the previous work. The extent of modification is a similar measure to our rate measurements in this study; they essentially imply a rate using two data points, the zero (unmodified) and the modified measurements. The values for extent of relative modification were uniformly scaled between the range of 0 and 1, with 1 corresponding to the maximal amount of modification. The resulting plot shows a positive and significant correlation (R2 = 0.54) similar to , and is shown in Figure S2. This analysis further supports our observation of a significant correlation between the SASA and reactivity and supports our assumptions of relatively equal reactivity of D and E for the purposes of analyzing the data. However, as for our data, some residues, such as E690 for Her4 monomer, deviate from the overall trend.
Comparison of CL techniques
HRF was recently used by Deperalta et al to study the structure of the monomer and dimer form of the therapeutic IgG1 mAb in order to map the dimer interface.Citation45 Here, these results from HRF on the monomer are compared to those of GEE labeling to understand the strengths and weaknesses of each approach.
As there are fundamental differences in the target residues in HRF and GEE labeling approaches, different results can be expected in any comparison of the techniques. HRF can label up to 18 of the 20 side-chain residues, although, due to the nearly 1000-fold variations in reactivity, the highly reactive sulfur containing and aromatic residues dominate the data.Citation36 As the reactivity of D and E are shown to be relatively equal, they should be observed at equal frequencies. The D and E residues tend to be on the protein surface due to their hydrophilic nature. In contrast, the HRF technique labels hydrophobic residues more readily because these residues are often protected from the bulk solvent. The mAb sequence revealed that there are 60 potential probes for GEE labeling (e.g., D, E, and the C-terminus) and 475 potential probes for HRF (including the 14 residues CFHIKLMPRSTVWY). This represents ˜9 and 71% of the total sequence coverage, respectively (out of 667 residues total), such that HRF has 8-fold greater potential coverage than GEE labeling. However, when considering the fraction of the target residues on the surface, we see that 67% (39 out of possible 58) have accessibility of one-third or more of their total possible SASA when examining D and E, while 43% of the potential HRF targets (203/475) have similar accessibility. This explains why 55% of the D and E residues were seen to be labeled (32/58) while only 21% of the possible HRF targets (100/475) were oxidized. For HRF, the modification of some low reactive and accessible residues can be difficult to prove because their low abundant signal is masked by highly reactive residues. Thus, although there are 8-fold more probes for HRF, the fraction of probes observed to be labeled across the entire sequence was only 3-fold higher.
A granular perspective is shown by results from selected peptides in . The first two rows of show contrasting cases where the rate constants are among the highest for HRF, whereas no labeling was observed for the case of GEE labeling experiments (columns 2–3 of ). This is supported by the highly reactive and solvent accessible profile of the constituent oxidative probes (column 4), while D/E residues are relatively less accessible as seen in column 5. HC peptide 262-280 shows an opposite result, exhibiting a relatively low rate of oxidation, while the rate of GEE labeling was one of the highest among all peptides (in relative terms). This is to be expected because it is composed of moderately reactive and solvent accessible oxidative probes (P263, 0.4 Å2; H274, 75.8 Å2; P277, 70.2 Å2), while it has four highly accessible D, E residues, leading to significant labeling by GEE.
Table 4. Comparison of labeling results of oxidative and carboxyl footprinting experiments. (A) Summary of sequence coverage using two approaches. (B) Examples of specific peptides showing different relative reaction kinetics in each case depending upon the solvent accessibility of the probes
Results from model peptides suggest that the relative reactivity of the target probes are similar. This can be particularly helpful for estimating the relative solvent accessibility area directly from the observed rate constants without the need to model the reactivity of the individual probes. This is in contrast with HRF, where there is wide variation in the relative reactivity of the probes, requiring complex modeling approaches to account for such variation. The drawbacks of the GEE-labeling approach include the limited number of probes available and long reaction times, rendering it of limited utility to study biomolecular dynamics. Because D and E residues are less reactive in the case of HRF versus being the prime targets in the case of GEE labeling, the two approaches clearly provide complementary information.
Discussion
This work shows the application of carboxyl footprinting on a glycosylated mAb, representing the structural characterization of a very important class of biotherapeutic proteins. It also shows quantitative characterization of GEE labeling by mean of dose response plots. This assessment of reproducibility indicates that structure assessment of a biologic using peptide level resolution can be characterized with a variation of <2%. Homology models were generated, and the corresponding solvent accessibility values were found to be positively and significantly correlated with SASA. The results provide complementary information to oxidative labeling, increasing the total sequence coverage map of the protein by ˜10%. Although oxidative labeling provides eight times more probes than the GEE labeling, the fraction of probes observed to be labeled across the entire sequence was only three-fold higher than the GEE labeling method. The DR curves are observed to be linear up to 45% labeling of individual peptides, indicating minimal structural perturbation. In contrast to HRF, results from peptides suggest similar reactivity of the D/E/C-terminal probes, which can be particularly beneficial to directly compare the rate constants from the peptides and their individual probes to draw solvent accessibility comparisons. Reduction in labeling efficiency of the N-terminal D and E is likely due to positive charge for both and intra-molecular cyclization for E. Overall, GEE labeling is a promising technology for structure assessment of mAbs.
Materials and Methods
Assessment by size-exclusion chromatography
The monomeric form of the therapeutic IgG1 mAb drug substance was enriched via fractionation from size-exclusion chromatography (SEC). The enriched monomer sample was determined to be 98% pure by SEC. Final concentration for the monomer was approximately 30 mg/mL.
Labeling, digestion and deglycosylation
The enriched monomer (98% pure) was diluted to 1 mg/mL in 1X phosphate-buffered saline (PBS) pH 7.4 (1.5 mM potassium phosphate monobasic, 155.2 mM sodium chloride, 2.7 mM sodium phosphate dibasic) (Invitrogen, Grand Island, NY) and 10 ug was taken for labeling reactions. Stock solutions of glycine ethyl ester (GEE) and 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) (Thermo Fisher Scientific, Rockford, IL) were made in 1X PBS pH 7.4 at a concentration of 1M and 0.025 M, respectively. GEE and EDC were added to the reaction vial so their final concentration was 240 mM and 5 mM, respectively. The labeling reaction was carried out at room temperature for 0, 1.5, 3, 6.5, and 10 minutes. The reaction was quenched by the addition of 1% formic acid (Thermo Scientific, Rockford, IL) to a final concentration of 0.1% (v:v). Next, the labeled monomer was buffer exchanged two times with 20 volumes excess of 8 M urea using a 0.5 mL 3K MWCO filter (Millipore, Billerica, MA). The monomer was deglycosylated at 37°C for 1 hour on the filter using 2 uL of undiluted PNGaseF (New England Biolabs, Ipswich, MA). The deglycosylation process allows for easier MS2 identification of the glycopeptide and its labeled forms, thereby increasing sequence coverage and structural resolution. The mAb was then reduced in 200 uL of 10 mM dithiothreitol (Acros, Rockford, IL) in 8 M Urea at 37°C for 1 hour on the filter and alkylated with 200 uL of 25 mM iodoacetic acid (Fluka Analytical, St. Louis, MO) in 8 M urea on the filter at room temperature in the dark for 30 minutes. Next, the sample was concentrated to a final volume of approximately 50 uL and then water (HPLC grade) was added to adjust the concentration of 8 M urea to 4 M urea. The monomer was digested on the filter with lysyl endopeptidase (Lys-C) Mass Spectrometry Grade (Wako Chemicals, Richmond, VA) in an enzyme: substrate ratio of 1: 20 for 30 minutes at 37°C. Next, the 4 M urea was buffer exchanged with 25 mM ammonium bicarbonate, pH 7.4, two times with 20 volumes excess and contents of the filter was transferred to an Eppendorf tube for digestion with sequencing grade trypsin (Promega, Madison, WI) with an enzyme:substrate ratio of 1:20 at 37°C overnight. Finally, samples were diluted in 0.1% formic acid (Thermo Scientific, Rockford, IL) prior to LC-MS/MS analysis. Triplicate samples with a relative molar concentration of EDC:mAb of 633:1 with labeling reaction times of 0, 1.5, 3, 6, and 10 minutes were analyzed by MS in order to generate quantitative dose response plots, and to assess the reproducibility and linearity of the labeling reaction. Another set of experiments used a relative molar concentration of EDC:mAb of 6330:1 for examining the effect of excessive EDC conditions over the native conformation of the mAb.
Stock synthetic peptides (Thermo Scientific, Rockford, IL) were diluted to 1 mg/mL (˜ 1 mM) in HPLC grade water. Next, 10 µg of peptide was labeled using the same protocol as for the intact mAb. Finally, samples were diluted in 0.1% formic acid prior to LC-MS/MS analysis.
LC-MS analysis
Data were acquired on an Orbitrap Elite mass spectrometer (Thermo Electron, San Jose, CA) interfaced with a Waters nanoAcquity UPLC system (Waters, Taunton, MA). For intact chain analysis, full scan MS1 data was collected in the Orbitrap detector at a resolution of 120000 followed by 3 successive ion trap (IT) scans. Samples were desalted on a trap column (180 μm ċ 20 mm packed with C18 Symmetry, 5 μm, 100 Å (Waters, Taunton, MA)) and subsequently resolved on a reversed phase column (75 μm × 250 mm nano column, packed with C18 BEH130, 1.7 μm, 130 Å (Waters, Taunton, MA)) using a gradient of 10 to 70% mobile phase B (0.1% formic acid and acetonitrile (ACN)) and mobile phase A (100% water/0.1 % formic acid) over a period of 30 minutes at ambient temperature and a flow rate of 300 nl/min. A total of 2 pmol of peptides were loaded on column in a 7 uL injection. Peptides eluting from the column were introduced into the nano-electrospray source with a capillary voltage of 2.5 kV. For all peptide analyses, chromatographic resolution was performed using a 2 to 42% acetonitrile gradient over a period of 40 minutes based on a 1% organic gradient at 37 °C. A full scan was recorded for eluted peptides (m/z range of 380–1800) in the FT mass analyzer at resolution R of 60,000 followed by MS/MS of the eight most intense peptide ions scans and by two MS/MS scans using an inclusion list. The m/z values in the inclusion list were derived from the theoretical digestion of the two largest peptides (HC 154-211 and LC 62-103) using the mAb sequence. MS/MS spectra were generated for peptides with a minimum signal of 2000 by collision-induced dissociation of the peptide ions at normalized collision energy of 35%, an isolation width of 2.5, and an activation time of 20 msec.
The resulting MS data were analyzed using ProtMapMS v.2.0, a commercial software package developed by NeoProteomics, Inc. specifically for the automated analysis of CL-footprinting data.Citation37 The data were searched for tryptic peptides of mAb using accuracy values of 10 ppm and 0.35 Daltons for MS1 and MS2 scans, respectively, with the allowed variable modifications of 85.0528 and 57.0215 Daltons, corresponding to the mass of GEE labeled amide form of the mAb and its hydrolyzed product, respectively. For each sample and labeling time ProtMapMS identified the labeled peptides and their unmodified forms using the highly accurate precursor ion masses in conjunction with the MS2 product ion spectra. Extracted ion chromatograms (EICs) of the labeled and original peptide forms were derived from MS1 data, and the corresponding peak areas were integrated. The fraction of the unlabeled peptide, compared to the sum total area of all its detected forms, was calculated at each reaction time. Experimental dose response plots were generated from the triplicate experiments, and fit to a first order exponential equation, providing rate constants for the labeling reaction for each detected peptide.
Structural model for mAb and surface accessibility calculations
The structure selected as a template to model the mAb in this study is from an anti-phenobarbital, subclass IgG1 antibody.Citation22 It consists of two HCs and two LCs, and displays a distorted Y-shaped molecule. As this crystal structure demonstrates a moderate, roughly symmetrical conformation, it has been selected to serve as a template to construct a structural model for the mAb in this work. A stretch of 13 residues was disordered and missing in the HC of 1BJ1 and this patch was built into the Fab structural model through homology modeling using the Swiss-Model, an automated protein homology-modeling server.Citation65 To generate a full model of the mAb, a 3.2 Å resolution crystal structure of an intact IgG1 monoclonal antibody was used as a template (PDB ID: 1IGYCitation22). A structural model containing two HCs in the correct sequence was generated based on 1IGY using the automated Swiss-Model. The template (1IGY) was five residues shorter at the C-terminus compared to the mAb in this study, so the last five residues of the HC were not incorporated into the structural model. Two copies of the Fab structural model were overlaid onto the two HCs using the alignment of the CH1 domains (residues 115-224). The connective regions (residues 221-227) were regularized and energy minimized in program COOT.Citation66 The final mAb structure model contains two copies Fab domain based on 1BJ1, and the hinges and the Fc domain based on 1IGY. Solvent accessibility calculations on the homology model were performed using VADAR.Citation37,67 Pymol was used to visualize the homology model and map the GEE labeled residues.Citation68 The relationship between the observed extent of modification and the solvent accessible area of the corresponding residues was calculated using Pearson's correlation coefficient.Citation60
Disclosure of Potential Conflicts of Interest
Dr. Chance and Dr. Kaur acknowledge that they are consultants of the company NeoProteomics, Inc., which markets the software product ProtMapMS and provides services in the field of monoclonal antibody structure assessment.
Acknowledgments
We would like to thank Prof. Masaru Miyagi and Dr. Sasa Belic for scientific discussions, Dr. Serguei Ilchenko for experimental help, and Sean Maxwell for helping with ProtMapMS.
Supplemental_Material.zip
Download Zip (310 KB)Funding
This work was supported in part by NIH grants R01-EB-09998 and P30-EB-09866 (to MRC), and in part by Genentech (to NeoProteomics, Inc.).
Supplementary Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Rader RA. (Re)defining biopharmaceutical. Nat Biotechnol 2008; 26:743–51.
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol 2010; 28:917--24.
- Harris M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol 2004; 5:292–302.
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9:423–30.
- McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs 2011; 3:209–17.
- Kaltashov IA, Bobst CE, Abzalimov RR, Wang G, Baykal B, Wang S. Advances and challenges in analytical characterization of biotechnology products: mass spectrometry-based approaches to study properties and behavior of protein therapeutics. Biotechnology advances 2012; 30:210–22.
- Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nature methods 2007; 4:798–806.
- Kueltzo LA, Wang W, Randolph TW, Carpenter JF. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze-thawing. J Pharm Sci 2008; 97:1801–12.
- Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov 2005; 4:298–306.
- Sali A, Glaeser R, Earnest T, Baumeister W. From words to literature in structural proteomics. Nature 2003; 422:216–25.
- Hegyi H, Gerstein M. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol 1999; 288:147–64.
- Sadowski MI, Jones DT. The sequence-structure relationship and protein function prediction. Curr Opin Struct Biol 2009; 19:357–62.
- Anfinsen CB. Principles that govern the folding of protein chains. Science 1973; 181:223–30.
- Pelton JT, McLean LR. Spectroscopic methods for analysis of protein secondary structure. Anal Biochem 2000; 277:167–76.
- Garidel P, Hegyi M, Bassarab S, Weichel M. A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy. Biotechnol J 2008; 3:1201–11.
- Sreerama N, Venyaminov SY, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal Biochem 2000; 287:243–51.
- Laue TM. Analytical ultracentrifugation. Curr Protoc Protein Sci 2001; Chapter 7:Unit 7 5.
- Jiang Y, Li C, Nguyen X, Muzammil S, Towers E, Gabrielson J, Narhi L. Qualification of FTIR spectroscopic method for protein secondary structural analysis. J Pharm Sci 2011; 100:4631–41.
- Leavitt S, Freire E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr Opin Struct Biol 2001; 11:560–6.
- Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol 1991; 222:311–33.
- Schotte F, Lim M, Jackson TA, Smirnov AV, Soman J, Olson JS, Phillips GN, Jr., Wulff M, Anfinrud PA. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science 2003; 300:1944–7.
- Harris LJ, Skaletsky E, McPherson A. Crystallographic structure of an intact IgG1 monoclonal antibody. J Mol Biol 1998; 275:861–72.
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 2001; 293:1155–9.
- Guddat LW, Herron JN, Edmundson AB. Three-dimensional structure of a human immunoglobulin with a hinge deletion. Proc Natl Acad Sci U S A 1993; 90:4271–5.
- Konermann L, Tong X, Pan Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J Mass Spectrom 2008; 43:1021–36.
- Liu T, Pantazatos D, Li S, Hamuro Y, Hilser VJ, Woods VL, Jr. Quantitative assessment of protein structural models by comparison of H/D exchange MS data with exchange behavior accurately predicted by DXCOREX. J Am Soc Mass Spectrom 2012; 23:43–56.
- Houde D, Berkowitz SA, Engen JR. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J Pharm Sci 2011; 100:2071–86.
- Marcsisin SR, Engen JR. Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal Bioanal Chem 2010; 397:967–72.
- Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev 2009; 28:785–815.
- Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct 2006; 35:251–76.
- Maleknia SD, Ralston CY, Brenowitz MD, Downard KM, Chance MR. Determination of macromolecular folding and structure by synchrotron x-ray radiolysis techniques. Anal Biochem 2001; 289:103–15.
- Mendoza VL, Vachet RW. Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal Chem 2008; 80:2895–904.
- Stadtman ER, Berlett BS. Fenton chemistry. Amino acid oxidation. J Biol Chem 1991; 266:17201–11.
- Brunner J, Semenza G. Selective labeling of the hydrophobic core of membranes with 3-(trifluoromethyl)-3-(m-[125I]iodophenyl)diazirine, a carbene-generating reagent. Biochemistry 1981; 20:7174–82.
- Hoare DG, OlsonA, Koshland DE, Jr. The reaction of hydroxamic acids with water-soluble carbodiimides. A Lossen rearrangement. J Am Chem Soc 1968; 90:1638–43.
- Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev 2007; 107:3514–43.
- Kaur P, Kiselar JG, Chance MR. Integrated algorithms for high-throughput examination of covalently labeled biomolecules by structural mass spectrometry. Anal Chem 2009; 81:8141–9.
- Watson C, Sharp JS. Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting. The AAPS journal 2012; 14:206–17.
- Xu G, Kiselar J, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Analytical chemistry 2005; 77:3029–37.
- Federici M, Lubiniecki A, Manikwar P, Volkin DB. Analytical lessons learned from selected therapeutic protein drug comparability studies. Biologicals 2013; 41:131–47.
- Wen J, Zhang H, Gross ML, Blankenship RE. Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc Natl Acad Sci U S A 2009; 106:6134–9.
- Zhang H, Shen W, Rempel D, Monsey J, Vidavsky I, Gross ML, Bose R. Carboxyl-group footprinting maps the dimerization interface and phosphorylation-induced conformational changes of a membrane-associated tyrosine kinase. Mol Cell Proteomics 2011; 10:M110 005678.
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 2000; 29:291–325.
- Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure 1998; 6:1153–67.
- Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, Ling V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. mAbs 2013; 5:86–101.
- Brenowitz M, Chance MR, Dhavan G, Takamoto K. Probing the structural dynamics of nucleic acids by quantitative time-resolved and equilibrium hydroxyl radical “footprinting”. Curr Opin Struct Biol 2002; 12:648–53.
- Dhavan GM, Crothers DM, Chance MR, Brenowitz M. Concerted binding and bending of DNA by Escherichia coli integration host factor. J Mol Biol 2002; 315:1027–37.
- Ralston CY, Sclavi B, Sullivan M, Deras ML, Woodson SA, Chance MR, Brenowitz M. Time-resolved synchrotron X-ray footprinting and its application to RNA folding. Methods Enzymol 2000; 317:353–68.
- Liu R, Guan JQ, Zak O, Aisen P, Chance MR. Structural reorganization of the transferrin C-lobe and transferrin receptor upon complex formation: the C-lobe binds to the receptor helical domain. Biochemistry 2003; 42:12447–54.
- Kiselar JG, Janmey PA, Almo SC, Chance MR. Structural analysis of gelsolin using synchrotron protein footprinting. Mol Cell Proteomics 2003; 2:1120–32.
- Chance MR. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem Biophys Res Commun 2001; 287:614–21.
- Guan JQ, Takamoto K, Almo SC, Reisler E, Chance MR. Structure and dynamics of the actin filament. Biochemistry 2005; 44:3166–75.
- Maleknia SD, Kiselar JG, Downard KM. Hydroxyl radical probe of the surface of lysozyme by synchrotron radiolysis and mass spectrometry. Rapid Commun Mass Spectrom 2002; 16:53–61.
- Kaur P, O'connor PB. Algorithms for automatic interpretation of high resolution mass spectra. J Am Soc Mass Spectrom 2006; 17:459–68.
- Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 2012; 4:17–23.
- Parker C MV, Mocanu M, Dicheva N, and Warren M. Mass Spectrometry for Post-Translational Modifications. Neuroproteomics: Boca Raton (FL): Taylor & Francis, 2010.
- Zhang H, Wen J, Huang RY, Blankenship RE, Gross ML. Mass spectrometry-based carboxyl footprinting of proteins: method evaluation. Int J Mass Spectrom 2012; 312:78–86.
- Vahidi S, Stocks BB, Liaghati-Mobarhan Y, Konermann L. Submillisecond protein folding events monitored by rapid mixing and mass spectrometry-based oxidative labeling. Anal Chem 2013; 85:8618–25.
- Chen JW, Rempel DL, Gross ML. Temperature Jump and Fast Photochemical Oxidation Probe Submillisecond Protein Folding. J Am Chem Soc 2010; 132:15502–04.
- Rodgers JL, Nicewander WA. 13 Ways to Look at the Correlation-Coefficient. American Statistician 1988; 42:59–66.
- Miller S, Janin J, Lesk AM, Chothia C. Interior and surface of monomeric proteins. J Mol Biol 1987; 196:641–56.
- Liu YD, Goetze AM, Bass RB, Flynn GC. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J Biol Chem 2011; 286:11211–7.
- Yu L, Vizel A, Huff MB, Young M, Remmele RL, Jr., He B. Investigation of N-terminal glutamate cyclization of recombinant monoclonal antibody in formulation development. J Pharm Biomed Anal 2006; 42:455–63.
- Moorhouse KG, Nashabeh W, Deveney J, Bjork NS, Mulkerrin MG, Ryskamp T. Validation of an HPLC method for the analysis of the charge heterogeneity of the recombinant monoclonal antibody IDEC-C2B8 after papain digestion. J Pharm Biomed Anal 1997; 16:593–603.
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006; 22:195–201.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66:486–501.
- Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 2003; 31:3316–9.
- DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol 2002; 12:14–20.