
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
To harness the potent tumor-killing capacity of T cells for the treatment of CD19+ malignancies, we constructed AFM11, a humanized tetravalent bispecific CD19/CD3 tandem diabody (TandAb) consisting solely of Fv domains. The molecule exhibits good manufacturability and stability properties. AFM11 has 2 binding sites for CD3 and 2 for CD19, an antigen that is expressed from early B cell development through differentiation into plasma cells, and is an attractive alternative to CD20 as a target for the development of therapeutic antibodies to treat B cell malignancies. Comparison of the binding and cytotoxicity of AFM11 with those of a tandem scFv bispecific T cell engager (BiTE) molecule targeting the same antigens revealed that AFM11 elicited more potent in vitro B cell lysis. Though possessing high affinity to CD3, the TandAb mediates serial-killing of CD19+ cells with little dependence of potency or efficacy upon effector:target ratio, unlike the BiTE. The advantage of the TandAb over the BiTE was most pronounced at lower effector:target ratios. AFM11 mediated strictly target-dependent T cell activation evidenced by CD25 and CD69 induction, proliferation, and cytokine release, notwithstanding bivalent CD3 engagement. In a NOD/scid xenograft model, AFM11 induced dose-dependent growth inhibition of Raji tumors in vivo, and radiolabeled TandAb exhibited excellent localization to tumor but not to normal tissue. After intravenous administration in mice, half-life ranged from 18.4 to 22.9 h. In a human ex vivo B-cell chronic lymphocytic leukemia study, AFM11 exhibited substantial cytotoxic activity in an autologous setting. Thus, AFM11 may represent a promising therapeutic for treatment of CD19+ malignancies with an advantageous safety risk profile and anticipated dosing regimen.
Abbreviations
| TandAb | = | tandem diabody |
| BiTE | = | bispecific T cell engager |
| ORR | = | overall response rate |
| NHL | = | non-Hodgkin lymphoma |
| CAR | = | chimeric antigen receptor |
| NK | = | natural killer |
| Fc | = | fragment crystallizable |
| Fv | = | variable fragment |
| scFv | = | single-chain variable fragment |
| B-ALL | = | B-precursor acute lymphoblastic leukemia |
| t1/2 | = | terminal elimination half-life |
| CNS | = | central nervous system |
| CD | = | cluster of differentiation |
| VH | = | variable heavy |
| VL | = | variable light |
| FR | = | framework region |
| IgG | = | immunoglobulin G |
| KD | = | dissociation constant |
| ka | = | association rate constant |
| kd | = | dissociation rate constant |
| CDR | = | complementarity determining region |
| SPR | = | surface plasmon resonance |
| His | = | histidine |
| CHO | = | Chinese hamster ovary |
| FACS | = | fluorescence-activated cell sorting |
| EC50 | = | half maximal effective concentration |
| CLL | = | chronic lymphocytic leukemia |
| SABC | = | standardized antibody binding capacity |
| PBMC | = | peripheral blood mononuclear cell |
| HSA | = | human serum albumin |
| IL | = | interleukin |
| TNF | = | tumor necrosis factor |
| IFN | = | interferon |
| FcgR | = | Fc gamma receptor |
| FcRn | = | neonatal Fc receptor |
| s.c. | = | subcutaneous |
| i.v. | = | intravenous |
| CL | = | clearance |
| Vss | = | volume of distribution at steady state |
| AUCtot | = | total area under the curve |
| Cmax | = | maximal concentration |
| WBA | = | whole body autoradiography |
| MSD | = | MesoScale Discovery |
| ECL | = | electrochemiluminescence |
| NOD/scid | = | nonobese diabetic/severe combined immunodeficiency |
| BBB | = | blood-brain barrier |
| DMSO | = | dimethyl sulfoxide |
| CCS | = | cell culture supernatant |
| PES | = | polyethersulfone |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEC | = | size exclusion chromatography |
| MWCO | = | molecular weight cut-off |
| SE-HPLC | = | size exclusion high-pressure liquid chromatography |
| FCS | = | fetal calf serum |
| Fab | = | fragment antigen-binding |
| PBS | = | phosphate buffered saline |
| PI | = | propidium iodide |
| E:T | = | effector:target |
| F | = | fluorescence |
| SD | = | standard deviation |
| ctrl. | = | control |
| w/o | = | without |
| HMF | = | high molecular weight forms |
| LMF | = | low molecular weight forms |
| PHA | = | phytohemagglutinin |
Introduction
The treatment of B cell malignancies was transformed by the introduction of therapeutic antibodies.Citation1 Overall response rates (ORR) for a subset of non-Hodgkin lymphoma (NHL) are now greater than 90% under regimens employing the first generation of anti-CD20 antibodies.Citation2 However, several NHL subtypes are not as responsive to these therapies, and the majority of patients with responsive NHL eventually relapse after the standard combined immunotherapy/chemotherapy regimen. Thus, both new first-line therapies and new salvage regimens are required for these unmet needs.
A number of innovative antibody-based therapeutics are under development for B cell malignancies. The principal optimizations pursued are enhancement of effector function and selection of tumor-targeting cellular marker. The Fc-optimization strategy relies upon engineering the Fc-domain to achieve more potent interaction with the Fcγ receptors expressed on endogenous immune effector cells; Fc-optimized antibodies, targeting CD20, obinutuzumab,Citation3 and targeting CCR4, mogamulizumab,Citation4 have been approved, and another targeting CD19, MOR208,Citation5 has entered clinical trials.
T cells are the most potent tumor-killing effector cells, but they cannot be recruited by canonical antibodies. Recently, 2 approaches to harness T cells for killing tumor cells have been developed. The first approach depends upon the introduction into T cells of an engineered chimeric antigen receptor (CAR) consisting of an extracellular domain targeting a desired antigen and intracellular signaling domains to trigger immune cell activation upon target engagement. A number of these CAR T cells have entered trials for B cell malignancies.Citation6 The second approach utilizes T cell recruitment via an engineered bispecific antibody, either full-length or fragment, possessing affinity for both a tumor-targeting cellular marker and, commonly, the activating receptor CD3. One such full-length bispecific antibody that recruits T, and natural killer (NK), cells, anti-Epcam/CD3 catumaxomab,Citation7 is approved for malignant ascites while another, anti-CD20/CD3 FBTA05Citation8 is in a clinical trial for B cell malignancies. However, the most promising bispecific antibody to date is the bispecific T cell engager (BiTE) blinatumomab, a tandem single-chain variable fragment (scFv) antibody targeting CD19/CD3 that was recently approved for the treatment of the B cell malignancy B-precursor acute lymphoblastic leukemia (B-ALL).Citation9
Blinatumomab possesses 2 monovalent domains: a high affinity CD19 scFv and a low-affinity CD3 scFv. It has demonstrated impressive efficacy in the clinic, exhibiting high clinical response rates in patients with relapsed/refractory B-ALL and NHL,Citation10 but the molecule has limitations. Its molecular weight (∼55 kDa) is below the glomerular filtration threshold, which results in a very short terminal elimination half-life (t1/2) of slightly over 1 hour.Citation11 To maintain suitable drug concentrations, blinatumomab is administered by continuous infusion via pumps, which results in substantial inconvenience for patients and an increased possibility of treatment-related adverse events (e.g., infections at site of implanted port). Also, up to 70% of patients exhibit symptoms consistent with transient cytokine release.Citation12 Finally, 15–20% of patients treated with blinatumomab experience severe central nervous system (CNS) adverse events, although these have always resolved upon suspension of drug administration.Citation12
To develop a highly potent bispecific immune effector cell-recruiter for B cell malignancies, and to demonstrate the applicability of the TandAb format in constructing potent immune cell-recruiting bispecific antibodies with good drug like properties, we employed the TandAb platform to engineer the CD19/CD3 TandAb AFM11. We hypothesized that it would exhibit higher potency than the BiTE because the TandAb format is bivalent for each specificity, yet permits a similarly close apposition of the target and effector cell membranes that is implicated in conferring high potency to bispecific fragment antibodies.Citation13 Due to the TandAb's molecular weight (∼105 kDa), it is not subject to glomerular filtration; our NK cell-recruiting (anti-CD30/CD16A) TandAb possesses a t1/2 of almost 20 h in man (data not shown). Thus we anticipate the ability to dose AFM11 by bolus infusion. Here, we describe the in vitro and in vivo characterization of AFM11 in a panel of biophysical, functional activity, human ex vivo, and safety risk-profiling assays.
Results
Humanization, affinity maturation, and germlining of variable domains derived from UCHT1 and HD37
Humanization of the anti-human CD3 mAb UCHTCitation14 and the anti-human CD19 mAb HD37Citation15 was performed by chain-shuffling via phage display. Synthetic genes encoding the respective variable heavy and light chains were designed and cloned into the pEXHAM phage display vector.Citation16 In the first step, the murine light chains were substituted by a repertoire of human κ and λ chains, and the resulting libraries were selected on CD3+ Jurkat cells or recombinant CD19-Fc, respectively, yielding chimeric clones containing the murine VH chain in combination with human VL chains.
In the second step, the murine heavy chains were substituted with a repertoire of human VH from FR1 through FR3 fragments, including CDR1/CDR2, while retaining the original murine VH CDR3 sequences. The murine JH elements were replaced by human JH1. For the CD3 domain, panning was performed on Jurkat cells with prior depletion on the CD3− Jurkat-derived cell line JRT3-T3.5. A single CD3-specific clone (C4) was strongly enriched after 3 rounds of selection and competitive elution with the IgG UCHT1. In the case of CD19, the scFv libraries were screened on HEK293 cells transfected with human CD19 after prior depletion on un-transfected HEK293 cells. Competitive elution using the IgG HD37 yielded 2 individual clones with highly homologous sequences; the clone (H22) with higher affinity was selected for further affinity maturation.
To further improve binding properties of the humanized anti-CD19 and anti-CD3 clones, affinity maturation was performed by permutation of the contact residues in CDR1/2 of the VH and CDR1/2/3 of the VL chain, respectively. The positions in the VH and VL chains selected for permutation were selected based on an analysis of Honegger and PlückthunCitation17 and according to Padlan et al.Citation18 Synthetic gene fragments containing permutations in the selected positions were synthesized and the respective libraries were cloned into the phage display vector pEXHAM.
The anti-CD3 scFv libraries were screened on Jurkat cells to isolate clones with increased affinity employing flow cytometric affinity titration. Thus, we identified clone LCHC21 as the best candidate for further development. Monovalent binding affinity of this scFv was improved by 8-fold, KD (LCHC21) = 1 nM (Jurkat cells) versus KD (C4) = 8 nM (Jurkat cells).
Affinity matured anti-CD19 scFv were identified by screening the libraries on HEK293 cells transfected with human CD19. The stringency of the selection process was further enhanced by introducing the parental clone as a competitor. A panel of clones with substantially improved affinities, compared with the parental humanized clone H22 (KD = 10 nM), were identified by surface plasmon resonance (SPR) and confirmed by titration on cells and measurements of cell-surface retention. The clone M13 (KD = 0.9 nM) was selected for further development.
Finally, the variable domains of the anti-CD3 scFv LCHC21 and the anti-CD19 scFv M13 were subjected to a germlining process since we identified several amino acid residues in the framework regions of these scFvs that deviated from the germline sequence. These positions were back-mutated, yielding clones LCHC21-ktay and M13-sm, without altering the affinity of both binding domains, and in the final TandAb format each domain retained its binding properties to the nominal target.
Construction of the AFM11 TandAb and comparators
We prepared mammalian expression vectors to compare the binding and T cell-mediated cellular cytotoxicity of: i) AFM11, a tetravalent TandAb containing the anti-CD19 Fv M13-sm and anti-CD3 Fv LCHC21-ktay described above; ii) an scFv2, or BiTE, also containing these Fvs; and iii) the BiTE blinatumomab, constructed as described,Citation19 utilizing the Fv domains of the anti-CD3 antibody TR66Citation20 and those of HD37. We also constructed a control TandAb in which the CD19 targeting domains were substituted with domains specific for human serum albumin. A hexa-histidine-tag (His6-tag) was added to the carboxyl terminus of each construct to aid in the purification and detection of the protein products. The gene organization and gene product for the TandAb is shown in . The tagged version of AFM11 is referred to as “AFM11-His.” The CD19M13-sm/CD3LCHC21-ktay scFv2 is referred to as “the CD19M13-sm/CD3LCHC21-ktay BiTE”; the latter scFv2 is referred to as “the CD19HD37/CD3TR66 BiTE,” a blinatumomab analog.
Figure 1. Design, production, and purification of recombinant AFM11-His. (A) Scheme of AFM11-His gene design. The gene construct is shown from 5′ to 3′-end encoding the monomeric TandAb subunit with N-terminal signal sequence, VH and VL domains separated by identical (Gly2Ser)2 linkers (L1, L2, L3), and the C-terminal hexa-histidine-tag (His6-tag). Upon expression, 2 polypeptide gene products dimerize in a head-to-tail fashion. (B) AFM11-His was expressed in 10-day fed-batch cultures of stably-transfected CHO cells which grow to high cell densities, maintain high viability, and secrete the AFM11-His product into the cell culture supernatant (CSS). (C) AFM11-His was purified by Ni-NTA Superflow chromatography followed by preparative size exclusion chromatography (SEC) on Superdex 200. High purity of the AFM11-His TandAb homodimer was demonstrated by analytical SEC. Purified AFM11-His runs in SEC as a single peak with a retention time that is consistent with its expected molecular weight of 106 kDa. (D) Integrity of the purified AFM11-His protein was analyzed by SDS-PAGE. Under denaturing, reducing and non-reducing conditions, AFM11-His monomers run at the expected size of ∼50 kDa. (E) Molecular forms of AFM11-His were analyzed by analytical SEC after incubation at 4°C, 21°C, or 37°C for the indicated numbers of days. High Molecular weight Forms (HMF) appear as a mixture of aggregates and species with an apparent molecular weight of ∼200 kDa, consistent with tetrameric forms; AFM11-His homodimer represents the main chromatographic peak under all conditions assayed; Low Molecular weight Forms (LMF) represent a minor fraction under all conditions and are consistent with the monomeric form of the AFM11 with molecular weights <60 kDa.
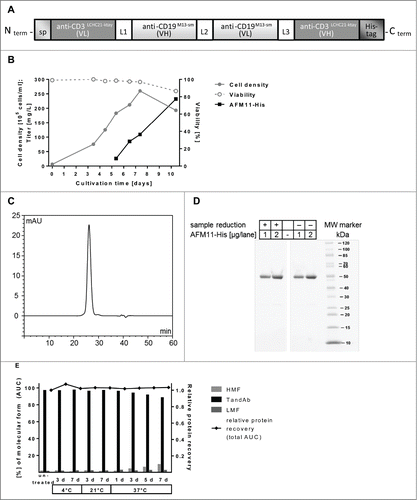
Production and biophysical characterization of AFM11-His
For the production of AFM11-His, suspension-adapted Flp-In Chinese hamster ovary (CHO) cells were transfected with the respective gene constructs and selected with Hygromycin B to yield stably transfected CHO cells having a single copy of the product gene site-specifically integrated into their genome. Stable cell pools typically recovered within 2–3 weeks after the transfection, and were eventually used for the production of secreted recombinant protein in shake flask fed-batch cultures. High cell densities of up to 2.5 × 107 cells/mL and product titers above 200 mg/L of AFM11-His (produced from a single gene copy/cell) were reached. Cell culture supernatants were harvested after 10 d at culture viabilities >80% (). Lab-scale production titers of untagged AFM11 using the same expression systems were very similar to those of AFM11-His (data not shown).
High purity and homogeneity of the AFM11-His TandAb homodimer (typically >90%) was demonstrated by SE-HPLC (). The purified product forms a single peak, and the peak retention time is consistent with an apparent molecular weight of ∼106 kDa.
Integrity of the purified AFM11-His protein was assayed by SDS-PAGE. Under denaturing, reducing, and non-reducing conditions, monomeric AFM11-His polypeptides run at ∼50 kDa ().
Purified AFM11-His protein exhibits very good thermal stability for up to 7 d in standard acetate buffer (). A slight increase in higher molecular weight forms, which are a mixture of tetramers of the AFM11-His polypeptide and larger aggregates (Fig. 1S), in accelerated thermal stability assays was resolved through the development of a stabilizing buffer formulation (data not shown).
AFM11-His exhibits avidity to both target antigens and cells
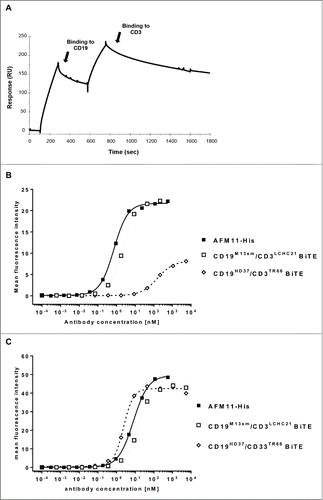
In , SPR sensograms of the binding of AFM11-His TandAb to immobilized recombinant human CD19-Fc and recombinant CD3γε are plotted as resonance units vs. time. Binding curves associated with different concentrations of AFM11-His, injected in subsequent measurement cycles, are normalized to the same baseline (data not shown). SPR measurements were performed with AFM11-His and both comparator molecules at 8 different concentrations ranging from 100 nM to 0.78 nM (). The anti-CD19 domain of AFM11-His possessed a KD of 0.4 nM, and that of anti-CD3 was 1.2 nM. Binding of the BiTE with domains identical to AFM11-His (the CD19M13-sm/CD3LCHC21-ktay BiTE) revealed 7.5-fold (3.0 nM) and 8.3-fold (10 nM) lower affinities to CD19 and CD3, respectively. The significant increase in avidity of the TandAb indicates that each domain is competent to bind its cognate target. KD of the CD19HD37/CD3TR66 BiTE to CD19 and CD3 were 2.1 nM and 110 nM, respectively, and consistent with previous data,Citation21,22 indicating a substantially lower affinity for CD3 than AFM11-His. Finally, the SPR experiments suggest the possibility of simultaneous binding to the CD19 and CD3 antigens, which is a requirement for the mode-of-action of the TandAb. Since the extrapolation of biological effect levels (e.g., receptor occupancy) based on recombinant protein assays should always be cross-referenced with binding data from flow cytometry, we assayed the binding of AFM11-His, the CD19M13-sm/CD3LCHC21-ktay BiTE, and the CD19HD37/CD3TR66 BiTE to CD3+ Jurkat cells and CD19+ Raji cells by flow cytometry (). The apparent CD3 affinity of AFM11-His (KD = 0.7 nM) was 4.5-fold higher than the affinity of the CD19M13-sm/CD3LCHC21-ktay BiTE (KD = 3.1 nM); the CD19HD37/CD3TR66 BiTE exhibited a substantially lower apparent affinity (KD = 154 nM) to CD3+ cells as was previously described.Citation22–24 The apparent CD19 affinity of AFM11-His (KD = 8.1 nM) was 2-fold higher than that of the CD19M13-sm/CD3LCHC21-ktay BiTE (KD = 15.1 nM); the CD19HD37/CD3TR66 BiTE exhibited an apparent affinity (KD = 2.4 nM) to CD19+ cells consistent with that previously describedCitation22–24 and approximately 3-fold higher than that of AFM11-His.
Table 1. Summary of the SPR measurements for AFM11-His and the comparator molecules. SPR measurements for binding to recombinant huCD19-Fc and huCD3γε; CD19HD37/CD3TR66 BiTE was measured employing the same protocol as described.Citation21 The tabulated dissociation constant (KD), association rate constant (ka), and dissociation rate constant (kd) were calculated using the global fit and the 1:1 Binding model across 6 concentration curves (0, 12.5, 25, 50, 100 and 200 nM) for AFM11-His and both comparator molecules
Figure 2. Binding of AFM11 and CD19/CD3 BiTEs to recombinant antigens and CD3+ cells. (A) Recombinant CD19-Fc was directly immobilized on the Sensor Chip (CM5). AFM11-His was injected (100 nM), and during the dissociation phase the second antigen (CD3) was injected. (B) Jurkat cells or (C) Raji cells were stained with the indicated concentrations of AFM11-His, CD19M13-sm/CD3LCHC21-ktay BiTE, and CD19HD37/CD3TR66 BiTE on ice. Cell-surface bound antibodies were detected by anti-His mAb followed by FITC-conjugated goat anti-mouse IgG. Mean fluorescence intensities determined by flow cytometry were modeled as a sigmoidal dose-response by non-linear regression to calculate KD.
AFM11-His-mediated cytotoxic activity in vitro is more potent than that of the BiTE
AFM11 was assayed for its ability to recruit T cells for the redirected lysis of CD19+ cells in different in vitro formats. To assess the kinetics of AFM11-His-mediated target cell lysis relative to that mediated by the CD19HD37/CD3TR66 BiTE, dose-response curves were measured in cytotoxicity assays with MEC-1 target cells and resting peripheral T cells as effector cells with incubation times between 30 min and 5 h (Fig. 2S). At each time point, EC50 and maximal antibody-mediated lysis were determined and plotted in . The results demonstrate rapid and efficacious cell lysis induced by AFM11-His. After 30 min incubation in the presence of AFM11-His, ∼30% target cell lysis was achieved with an EC50 range of 1–2 pM. During lengthier incubation, the potency of AFM11-His increased by roughly ten-fold and the EC50 reached ∼ 0.2 pM after 5 h. Over the range of experimental time courses assayed, the EC50 values measured for the CD19HD37/CD3TR66 BiTE were always between 16 and 34-fold higher than those measured for AFM11-His. The maximal target cell lysis in the presence of AFM11-His was only marginally higher (6%) than that induced by the CD19HD37/CD3TR66 BiTE. In summary, the potency of AFM11-His-mediated lysis of CD19+ target cells is substantially higher than that of the CD19HD37/CD3TR66 BiTE at every time point measured in the cytotoxicity assay.
Figure 3 (See previous page). Comparison of cytotoxic properties of AFM11-His and the CD19HD37/CD3TR66 BiTE on cell lines. (A) EC50 and (B) specific lysis were determined in cytotoxicity assays with calcein-labeled CD19+ MEC-1 target cells and primary human T cells as effector cells at an E:T ratio of 25:1 with incubation times ranging between 30 min and 300 min. At each time-point the EC50 (calculated by non-linear regression of the data modeled as a sigmoidal curve) and specific lysis for AFM11-His and CD19HD37/CD3TR66 BiTE were plotted as representative experiments. (C) AFM11-His-mediated target cell lysis by CD4+ and CD8+ T cell subsets. 4 and 23 h FACS-based cytotoxicity assays for AFM11-His and CD19HD37/CD3TR66 BiTE were performed with either unfractionated human T cells or enriched human CD4+ or CD8+ T cells as effector cells, and CD19+ NALM-6 cells as target cells at an E:T ratio of 5:1. The plotted values represent the mean and the SD of duplicate measurements. (D, E) Serial killing mediated by AFM11 and CD19HD37/CD3TR66 BiTE was demonstrated by FACS-based cytotoxicity assays. NALM-6 target cells and enriched human T cells were used at the indicated E:T ratios in the presence of increasing concentrations of AFM11-His and CD19HD37/CD3TR66 BiTE. After 23 h incubation, EC50 (D) and percentage of target cell lysis at 10 pM (E) were calculated for each E:T ratio and plotted as representative experiments.
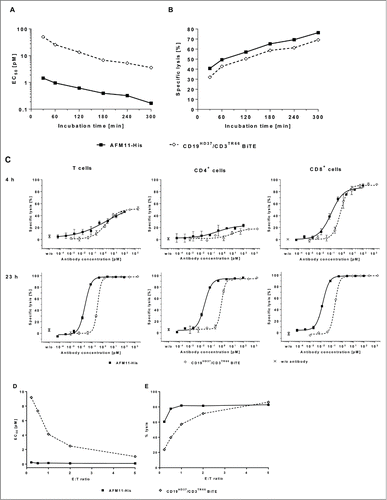
Unanswered is whether only CD8+ T cells are recruited by AFM11-His or whether AFM11-His also activates CD4+ T cells for redirected target cell lysis. To address this question unfractionated T cells, CD4+ T cells, and CD8+ T cells were enriched from a single blood donor and employed in a fluorescence-activated cell sorting (FACS)-based cytotoxicity assay with incubation times of 4 and 23 h. As depicted in and Table 2S, the results demonstrate that after 4 h incubation only enriched CD8+ T cells induce nearly complete (86%) AFM11-His-mediated target cell lysis. Lysis induced by enriched CD4+ cells (21%) or unfractionated T cells (54%) was substantially lower and a maximal lysis for both was reached only at high antibody concentrations. In contrast, the results of a 23 h assay demonstrate similar potency and efficacy of AFM11-His with CD4+ or CD8+ T cells. The CD19HD37/CD3TR66 BiTE exhibited similar cytotoxic characteristics under the conditions used in this assay, with similar maximal target cell lysis as AFM11-His, but it was up to 10-fold less potent than AFM11-His. To assess whether AFM11-His is able to mediate serial-killing, FACS-based cytotoxicity assays with serial dilutions of AFM11-His and BiTE at low E:T ratios were performed (Fig. 3S), and cytotoxic potency (EC50) and efficacy (maximal cell lysis) were calculated. The assays demonstrate the superior potency of AFM11-His at all E:T ratios tested () and higher efficacy at low E:T ratios (). The small difference between the observed maximal lysis at an E:T of 0.2:1 (60%) and that observed at E:T of 0.5:1 and higher (80%) indicate that AFM11-His engages T cells to lyse more than one target cell under these experimental conditions as has been previously reported for the BiTE.Citation25
Because tumor cells of disparate origins expressing antigen at different levels could show distinct susceptibilities toward AFM11-His-mediated lysis by T cells, 4 h duration cytotoxicity assays with a broad range of human CD19+ lymphoma and leukemia cells lines (hairy cell leukemia: JOK-1; Burkitt lymphoma: Raji, DAUDI, and Namalwa; CLL: MEC-1; ALL: VAL and RS4;11; follicular lymphoma: DOHH-2; CML: RPMI-8866; MCL: GRANTA-519) were performed. To obtain rank-ordered quantities of cell-surface expressed CD19 receptors, all cell lines were assayed for their standardized antibody binding capacity (SABC), using the anti-CD19 mAb HD37, and then employed as target cells in cytotoxicity assays in the presence of serial concentrations of AFM11-His. Figure 4S, summarizing the EC50 values relative to the CD19 density, demonstrates the high cytotoxic potency of AFM11 independent of CD19 density and origin of the tumor cell line. Further, experiments with cultures of peripheral blood mononuclear cells (PBMC) from healthy volunteers were performed to assess the specificity of AFM11-His-mediated lysis in an autologous setting. Although a high variability in the response rate was observed, Figure 5S shows that only B cell numbers (identified by CD20 positivity to avoid the possibility of interference from the CD19-binding TandAb) were substantially depleted from the PBMC cultures; however, CD20− cells, such as CD4+ and CD8+ T cells, CD56+ NK cells, and CD13+/CD14+ monocytes, were not affected.
Figure 4. Cytotoxic activity of AFM11-His in ex vivo autologous B-CLL cultures. AFM11-induced apoptosis in B-CLL autologous cultures of patient PBMC. PBMC from 4 patients with B-CLL were cultured either in the presence of 100 ng/mL AFM11-His or HSA/CD3 control TandAb. After 24 and 72 h, apoptosis in CD5+/CD23+ CLL blasts was determined by Annexin V staining and flow cytometric analysis. The specific apoptosis in AFM11-His-treated samples was calculated and plotted for all 4 patients (coded with 2 letters and 2 digits). The total fraction of T cells in these samples ranged between 2.0% and 11.1%, the fraction of CD5+CD23+ B-CLL cells ranged between 87.9% and 92.7%, and the E:T ratio ranged between 8 and 46.
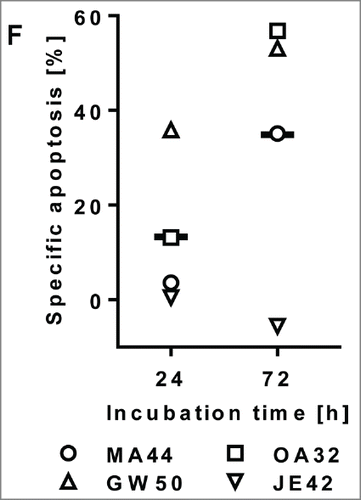
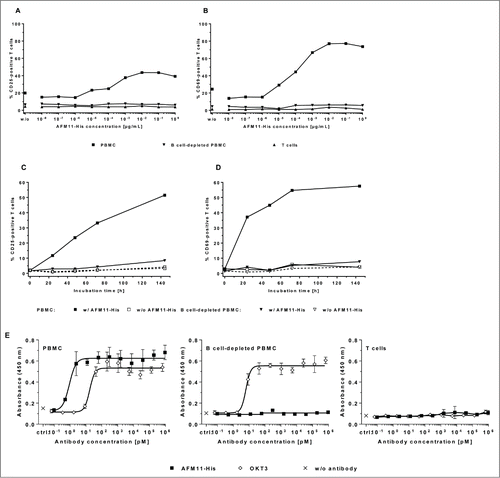
Figure 5. AFM11 does not facilitate activation of human T cells in the absence of CD19+ target cells. Dose-responsive induction of CD25 by AFM11-His (A) and CD69 (B) expression on human T cells was assayed in cultures of human PBMC, B cell-depleted PBMC, and enriched T cells after 48 h incubation. Unfractionated PBMC contained 3.8% CD19+ cells. B cell-depleted cultures possessed 0.6% CD19+ cells, and the enriched T cell cultures contained 0.1% CD19+ cells. The kinetics of CD25 (C) and CD69 (D) expression induced by AFM11-His (10 ng/mL) were determined in human PBMC cultures containing 3.5% CD19+ cells and in B cell-depleted PBMC cultures containing < 0.1% CD19+ cells. A through D present representative experiments. (E) No induction of T cell proliferation in the absence of target cells. Unfractionated PBMC, B cell-depleted PBMC, and enriched unstimulated human T cells were cultured in the presence of increasing concentrations of AFM11-His or OKT3 for 4 days, and then pulsed overnight with BrdU. Incorporation of BrdU was assayed by a BrdU ELISA. Control (ctrl.) cells were cultured in the absence of antibodies. Mean absorbance, and SD of triplicates, measured at 450 nm are plotted. The relative fractions of CD19+ cells were 8% in unfractionated human PBMC, 0.8% in B cell-depleted PBMC, and < 0.1% in enriched T cells.
AFM11-His facilitates apoptosis in ex vivo samples from B-cell chronic lymphocytic leukemia patients
Blood from 4 B-cell chronic lymphocytic leukemia (B-CLL) patients was collected, and PBMCs were isolated and frozen. Since B-CLL cells are exceptionally prone to spontaneous apoptosis, we utilized a control TandAb with unrelated targeting domains (HSA/CD3) and specific apoptosis was calculated as described. Employing FACS analysis, we determined that patient samples contained 2.0 – 11.1% (mean 5.6%) T cells and 87.9 – 92.7% B-CLL blasts (CD5+, CD23+) as a fraction of all PBMC. Three out of 4 B-CLL patient samples treated with AFM11-His exhibited substantial apoptosis after 24 and 72 h (). Specific apoptosis of B-CLL cells in all patients was on average 13.3% after 24 h and increased to 34.9% after 72 h (in responding patients the averages were 17.5% and 48.3%, respectively).
AFM11-His mediates strict CD19+ target-specific T cell activation
T cell activation is associated with the release of pro-inflammatory cytokines that could lead to cytokine release syndrome and severe adverse events in a clinical setting. Therefore the secondary pharmacodynamic effects of AFM11-His were investigated in several in vitro assays that assessed the activation, proliferation and cytokine release of T cells in the presence or absence of CD19+ target cells. demonstrates that AFM11-His induces the expression of the T cell activation markers CD25 and CD69 in a dose- and time-dependent manner only in the presence of CD19+ cells. After depletion of CD19+ cells, or after enrichment of T cells, no substantial T cell activation is observed. Analogous to the strict CD19-dependent activation of T cells by AFM11-His, it only induced T cell proliferation () and the release of interleukin (IL)-2, IL-4, IL-6, tumor necrosis factor (TNF), and interferon (IFN)-γ () in the presence of CD19+ cells, but not in B cell-depleted PBMC or enriched T cell cultures. Thus, these data suggest that bivalent high affinity binding to cell surface CD3 is not sufficient to trigger T cell activation in contrast to previous suggestions;Citation25 rather it requires cross-linking, or immobilization of the antibody by other cells as described previously.Citation26–28 Using the anti-CD3ε IgG OKT3 as a control in the proliferation and cytokine release assays, we observed that the requirement of cross-linking for efficient T cell activation is not restricted to bispecific antibodies that recruit T cells via CD3 like AFM11-His. This requirement is also true for anti-CD3 IgG antibodies such as OKT3: no activation of T cells was observed with bivalent anti-CD3 IgG in homogeneous T cell preparations; however, potent activation of T cells was observed in the presence of FcγR-expressing immune cells, which are capable of crosslinking T cells via binding to the Fc-domain of anti-CD3 IgG consistent with the observations of others.Citation29
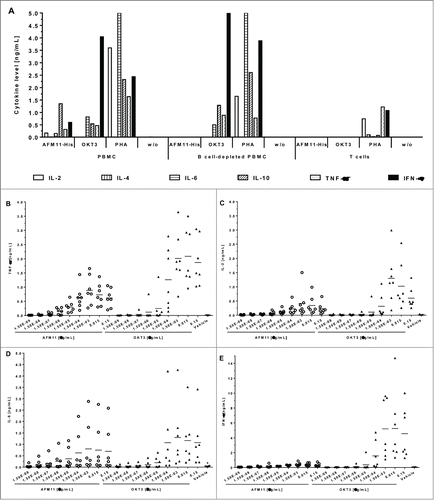
Figure 6. Cytokine release by AFM11-His is strictly dependent on the presence of CD19+ target cells. (A) Cytokine release in cultures of PBMC, B cell-depleted PBMC, and enriched human T cells. Unfractionated human PBMC, B cell-depleted PBMC, and enriched T cells were cultured in the presence of: i) 10 µg/mL AFM11-His or OKT3, ii) 100 µg/mL PHA, or iii) without antibodies (w/o). After 48 h incubation IL-2, IL-6, IL-10, TNF, and IFN-γ in the cell-free culture supernatant were quantified by multiplexing and the results of representative experiments were plotted. (B) TNF, (C) IL-2, (D) IL-6, and (E) IFN-γ levels in culture supernatants of PBMC from 7 individual donors. Cytokine levels were determined by Luminex upon stimulation with antibodies in solution. PBMC were either stimulated with AFM11 (open circles) or OKT3 (closed triangles) at the indicated concentrations. IL-2 and TNF levels were measured after 24 h, IL-6 and IFN-γ levels were measured after 48 h. Background (asterisks) was measured in cultures supplemented with vehicle (formulation buffer) corresponding to the volume of highest AFM11 concentration used. Horizontal bars indicate mean values across the 7 donors.
AFM11-induced cytokine release is less than that of OKT3 in a multi-donor study and in PBMC cultures spiked with Raji cells
The potential of AFM11 to induce cytokine release in vitro was determined, under several stimulation conditions (soluble, wet or dry immobilized test item), in PBMC cultures derived from 7 individual healthy donors. As part of the mode-of-action of AFM11, the linking of CD19+ target cells with T cells, subsequent T cell activation, and resulting substantial cytokine release are expected. The anti-CD3 antibody OKT3 served as a control at each concentration and stimulation condition. The levels of IL-2, IL-6, IL-10, TNF and IFN-γ in PBMC culture supernatants were determined by Luminex. Cytokine levels were determined after 48 h; in addition, for TNF and IL-2, for which peak levels are expected earlier, concentrations were also measured after 24 h. Upon stimulation with soluble AFM11 and OKT3, measurable levels of IL-2 and TNF were detectable only after 24 h stimulation, while all other cytokines exhibited the strongest response after 48 h. Generally, the mean peak levels of all cytokines observed upon stimulation with soluble AFM11 were markedly lower compared with OKT3 stimulation (by a factor of 1.5 to 3). The mean and individual cytokine levels induced by AFM11 and OKT3 are shown in .
As expected, the immobilization of AFM11 or OKT3 generally enhanced the response (data not shown). No differences between wet or dry immobilization were observed for most of the cytokines. In addition to the determination of the general potential of AFM11 to induce cytokine release in PBMC cultures, the effect of increased numbers of CD19+ cells in the cultures was assessed. This would mimic the situation in patients with a large tumor burden, e.g., reflecting the situation in ALL patients where the PBMC population may be dominated by CD19+ cells. The measurement was limited to PBMC from a single selected donor with known robust cytokine response upon AFM11 stimulation and was restricted to the pro-inflammatory cytokines IL-2, IL-6, TNF and IFN-γ. Undiluted PBMC cultures stimulated with soluble AFM11 revealed cytokine release levels comparable to those observed in the 7-donor study. Spiking PBMC cultures with CD19+ Raji cells at different ratios (5:1, 15:1 and 45:1) strongly enhanced the cytokine release of IL-2, TNF and IFN-γ. Figure 6S illustrates the effect of spiking the culture at a ratio of 5:1. An enhancement was also observed for IL-6, but was less pronounced. The intensity of the cytokine response was dependent on the amount of spiked Raji cells in all cases were AFM11 was employed for stimulation. In contrast, no dependence of the cytokine response upon the amount of spiked-in Raji cells was observed in OKT3-stimulated PBMC cultures. This suggests that the enhanced cytokine release is a consequence of the mode-of-action of AFM11, and is strongly correlated with the B cell load.
AFM11 mediates potent in vivo tumor growth inhibition
The pharmacodynamic properties of AFM11 in vivo were determined in a Burkitt lymphoma xenograft model in NOD/scid mice reconstituted with human PBMC. NOD/scid mice were xenotransplanted on day 0 by subcutaneous (s.c.) injection with a suspension of 2.5 × 106 of Raji cells pre-mixed with 1 × 107 purified human PBMC from healthy donors. To account for potential donor variability of the PBMC, each of the experimental groups was subdivided into 3 cohorts each receiving PBMCs from a single donor. Two groups without PBMC effector cells (Raji cells only, not shown) served as negative controls. One of the control groups was treated with vehicle; the other received only AFM11. All animals of the experimental groups transplanted with tumor cells and PBMC received an intravenous (i.v.) bolus on days 0, 1, 2, 3 and 4 of either vehicle (control) or AFM11 at 4 different dose levels as indicated (0.005, 0.05, 0.5 and 5 mg/kg). The mean tumor volume development is depicted in . All animals in the control groups reliably developed tumors. The fastest increase in tumor volume was observed in the group receiving tumor cells and vehicle only (data not shown). The engraftment of human PBMC plus vehicle (i.e., Raji cells, PBMC, vehicle) delayed tumor growth, indicating a slight graft-versus-tumor response in this group. Tumor growth in all AFM11-treated animal groups that received tumor cells and PBMC exhibited statistically significant tumor volume reduction (p ≤ 0.05) relative to the vehicle-treated control group, and a dose response was observed. A disproportionate mean tumor development was seen in the 0.5 mg/kg group due to a single animal that did not respond to the AFM11 treatment. In the AFM11 low dose group receiving 0.005 mg/kg, statistical significance was restricted to days 17, 20 and 22. All other treatment groups exhibited statistically significant differences from day 13 until day 34. The group treated with 5 mg/kg AFM11 exhibited complete tumor remission in all 9 animals, which was maintained until day 36 of the study; a single tumor reappeared on day 38 in one of 9 animals.
Figure 7. Xenograft models and tissue-distribution of AFM11 in tumor-bearing mice. (A) Effect of AFM11 on the growth of Raji Burkitt lymphoma in NOD/scid mice. Six experimental groups of immunodeficient NOD/scid mice were xenotransplanted by subcutaneous injection with a suspension of 2.5 × 106 of Raji tumor cells on day 0. Tumor cells were mixed with 1 × 107 human PBMC from healthy donors. To account for potential donor variability, each of the experimental groups was subdivided into 3 cohorts each receiving PBMCs from a single donor. Two groups without PBMC effector cells (tumor cells only, not shown) served as negative controls. One of the control groups was treated only with vehicle; the other received only AFM11. All animals of the experimental groups transplanted with tumor cells and PBMC received an intravenous bolus on days 0, 1, 2, 3 and 4 of either vehicle (control) or AFM11 at 4 different dose levels as indicated (0.005, 0.05, 0.5, and 5 mg/kg). All treatment groups receiving AFM11 exhibited statistically significant tumor growth inhibition, relative to the control group, from day 13 until day 34, excepting the low dose group (0.005 mg/kg) in which statistical significance was restricted to days 17, 20 and 22. (B) No effect of AFM11 dosing regimen on the growth of Raji Burkitt lymphoma in NOD/scid mice. NOD/scid mice were xenotransplanted by subcutaneous injection with a suspension of 2.5 × 106 of tumor cells on day 0. Tumor cells were mixed with 1 × 107 human PBMC from healthy donors. To account for potential donor variability, each of the experimental groups was subdivided into 3 cohorts each receiving PBMCs from a single donor. Animals received an intravenous bolus of AFM11 on days 0, 1, 2, 3, and 4, or a single injection on day 0, at a dose level of 0.5 mg/kg. The control group received 5 injections of vehicle. Both A and B present mean tumor volume and SD. (C) AFM11 tissue distribution in Raji xenograft-bearing NOD/scid mice. Whole body autoradiogram and the corresponding scanned section of a Raji xenograft-bearing NOD/scid mouse sacrificed at 24 h post injection of125I-labeled AFM11.
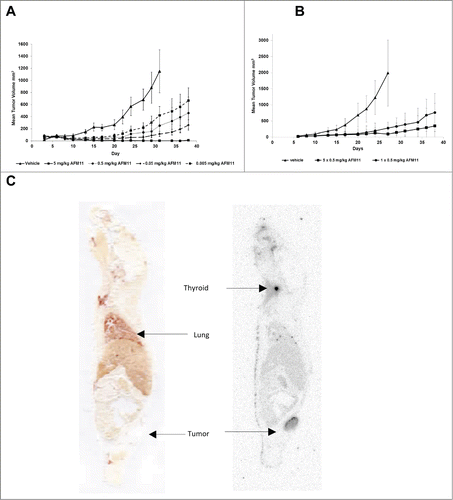
In an independent study, different dosing regimens were assessed in the same model at a dose of 0.5 mg/kg. Dosing of AFM11 on 5 consecutive days revealed the more efficient inhibition of tumor growth and 3/8 animals were tumor-free at the end of the observation period (). However, a single injection of AFM11 on day 0 was sufficient to efficiently delay tumor growth compared to the vehicle treated animals, and no statistically significant difference relative to 5 consecutive injections was observed.
AFM11 pharmacokinetics
The pharmacokinetic assessment focused entirely on studies in mice addressing the basic pharmacokinetic properties of AFM11 in an environment without human CD3 and CD19, due to the lack of non-human primate cross-reactivity for AFM11. Thus, the data generated cannot identify pharmacokinetic effects arising from the interaction of AFM11 with its nominal target molecules.
Basic pharmacokinetic parameters were determined after a single bolus i.v. injection, at 3 dose levels of AFM11, in CD1 mice. An additional group received a single s.c. administration at the highest dose level to determine the bioavailability of AFM11 via this route of administration.
Serum concentration declined in a biexponential manner with geometric mean t1/2 of ∼18 – 22 h in all dose groups. These t1/2 are supported by an independent tissue distribution study performed in tumor-bearing mice employing125I-labeled AFM11 described below.
After i.v. administration, a geometric mean apparent volume of distribution of AFM11 at steady state (Vss) of 2 mL was determined. This is approximately the total plasma volume of mice, indicating limited distribution of AFM11 into tissues. A clearance (CL) of ∼0.5 mL/h was measured for AFM11, which is considerably less than the hepatic and renal blood flow rates in mice; this suggests that neither glomerular filtration nor hepatic degradation dominate the elimination of AFM11. Systemic exposure to AFM11, as measured by AUCtot, and Cmax, increased in an approximately linear manner.
Upon s.c. administration of AFM11, the maximum serum concentration of AFM11 was observed after 8 h. An estimation of the bioavailability based on AUCtot indicates that AFM11 is ∼24% bioavailable after s.c. administration. Terminal elimination kinetics for both administration routes (i.v. and s.c.) appear identical.
AFM11 exhibits exclusive localization to tumor tissue
AFM11 was radiolabelled (125I) by direct electrophilic radioiodination. Binding assays performed on Jurkat and Raji cells revealed that radiolabelling of AFM11 with125I did not affect the affinity for CD3 and CD19 binding. The product had sufficient stability in buffer and mouse serum to be suitable for a distribution experiment (data not shown).
For the in vivo evaluation of tumor targeting, tissue distribution, blood pharmacokinetics, and elimination of radiolabelled AFM11, Raji tumor-bearing mice (tumor volume ∼239 ± 42 mm3) were dosed at 5 mg/kg via the i.v. route. The tissue distribution study was performed with 3 animals per time point (0.5, 2, 4, 10, 24 and 48 h), 1 animal was frozen and reserved for whole body autoradiography (WBA).
The data demonstrate substantial accumulation and retention of AFM11 in tumor tissue. shows a whole body autoradiogram and the corresponding scanned sections of a Raji xenograft-bearing NOD/scid mouse sacrificed at 24 h post-injection. The tumor retention of125I-AFM11, combined with its rapid clearance from normal tissues and blood, results in high tumor-to-normal organ ratios, generally greater than 4:1 at 24 and 48 h time points.
The pharmacokinetic parameters calculated using125I-AFM11 are consistent with those determined (presented above) employing the high-sensitivity MSD electrochemiluminescence (ECL) assay for unlabeled AFM11. Approximately 80% of the125I radioactivity is recovered in urine and feces at 48 h post-injection. The urinary elimination route accounts for 84% of the elimination process. The amounts of free125I in urine are > 99%. Thus, radioactivity in urine is associated with free125I and not125I-AFM11. Thus it is unsurprising that a minor accumulation of radioactivity is observed in the thyroid.
Discussion
While there has been dramatic progress in B cell malignancy treatment outcomes since the introduction of the first generation of therapeutic antibodies, additional treatment options are needed to address salvage therapy for non-responders, the population that experiences relapse subsequent to current standard-of-care therapy, and those with diseases that do not respond well to conventional anti-CD20 antibodies.
A second generation of antibody therapeutics, Fc-optimized antibodies, has exhibited substantial improvement in tumor-killing ability over conventional antibodies, and representative antibodies that target B cell malignancies are in development.Citation3-5 Nevertheless, the demand for even more efficacious treatments catalyzed the development of a new class of bispecific immune cell-recruiting antibodies. Such antibodies specifically, with extreme potency, recruit particular immune effector cells, and are able to recruit NK cells,Citation30 T cells,Citation23 or both.Citation7 Two such antibodies have been approved for marketing, catamuxomab in 2009 and blinatumomab in 2014, and others are in preclinical and clinical development.
Although the T cell-recruiting approach has demonstrated promising results in the clinic, the t1/2/persistence in human circulation of these molecules could be improved. Due to their size, fragment antibodies such as the BiTE possess very short t1/2 and require administration by continuous infusion, which presents significant challenges. Full-length antibodies containing the Fc domain are competent to interact with FcRn, and thus possess t1/2 in the range of 2–4 weeks. Therefore, rapid intervention for management of possible adverse events with full-length T cell-recruiting antibodies would be complicated due to their extended presence in circulation. Similarly, CAR T cells are expected to persist in patients for extended periods of time, and thus could also present significant risk in case of treatment-related complications. We rationalized that a T cell-recruiter that both possesses a longer t1/2 than the BiTE, but not as long as a full-length IgG, and maintains a rigid structure that may be more favorable for optimal target engagement,Citation21 and thus increased cytotoxicity, would be optimal for dosing and the management of adverse events. Furthermore, though response to blinatumomab is impressive, with 43% of relapsed and refractory ALL patients achieving complete response,Citation9 a higher response rate is needed.
Previously, we described a TandAb that can efficiently and potently recruit NK cells to kill Hodgkin lymphoma cells in vitro,Citation30 and its activity was evaluated in a Phase 1 clinical study that included patients refractory to current standard-of-care treatments.Citation31 In this study, we sought to expand the utility of the TandAb platform by developing a T cell-recruiting TandAb, AFM11, and thus demonstrate the applicability of the TandAb platform to the engineering of potent immune cell-recruiting bispecific antibodies with good drug-like properties. Thus, we constructed the CD19/CD3 TandAb AFM11, for the treatment of B cell malignancies, as a potentially more potent T cell-recruiting therapeutic with optimal t1/2 properties.
AFM11 possesses the same target specificity domains as blinatumomab; however, the design of AFM11 confers distinct features that differentiate it from the BiTE. The molecular weight of AFM11 is approximately twice that of blinatumomab, whose low molecular weight of 55 kDa endows a t1/2 of 1.25 h, requiring administration by continuous infusion. We hypothesized that AFM11's increased size, ∼105 kDa, which exceeds the passive glomerular filtration threshold, would alter the dosing paradigm for T cell-recruiting fragment antibodies.
To explore the effect of the design criteria that distinguish the CD19/CD3 TandAb from the CD19/CD3 BiTE blinatumomab, i.e., a tandem diabody that binds bivalently with high affinity for CD3 vs. an scFv2 format with a relatively modest affinity, we constructed scFv2 having either the Fv domains of AFM11 or those of blinatumomab. Binding and cytotoxicity assays were then employed to profile the 3 molecules. In the SPR and cell-based assays, comparison of the binding data from AFM11 and its scFv2 analog confirms that AFM11 binds to both antigens bivalently as the avidity effect increases the apparent binding affinities for both CD3 and CD19 by almost 10-fold. The binding assay data for the BiTE are consistent with those published;Citation22,23 thus the binding affinities of AFM11 for CD19 and CD3 are approximately 5-fold and 100-fold higher, respectively, than those of the BiTE. In addition, the SPR experiment with sequential injections of AFM11 and recombinant CD3 onto immobilized CD19-Fc demonstrates that the TandAb is capable of binding to both antigens simultaneously. The effect of this bivalent high affinity bispecific binding then reveals itself in the profile of cytotoxic activity. In the calcein-release cytotoxicity time-course experiments, AFM11 is approximately 2 orders of magnitude more potent (EC50) than the BiTE, but only slightly more efficacious (percent specific lysis). In the FACS-based assay of cytotoxicity as a function of effector:target ratio, both AFM11 and the BiTE mediate serial-killing; however, AFM11, unlike the BiTE, exhibits almost no dependence of potency or efficacy upon effector:target ratio over the range assayed. The large difference in efficacy between AFM11 and the BiTE observed in the calcein-release cytotoxicity assay relative to that observed in the FACS-based assay is probably attributable to the difference in assay duration (6 h versus 24 h) and cell type (MEC-1 vs. NALM-6). In an assay of the contributions of distinct T cell populations to cytotoxicity, at the early time point (4 h) CD8+ T cells are the major contributors, whereas at the later time point (23 h) the contributions from CD8+ and CD4+ T cells are approximately equal; this is consistent with previous observations.Citation21,22 Finally, we observe that AFM11 potency is not correlated with target cell CD19 density.
Having characterized the effect of the TandAb platform design choices, the functional properties and specificity of AFM11-mediated cytotoxicity, which affects safety risk profile, were further investigated independent of the BiTE. It has previously been asserted that high affinity, bivalent binding to CD3 could negatively affect the function of an immune effector-recruiting therapeutic. First, it was suggested that high affinity binding to CD3 would preclude the mediation of serial-killing.Citation25 However, as discussed above, AFM11 efficiently mediates serial-killing similar to the BiTE. Second, it was suggested that high affinity, bivalent binding to CD3 might lead to non-target—specific T cell activation.Citation25 To further explore this issue we assayed: i) the dose-response and kinetics of AFM11-mediated induction of the activation markers CD25 and CD69; ii) the AFM11-mediated induction of T cell proliferation; and iii) AFM11-mediated cytokine release in cultures of PBMC, B cell-depleted PBMC, and enriched T cells. The activation marker assays exhibit a dose-dependent induction of CD25 and CD69 only in unfractionated PBMC. In the T cell proliferation assay, which employed unfractionated PBMC, B cell-depleted PBMC, and enriched unstimulated human T cells that were cultured in the presence of increasing concentrations of AFM11 or OKT3, AFM11 only induced proliferation in unfractionated PBMC. However, OKT3 induced proliferation in both unfractionated PBMC and B cell-depleted PBMC while neither AFM11 nor OKT3 induced proliferation in purified T cells. This is significant because OKT3 retained the ability to induce T cell proliferation only in the presence of FcγR+ cells. These cells afforded the possibility of OKT3 cross-linking T cells with them via its Fc domain. The results of the cytokine release assay are similar to those of the T cell proliferation assay in that they reveal the propensity of OKT3 to induce effector cell activation and subsequent cytokine release (IL-2, IL-4, IL-6, IL-10, TNF, and IFN-γ) in the presence of T cell targets and Fc-responsive effectors while AFM11 elicited minimal release only in the presence of PBMC. Thus, these data indicate that high affinity, bivalent binding to CD3 does not lead to non-target—specific T cell activation; it simultaneously illuminates the requirement of cell-to-cell cross-linking, as opposed to mere antibody engagement of CD3, for activation of T cells.
Additional safety risk-profiling studies addressed the questions of the population-variability of the potential cytokine release induced by AFM11, the cytokine release induced by AFM11 in PBMC spiked with CD19+ tumor cells, which emulates an ex-vivo tumor assay, and the biodistribution of AFM11 in an in vivo tumor-burdened context. In the population-variability study, the cytokine release (IL-2, IL-6, TNF, and IFN-γ) induced by AFM11 in unfractionated PBMC is reduced relative to that induced by OKT3 across all of the cytokines, and only AFM11-induced IL-6 release approaches that of OKT3; this is consistent with the blinatumomab clinical experience.Citation32 In unstimulated PBMC spiked with CD19+ tumor cells at a ratio of 5:1, AFM11 induces a dose-responsive cytokine release consistent with its mode of action. Lastly, in the biodistribution study radiolabeled AFM11 exhibited excellent localization to tumor tissue with minimal distribution to normal tissue, excepting the spleen as expected.
The in vivo murine model studies reveal dose-responsive tumor-growth inhibition that at the highest dose afforded complete protection from tumor outgrowth. Moreover, the dosing regimen experiment seems to indicate that a single dose of AFM11 may have been sufficient to eliminate all of the initially implanted tumor cells. Alternatively, as previously hypothesized, the murine environment may have either cleared or rendered inactive the implanted T cells, resulting in the observed lack of effect of later AFM11 doses.Citation33 Finally, in the human ex vivo tumor study, AFM11 exhibited substantial cytotoxic activity against CLL cells. Thus, we confirm potent AFM11 activity not only in cell lines and in mice, but also on human tumors in an autologous setting, i.e., in a realistic context with an effector:tumor cell ratio of ∼1:10–50, consistent with the CLL disease state.
AFM11 not only exhibits excellent functional properties, a reasonable in vitro safety risk profile, and potent in vivo and ex vivo function, but it also affords ease of manufacture and good drug-like properties. It is readily manufacturable; the lab-scale stable pool expression titers, which were in excess of 200 mg/L, imply that a standard bioreactor run would yield enough material to treat thousands of patients (cell line titers in the course of GMP manufacture are typically in the range of 400–500 mg/L; data not shown). The final product is a highly stable homodimeric protein with no higher order oligomers or aggregates present; in an accelerated thermostability assay AFM11 preserves this fundamental biophysical property. Lastly, in vivo the murine pharmacokinetic properties have the signature of good drug-like properties, albeit the studies were performed in a non-crossreactive species. Unlike other fragment antibodies, AFM11 has a molecular weight above the glomerular filtration cutoff. Thus, it possesses a long murine t1/2, as measured in this study, which is similar to that observed with another TandAb in man, where target-mediated disposition effects are present.
Finally, the molecular weight of AFM11 may mitigate the CNS adverse events associated with blinatumomab treatment. The etiology of these CNS events is unclear. It has been hypothesized that they arise from cytokine-induced inflammation caused by blinatumomab-activated T cells during or after their transit into the CNSCitation23 for ordinary immune surveillance.Citation34 However, it cannot be excluded that this T cell activation occurs within the CNS since one of the most frequent sites of leukemic metastasis is the CNS due to the compromised blood-brain barrier (BBB).Citation35 The low molecular weight of blinatumomab, which would facilitate passive diffusion across a compromised BBB, may play a role in this process. Indeed, to date there are no reports of similar CNS events from the trial of the full-length IgG (CD20/CD3) FBTA05. Moreover, due to its increased size, AFM11 may be less able to diffuse across the compromised BBB than the BiTE, affording a more beneficial CNS safety risk profile.
This study demonstrates that AFM11 is a potent T cell-recruiting therapeutic. It possesses an optimal t1/2 relative to other antibodies, and may overcome the challenges associated with the dosing regimen of the CD19/CD3 BiTE. The ongoing clinical studies with AFM11 will determine whether, due to its increased size, this therapeutic also exhibits a reduced risk of CNS complications.
Methods
Humanization and affinity maturation
The DNA coding for the murine anti-CD3 antibody UCHT1Citation14 and the murine anti-CD19 antibody HD37Citation15 Fv domains were ordered as synthetic DNA fragments (Geneart, Regensburg, Germany), and were cloned into the pEXHAM vector.Citation16
For the generation of Vλ and Vκ light chain-shuffled libraries, plasmid DNA derived from the high complexity naïve IgM scFv phage display libraryCitation16 was subjected to restriction digest for removal of the VH chain coding sequences. These were either removed by EcoRI/MluI (λ chain containing plasmids) or by NcoI/HindIII (κ chain containing plasmids). Either the HD37 or the UCHT1 coding VH chain was cloned into the linearized vectors and transformed by electroporation into electrocompetent E. coli XL1 blue, resulting in rearranged libraries with a complexity of ∼10Citation8 for both libraries.
To construct vectors for the shuffling of the VH CDR1/2 chains, the VH chains of the chimeric clones were replaced (NcoI/MluI) by a synthetic truncated VH fragment composed of the respective VH CDR3 sequence with a BssHII restriction site 5′ flanking the CDR3, followed by a respective human VJ element. VH CDR1/2 fragments were amplified by PCR using a vector-specific 5′ primer in combination with a set of reverse primers annealing to the VH CDR3 adjacent framework coding region. DNA from Affimed's naïve libraries served as a template. The fragments were cloned via the NcoI/BssHII restriction sites yielding libraries with a complexity of ∼108.
Libraries for affinity maturation by permutation of contact residues either in the CDR1/2 of the VH chain (NcoI/BssHII) or in the CDR1/2/3 of the VL chain (NcoI/HindIII) were constructed by cloning synthetic permuted DNA fragments (GeneArt).
Panning for the selection of anti-CD3 binders was performed on Jurkat cells; the libraries were depleted on 2 × 107 JRT3-T3.5 cells for 2 h before transfer to 107 Jurkat cells. Bound phage were eluted by competition with UCHT1. The anti-CD3 scFv with the highest binding identified by this procedure was denoted LCHC21. Panning for the selection of anti-CD19 binders was performed either on a recombinant protein (CD19-Fc immobilized on Maxisorp (NUNC, Denmark) microtiter plates) or on HEK293 cells transfected with the pcDNA3.1(+) plasmid encoding human CD19 (a kind gift of Dr. Bernhard Stockmeyer, University of Erlangen). For the latter, the libraries were depleted on 2 × 107 HEK293 cells for 2 h before transfer to 107 HEK293-CD19 cells. Bound phage were eluted by competition with HD37. The anti-CD19 scFv with the most optimal binding was denoted M13.
Several deviations from the germline sequences were identified in both anti-CD3 and anti-CD19 domains employing an IgBLASTCitation36 search and were back-mutated. In the VL chain of the anti-CD3 clone LCHC21, the positions E39, A72, V84, and V87 were back-mutated to K39, T72, A84, and Y87 (Kabat numbering); the resultant clone is denoted LCHC21-ktay. In the VH of the anti-CD19 clone M13, the amino acid residues T82b and T89 were reverted to the germline S82b and M89, and the resultant clone is denoted M13-sm. Mutagenesis was performed employing synthetic gene fragments (Geneart).
Constructs
AFM11-His, the CD19/CD3 TandAb, is a research derivative that, unlike AFM11, contains a His6-tag at the C-terminus to facilitate purification and detection. Its design, anti-CD19, and anti-CD3 domains are identical to those of AFM11. The construct design for the CD19/CD3 TandAb consists of alternating VL and VH of anti-CD3 and anti-CD19 domains connected with 3 identical 6 amino acid linkers (L1, L2, and L3) and conforming to the following topology: VLCD3(LCHC21-ktay)-(G2S)2-VHCD19(M13-sm)-(G2S)2-VLCD19(M13-sm)-(G2S)2-VHCD3(LCHC21-ktay). Gene sequences encoding the VL and VH domains of anti-CD3LCHC21ktay separated by a peptide linker (VL-(G2S)2-VH) were synthesized (Geneart) and subcloned into the mammalian expression vector pcDNA5/FRT (Life Technologies, Carlsbad, US) to facilitate expression of AFM11-His in CHO cells. The resulting construct was digested to separate the VH and VL coding sequences utilizing a recognition site located within the linker sequence (BamHI), and ligated with a fragment encoding the VH-(G2S)2-VL of anti-CD19M13-sm to yield the final CD19/CD3 TandAb construct. A schematic of the AFM11-His polypeptide is shown in . To facilitate antibody secretion, the expression construct was designed to contain coding sequences for an N-terminal signal peptide. Sequences of all constructs were confirmed by DNA sequencing (Eurofins Genomics, Ebersberg, Germany) using the primer pair 5′-TAATACGACTCACTATAGGG-3′and 5′-TAGAAGGCACAGTCGAGG-3′.
Two comparator molecules in the BiTE format were also constructed: the CD19HD37/CD3TR66 BiTECitation19,20,37 and the CD19M13-sm/CD3LCHC21-ktay BiTE (anti-CD19 and anti-CD3 as in AFM11-His). Both genes were synthesized (Geneart) and subcloned into the pcDNA5/FRT mammalian expression vector for CHO-expression. However, the CD19M13-sm/CD3LCHC21-ktay expression construct contained the linkers L1: (G2S)5, L2: G4S, L3: (G2S)5.. Both constructs also contained coding sequences for an N-terminal signal peptide and a C-terminal His6-tag, as in the case of AFM11-His. Sequences of both constructs were confirmed by DNA sequencing employing the above primers.
Human CD19 antigen was constructed as a C-terminal Fc fusion. The sequence encoding the complete extracellular domain of CD19 was synthesized (GeneArt) and subcloned into the mammalian expression vector pcDNA5-FRT. Human CD3 antigen was constructed as a CD3γε fusion protein as described.Citation38 The sequence encoding the fusion construct was synthesized (GeneArt) and subcloned into the bacterial expression vector pET26. Sequence confirmation of both CD19-Fc and CD3γε fusion proteins was performed as described for the TandAb constructs with the exception that for CD3 the reverse primer was 5′- GCTAGTTATTGCTCAGCGG-3′.
Expression and purification of recombinant antibodies and antigens
Flp-In CHO cells (Life Technologies) adapted to suspension growth were cultured in shaker incubators equilibrated at 37°C, 5% CO2, 140 rpm in HyClone CDM4 CHO medium (GE Healthcare, Freiburg, Germany) containing 2 mM Glutamine (Life Technologies) and HT supplement (Life Technologies).
Stable, recombinant protein-expressing cells were generated by co-transfection of Flp-In CHO with pcDNA5/FRT vectors containing the gene of interest and the Flp recombinase vector pOG44 (Life Technologies) using PEI (Polysciences, Eppelheim, Germany) as transfection reagent and subsequent selection for stable cell pools with Hygromycin B (Life Technologies). In brief, 2.5 µg vector DNA and PEI were combined at a ratio of 1:3 (µg/µg) in a total of 100 µL OptiMEM I medium (Life Technologies), incubated for 10 min, and added to 2 × 106 Flp-In CHO cells suspended in 1 mL CHO-S-SFM II medium (Life Technologies). Following 24 h incubation, cultures were diluted to a density of 0.1 × 106 viable cells/mL in CHO-S-SFM II medium, and selection for stably transfected cells was started by addition of 500 µg/mL Hygromycin B. During the selection, viable cell densities were measured twice a week, and cells were centrifuged and resuspended in fresh selection medium at a maximal density of 0.1 × 106 viable cells/mL. Stable cell pools were transferred into HyClone CDM4 CHO medium (GE Healthcare) containing 2 mM glutamine (Life Technologies) and HT supplement (Life Technologies), cultured in shake flasks, and cryopreserved in medium containing 50% ProFreeze (Lonza, Basel, Switzerland) and 7.5% dimethyl sulfoxide (DMSO).
Recombinant proteins were produced in fed-batch cultures of stably transfected CHO cells seeded at a density of 6 × 105 cells/mL medium in polycarbonate Erlenmeyer flasks with gas permeable caps (Corning, Amsterdam, The Netherlands). The cultures were supplemented with either 4% (v/v) ActiCHO Feed A or 0.4% (v/v) ActiCHO Feed B (GE Healthcare) on day 0 (starting day), and double amounts on days 3, 5, and 7. Samples were collected from the production cultures prior to feeding, and cell density and viability was assessed. Cell culture supernatants (CCS) were harvested after 10 days, cleared by centrifugation and vacuum filtration using 0.22 µm polyethersulfone (PES) Membrane Filters (Millipore) before further use.
Protein expression titers and product integrity in CCS were analyzed by SDS-PAGE on days 5, 7 and 10 using Criterion Stain-Free technology (Bio-Rad, Munich, Germany). Product titers were determined semi-quantitatively by comparison with a reference TandAb of known concentration. AFM11-His or BiTE molecules were purified from the CCS by HisTrap FF 5mL column (GE Healthcare, cat.: 17–5255–01) chromatography followed by preparative size exclusion chromatography (SEC) on Superdex 200 (GE Healthcare). Purified AFM11-His was buffer-exchanged into 10 mM Na-acetate pH 5 using PD-10 desalting columns (GE Healthcare), concentrated by centrifugational ultrafiltration using Amicon Ultra 15, MWCO 10 kDa (Millipore) to a concentration of 1.0–1.5 mg/mL, and stored at −80°C. Protein concentration was determined by UV-spectroscopy (280 nm).
The CD19-Fc fusion was expressed in CHO cells and purified via protein A and SEC. The CD3γε fusion was expressed in E.coli in inclusion bodies. Inclusion bodies were isolated, solubilized, refolded and purified via immunoaffinity chromatography, followed by SEC as described.Citation38
Analysis of purity and stability of AFM11-His
The purity, and homogeneity of product samples were assessed by analytical size exclusion chromatography (SE-HPLC) as well as by SDS-PAGE under reducing and non-reducing conditions followed by immunoblotting with His-tag–specific antibodies. Endotoxin content of purified protein was determined by LAL test (Endosafe, Charles River, Wilmington, US). Purified proteins were stored as aliquots at −80°C until use.
Thermal stability of purified AFM11-His was assayed under accelerated conditions for up to 7 d at 2–8°C and 37°C, respectively, in storage buffer (10 mM sodium acetate pH 5). At defined time points incubation was stopped and samples were either assayed using SDS-PAGE, SE-HPLC, and UV-spectroscopy or stored at −20°C for subsequent analysis.
Surface plasmon resonance analysis
SPR analysis for AFM11-His and the CD19M13-sm/CD3LCHC21-ktay BiTE was performed (BIAcore X100, GE Healthcare) on CD3γε and CD19-Fc fusion proteins that were covalently coupled (Amino Coupling Kit, GE Healthcare) to a CM5 chip (GE Healthcare) according to manufacturer instructions. The antigens were diluted in the immobilization buffer (10 mM sodium acetate, pH 5.5) to a concentration of 25 µg/mL. For the coupling procedure, the predefined method “Amine” of the Biacore X100 control software was employed, and a target level of 500 RU was selected. The surface of the chip was activated with EDC/NHS before injecting the recombinant protein solutions. Free binding sites on the surface were blocked with ethanolamine. 1 × HBS-EP+ was selected as a running buffer for the entire coupling procedure.
AFM11-His concentration was assayed prior to SPR measurement by A280 determination using Nanodrop 2000c (Thermo Fisher Scientific) with cuvette. The concentration of the stock solution (c) was calculated according to the equation:where the extinction coefficient of 2.25 was calculated employing the 'Expasy Protparam' prediction tool.Citation39 For calculation of the molar concentration, a molecular weight of 103.4 kDa was assumed. AFM11-His was adjusted to final concentrations of 100, 50, 25, 12.5, 6.25, 3.125, 1.56, and 0.78 nM by serial dilution in 1 × HBS-EP+ buffer supplemented with 1 mg/mL NSB reducer (GE Healthcare). 1 × HBS-EP+ was used as running buffer for the entire procedure. AFM11-His was injected at a flow rate of 30 µL/min for 180 s, representing the longest possible contact time for this device. Signals measured without antigen, were subtracted from signals measured with antigen to correct the binding curves for background binding. After 720 s dissociation time, the recombinant CD3γε and CD19-Fc chips were regenerated by injection of either 10 mM Glycine-HCl pH 1.75 or 3.5 M MgCl2 (50 s, 10 µL/min), respectively. The regeneration was complete since baseline levels identical to those observed before AFM11 injection were recovered. Prior to each measurement, 3 “start-up cycles” were performed without the analyte. AFM11-His solutions were assayed from lowest to highest concentration, and one cycle with neat running buffer (0 µM) was included. The kinetic evaluation exhibited low variability for both molecules, with an intra-assay variation < 2%. The highest AFM11-His concentration was excluded from the fit, since the Residual Plot Analysis and the SE indicated unreliable fitting at this concentration. Apparent binding affinities of AFM11-His to the recombinant human target molecules were calculated by applying the 'Langmuir 1:1 interaction' model.
To facilitate the comparison of the properties of the CD19HD37/CD3TR66 BiTE with those presented in the literature, the SPR protocol describedCitation21 was applied with the following modifications: the concentrations of the analyte were 0, 0.78 1.56, 3.125, 6.25, 12.5, 25, 50, 200 and 400 nM, and association and dissociation times were 180 s and 720 s, respectively.
Cell lines and cell culture
MEC-1 cells (cat.: ACC 497), RS4;11 cells (cat.: ACC 508), JRT3-T3.5 cells (cat.: TIB-153) GRANTA-519 cells (cat.: ACC 342), VAL cells (cat.: ACC 586), NAMALWA.pnt cells (cat.: ACC 69), SU-DHL-6 cells (cat.:ACC 572), RPMI-8866 cells (cat.: ACC 572), DOHH-2 cells (cat.: ACC 47), and NALM6 cells (cat.:ACC 128) were purchased (DSMZ). Daudi, Raji, and JOK-140 were provided by Dr. G. Moldenhauer (DKFZ Heidelberg). All cells were cultured under standard conditions in DMEM (cat.: 41965–039), IMDM (cat.: 12440–053), or RPMI 1640 (cat.: 21875–034) medium supplemented with: 10% heat-inactivated fetal calf serum (FCS) (cat.: 10270–106), 100 U/mL penicillin G, 100 µg/mL streptomycin (cat.: 1540–122), and 2 mM L-glutamine (cat.: 25030–024; all Life Technologies).
Isolation of PBMC and enrichment or depletion of PBMC subsets
PBMCs were isolated from healthy volunteers’ buffy coat (German Red Cross, Mannheim, Germany) by density gradient centrifugation. The buffy coat samples were diluted with a 2-to-threefold volume of PBS (Life Technologies, cat.: 14190–169), layered on a cushion of Histopaque-1077 (Sigma-Aldrich, St. Louis, MO, USA, cat.: H8889), and centrifuged at 800 × g for 25 min at room temperature without brake. PBMC located at the interface were collected and washed 3 times with PBS before they were used for the cytotoxicity assay, the enrichment of PBMC subsets, or flow cytometric analysis. Negatively-selected unfractionated human T cells were immunomagnetically isolated from the PBMC (EasySep™ Human T Cell Enrichment Kit and the Big Easy EasySep™ Magnet, Stem Cell Technologies, Grenoble, France, cat: 19051) according to manufacturer's instructions. CD4+ and CD8+ T cells were enriched from isolated PBMC (EasySep™ Human CD4+ and CD8+ T cell enrichment kit, Stem Cell Technologies, Grenoble, France, cat: 18001) according to manufacturer's instructions. B cells were depleted from isolated PBMC (EasySep™ Human CD19 Positive Selection Kit, Stem Cell Technologies, Grenoble, France, cat: 18054) according to manufacturer's instructions.
Assay of cell surface CD19 expression density on cell lines
The expression density of CD19 on the surface of different cell lines was determined using QIFIKIT (Dako via Biozol, Eching, Germany, cat.: DAK-K007811–8) and mAb HD37 (Merck-Millipore, Schwalbach, Germany, cat.: CBL 582) according to the manufacturer's instructions. In brief, aliquots of 1 × 106 cells were stained with 100 µg/mL mAb HD37 followed by the F(ab')2 fragment of FITC-conjugate goat anti-mouse antibody. As a negative control, aliquots of 1 × 106 cells were stained with 100 µg/mL mAb 9E10 (Acris, Herford, Germany, cat.: SM1863P) followed by the F(ab')2 fragment of FITC-conjugate goat anti-mouse antibody (Dako). To calculate the specific antibody binding capacity, calibration beads representing 5 populations bearing different distinct numbers of mAb molecules were stained with the F(ab')2 fragment of FITC-conjugate goat anti-mouse antibody. From the resulting mean fluorescence intensities a calibration curve was calculated. This calibration curve was used to calculate the specific antibody binding capacity of HD37 for the different cell lines.
Cell-binding assays and flow cytometric analysis
Aliquots of Jurkat or Raji cells were incubated with 100 µL of serial dilutions of AFM11-His, starting from 100 µg/mL, in FACS buffer (PBS, Life Technologies, cat.: 14190–169) containing 2% heat-inactivated FCS (Life Technologies, cat.: 10270–106), 0.1% sodium azide (Roth, Karlsruhe, Germany, cat.: A1430.0100) supplemented with 1 mg/mL polyclonal human antibody (Gammanorm, Octapharma, Langenfeld, Germany, cat.: PZN-2451445), for 45 min on ice. After repeated washing with FACS bufffer, cell-bound antibodies were detected with 10 µg/mL anti-His mAb 13/45/31–2 (Dianova, Hamburg, Germany, cat.: DIA910–1MG) followed by 15 µg/mL FITC-conjugated goat anti-mouse antibody (Dianova, cat.: 115–095–062). The cells were then washed again and resuspended in 0.2 mL of FACS buffer containing 2 µg/mL propidium iodide (PI) (Sigma, cat.: P4170) in order to exclude dead cells. The fluorescence of 5 × 103 living cells was measured using a Beckman-Coulter FC500 MPL flow cytometer using the MXP software (Beckman-Coulter, Krefeld, Germany). Mean fluorescence intensities of the cell samples were calculated using CXP software (Beckman-Coulter). After subtracting the fluorescence intensity values of the cells stained with the secondary and tertiary reagents alone, the values were used for analysis with GraphPad Prism (version 6.00 for Windows, GraphPad Software, La Jolla California USA). For the calculation of KD, the equation for one-site-binding (hyperbolic) was used. For better graphical visualization, the equation of a sigmoidal-dose-response, identical to the 4 parameter logistic equation, was used for calculation of the non-linear regression curve.
Cytotoxicity assays
For the calcein-release assay, target cells were labeled with 10 µM calcein AM (Life Technologies, cat.: C3100MP) for 30 min in RPMI medium at 37°C, washed, and 1 × 104 cells were seeded, in individual wells of a 96-well micro plate, together with effector cells in a total volume of 200 µL at the indicated effector:target (E:T) ratios in the presence of increasing antibody concentrations. After incubation for the indicated time periods at 37°C in a humidified 5% CO2 atmosphere, the fluorescence (F) of calcein released into the supernatant was measured by a plate reader at 520 nm (Victor 3, Perkin Elmer, Turku Finland, cat.: 1420–012). The specific cell lysis was calculated as: [F(sample)–F(spontaneous)]/[F(maximum)–F(spontaneous)] × 100%. F(spontaneous) represents fluorescence released from target cells in the absence of effector cells and antibodies, and F(maximum) represents that released after total cell lysis induced by addition of 1% Triton X-100 (Roth, cat.: 3051.2). Regression curves were fit to calculate EC50 (GraphPad Prism).
For the FACS-based cytotoxicity assay, target cells were harvested, washed twice with RPMI medium without FCS, and resuspended in diluent C (PKH67 Green Fluorescent Cell Linker Mini Kit, Sigma, cat.: MINI67) to a density of 2 × 107/mL. The cell suspension was then mixed with an equal volume of a double-concentrated PKH67-labeling solution (e.g., 1 µL PKH67 in 250 µL diluent C) and incubated for 2 – 5 min at room temperature with periodic mixing according to the manufacturer's instructions. The staining reaction was stopped by adding an equal volume of FCS and incubation for 1 min. After washing the labeled target cells with complete RPMI medium, cells were counted and resuspended to a density of 2 × 105/mL in complete RPMI medium. 2 × 104 target cells were then seeded together with effector cells at the indicated E:T ratio and the indicated antibodies in individual wells of a round-bottomed 96-well microtiter plate in a total volume of 200 µL/well. Spontaneous cell death and killing of targets by effectors in the absence of antibodies were determined in at least 3 replicates on each plate. TandAb-mediated killing was usually determined in duplicates.
After centrifugation for 2 min at 200 × g at room temperature, the assay plates were incubated for the indicated periods of time at 37°C in a humidified atmosphere with 5% CO2. After incubation, cultures were washed once with FACS buffer and then resuspended in 150 µL FACS buffer supplemented with 2 µg/mL PI. The absolute number of living target cells, characterized as positive for green PKH67 staining but negative for PI staining, was quantified using a Beckman-Coulter FC500 MPL flow cytometer (Beckman-Coulter).
Based on the measured remaining living target cells, the percentage of specific lysis was calculated according to the formula: [1-(Nliving targets(sample)) / (Nliving targets(spontaneous))] × 100%. EC50 values were calculated by non-linear regression of the data modeled as sigmoidal dose-response curves using GraphPad Prism (GraphPad Software).
Autologous PBMC cultures
1.5 × 106 PBMC from 10 healthy donors were seeded, in the presence of 100 ng/mL or 1 ng/mL of the indicated antibodies, in individual wells of a flat-bottomed 96-well microtiter plate in a total volume of 200 µL/well. As a control, PBMC were cultured in the absence of antibodies. After incubation for 22 – 24 h at 37°C in a humidified atmosphere with 5% CO2, cells were harvested, washed twice with PBS, resuspended in 300 µL FACS buffer supplemented with 1 mg/mL polyclonal human IgG (Gammanorm, Octapharma, cat.: PZN-2451445) and divided into 3 aliquots for staining of T cells (CD4+, CD8+), monocytes (CD13+/CD14+), B cells (CD20+) and NK cells (CD56+) The cells were incubated for 15 min in the dark with the antibodies (CD4 (Beckman-Coulter, cat.: A07750), CD8 (Beckman-Coulter, cat.: A07756), CD13 (BD Biosciences, Heidelberg, Germany, cat.: 561599), CD14 (BD Biosciences, cat.: 555397), CD20 (Biozol, cat.: 302304), CD56 (Beckman-Coulter, cat.: A21692)) at concentrations according to manufacturer recommendations. Following incubation, samples were washed once with 200 µL FACS Buffer and measured on a Beckman-Coulter FC500 MPL flow cytometer (Beckman-Coulter) using protocols established for PBMC and multicolor stainings (CD4 CD8 CD25 CD69; CD14 CD13; CD20 CD56) in the MXP acquisition software (Beckman-Coulter). CXP analysis software (Beckman-Coulter) was used for determination of the percentages of CD4/CD8+ cells, CD13+/CD14+, CD20+, and CD56+ cells. For the calculation of AFM11-His mediated changes (reduction or increase) in the relative amounts of CD4/CD8+, CD13+/CD14+, CD20+, and CD56+ cells, the percentages were compared in samples with AFM11-His or with control TandAb (HSA/CD3) treatment. The values were analyzed and plotted using GraphPad Prism (GraphPad Software).
Ex vivo apoptosis in B-CLL patient samples
Human studies were performed after written informed consent of the study participants, in accordance with the Declaration of Helsinki, and were approved by the institutional review board of the University hospital Würzburg (#232/14). PBMC were isolated by density gradient centrifugation using Ficoll/Hypaque (Biochrom, Berlin, Germany) from 4 B-CLL patients with more than 90% B-CLL cells in the separated fraction. RPMI 1640 media (Lonza, Basel, Switzerland) supplemented with 10% heat inactivated pooled human serum and 1% Pen Strep (Thermo Fisher Scientific) was used for apoptosis assays. Prior to the start of experiments, cells were stored in liquid nitrogen after resuspension in freezing medium (RPMI 1640 (Lonza) supplemented with 10% heat inactivated pooled human serum, 1% Pen Strep (Thermo Fisher Scientific) and 10% DMSO (Sigma-Aldrich, St. Louis, MO, USA) at a concentration of 2 × 107 cells/mL.
The following anti-human mAb were used for phenotypic analysis: CD4-PE (SK3), CD8-FITC (SK1) (both BD Bioscience), CD5-PerCP (Clone UCHT2), CD23-BV421 (Clone EBVCS-5), Annexin V-Alexa 647 (all Biolegend, San Diego, USA). For analysis of surface markers, cells were incubated with mAb at 4°C for 20 min. Staining was performed in PBS (Lonza) supplemented with 0.2% bovine serum albumin or Annexin V Binding Buffer (BD Bioscience). 2 × 106 thawed PBMCs from B-CLL patients were incubated with AFM11 and control antibody (HSA/CD3 TandAb) at a concentration of 100 ng/mL for 72 h in 1000 µL medium in a 48-well plate. Apoptosis of PBMC resuspended in PBS was measured by Annexin V staining (Biolegend). Cells were analyzed by FACS-Canto II flow cytometry (BD Biosciences) with FlowJo software (Tree Star, Inc., Ashland, OR, USA). The specific apoptosis was calculated as % specific apoptosis =100 × (% induced apoptotic cells –% spontaneous apoptotic cells)/(100 –% spontaneous apoptotic cells). Data were plotted using GraphPad Prism (GraphPad Software).
Proliferation assay
Before the cultures for the BrdU incorporation assay were initiated, all assay plates were blocked with RPMI 1640 medium supplemented with 10% heat-inactivated human type AB serum (Sigma, cat.: H4522), 4 mM L-glutamine (Life Technologies, cat.: 25030–024), 100 U/mL penicillin G sodium, 100 µg/mL streptomycin sulfate (Life Technologies, cat.: 15140–122), 1 mM sodium pyruvate (Life Technologies, cat.: 11360–039), 0.05 mM β-mercaptoethanol (Sigma, cat.: M3148–25ML) (herein referred to as complete RPMI medium) for 2 h at room temperature to prevent non-specific binding of antibodies to the plastic surface. 4 × 105 PBMC, B cell-depleted PBMC, or enriched T cells were seeded in complete RPMI medium in individual wells of flat-bottomed 96-well microtiter plates together with the indicated concentrations of test and control items in a total volume of 200 µL/well in triplicate. To assess spontaneous proliferation, cells were cultured in the absence of antibodies in 6 replicates. Plates were then incubated for 4 d at 37°C in a humidified 5% CO2 atmosphere. Cell cultures were pulsed with 100 µM BrdU 18 h prior to the end of incubation. Incorporated BrdU was quantified (BrdU Proliferation ELISA kit, Roche, Mannheim, Germany, cat.: 11647229001) according to manufacturer's instructions. After adding 100 mM H2SO4 to stop the color development, the absorption at 450 nm was measured with a multilabel plate reader (Victor 3). EC50 values were determined by non-linear regression (GraphPad Prism, GraphPad Software) of the absorbance data modeled as a sigmoidal dose-response. For graphical presentation mean, standard deviation (SD) of replicates, and regression curves were plotted using GraphPad Prism.
T cell activation marker assay
1 × 106 PBMC, B cell-depleted PBMC or enriched T cells were seeded, together with the indicated antibodies, in individual wells of a round-bottomed 96-well microtiter plate in a total volume of 200 µL/well. After incubation for the indicated time period at 37°C in a humidified 5% CO2 atmosphere, the cells were pelleted by centrifugation for 5 min at 500 × g. They were then resuspended in 200 µL FACS buffer, supplemented with 1 mg/mL polyclonal human IgG (Gammanorm, Octapharma, cat.: PZN-2451445), together with the following antibodies and the appropriate isotype controls: CD4-FITC (cat.: A07750), CD8-FITC (cat.: A07756), CD25-PE (cat.: A07774), and CD69-PC5 (cat.: IM2656, all Beckman Coulter) according to the procedure recommended by the manufacturer. After incubation, cells were assayed on a Beckman-Coulter FC500 MPL flow cytometer (Beckman-Coulter) using protocols established for PBMC and multicolor labeling employing the MXP acquisition software (Beckman-Coulter). For the determination of the percentages of CD25+ and CD69+ T cells, the CXP analysis software (Beckman-Coulter) was used.
Cytokine release assays
Before cells were seeded for the cytokine release assay, all assay plates were blocked with RPMI medium supplemented with 5% heat-inactivated FCS overnight at 4°C in order to prevent non-specific binding of antibodies to the plastic surface. After removal of the blocking medium, 5 × 105 PBMC, B cell-depleted PBMC, or enriched T cells were seeded in RPMI 1640 medium supplemented with 2 mM L-glutamine, 100 U/mL penicillin G sodium, 100 µg/mL streptomycin sulfate, and 5% heat-inactivated FCS in individual wells of flat-bottomed 96-well microtiter plates in a total volume of 200 µL/well together with the indicated concentration of AFM11-His and OKT3 (Biolegend, cat.: 317304). Assay plates were incubated for 48 h at 37°C in a humidified 5% CO2 atmosphere. After centrifugation at 70 × g for 2 min at room temperature, cell-free culture supernatants were harvested and frozen in aliquots at −80°C for cytokine quantification. IL2, IL4, IL6, IL10, TNF, and IFNγ were quantified (Th1/Th2 CBA kit, BD Biosciences, Heidelberg) at Bioassay GmbH (Heidelberg, Germany).
Cytokine release assays (7 donor study)
Cryopreserved PBMCs were pre-incubated at high cell density overnight (RPMI 1640, 10% FBS, 1% Pen/Strep, 2 mM L-Glutamine, 37°C, 5% CO2). The test items were added in soluble form or immobilized to the high binding tissue culture plate (96 Well Flat Bottom, Corning, cat# 3361) at the desired concentrations by air dry binding or wet coating. Cells were transferred from polypropylene plate to the plate with bound test compounds for immobilized approaches; alternatively, soluble test item was added to the cells directly into the tissue culture plate. After 48 h incubation, the plates were centrifuged at 1100 rpm (300 × g) for 10 min. Sample supernatants were collected and stored at −80°C for subsequent cytokine detection. For IL-2 and TNF measurements an additional plate was harvested after 24 h. Cytokine levels were determined by Luminex using Affymetrix cytokine and chemokine custom multiplex kits in the laboratories of Eurofins Panlabs Inc. (Bothell, WA) according to their validated protocols.
In vivo xenograft models
NOD/scid mice (NOD/MrkBomTac-Prkdcscid, Taconic, Denmark) were xenotransplanted with a suspension of 2.5 × 106 of Raji tumor cells by subcutaneous injection on day 0. The tumor cells were mixed with 1 × 107 human PBMC from healthy donors immediately prior to injection. To account for potential donor variability of the PBMC, each of the experimental groups was subdivided into 3 cohorts each receiving PBMCs from a single donor. Animals received i.v. doses of the test items in 5 daily injections: 2 h post tumor cell inoculation and subsequently after 24, 48, 72, and 96 h. There were 4 dose levels: 0.005, 0.05, 0.5, and 5.0 mg/kg. Tumor volume was monitored 3 times per week by measuring the large and small diameters of each tumor with a caliper. Tumor volumes were calculated from diameters according to the formula: V = (small diameter)2 × (large diameter) × 0.5.Citation41
Determination of pharmacokinetic parameters
A total of 80 CD1 mice were enrolled in 4 treatment groups. Animals received an i.v. single administration of 20, 4, and 0.8 mg/kg of AFM11. A satellite group for bioavailability assessment was subcutaneously dosed with 20 mg/kg of AFM11. Blood was collected over 96 h at −72 h, 5, 10, 30 min, and 1, 2, 4, 6, 8, 24, 48, 96 h post dose in staggered sampling groups. AFM11 serum concentrations were determined using the MSD ECL technology. The pharmacokinetic parameters (Cmax, C0, Tmax, CL, Vss, MRT, T1/2, AUC) were calculated by a non-compartmental analysis using Kinetica pharmacokinetic software, v.4.2 (Thermo Fisher Scientific).
Tissue distribution in mice
AFM11 was radiolabelled (125I) by direct electrophilic radioiodination, using Iodogen as oxidant, to a final specific activity of about 0.1 mCi/mg (3.7 MBq/mg). The radiolabelled product had a radiochemical purity of 97.5%. SE-HPLC analysis and SDS-PAGE suggest that the radioiodination process did not affect the integrity of AFM11 protein.
A total of 34 NOD/scid mice were subcutaneously xenografted in the right flank with 5 × 106 Raji tumor cells in 200 µL of PBS. Tumor-bearing mice with a tumor volume of about 239 ± 42 mm3 were injected intravenously in the tail vein at a dose level of 5 mg/kg. The tissue distribution study was performed on 3 animals per time point, 1 animal was frozen and reserved for whole body autoradiography.
At the time of sacrifice, organs of interest were excised, rinsed of residual blood and weighed. The selected tissues were: blood, tumor, liver, kidneys, heart, lung, spleen, brain, thyroid, bone (femur), muscle, stomach, ileum (with intestine contents). The counting of tissue radioactivity was performed in an automatic gamma counter (Wallace Wizard 2470, Perkin Elmer, Waltham, MA, USA). The concentration of radioactivity is expressed as percentage of the injected dose per gram of tissue (%ID/g). In addition, the ratio between the radioactivity retained in blood and the radioactivity in tumor and normal tissues was calculated (ratio tumor/organ) as well as the ratio between the radioactivity retained in the tissues and the radioactivity in blood (ratio organ/blood). For blood, this was approximated assuming that blood accounts for 70–80 mL/kg (1.64 mL of total blood for a 21–22 g mouseCitation42).
The distribution of radioactivity in excretion was assayed using 3 mice individually housed for 48 h in metabolism cages. The urine and feces were collected at 24 and 48 h. The radioactivity in excretion is expressed as percentage of the injected dose. The contribution of urinary and fecal routes was assessed and expressed as total excreted radioactivity. Assays for radiochemical purity of urinary samples were performed using instant thin layer chromatography to quantify the percentage of free iodine released from AFM11.
Disclosure of Potential Conflicts of Interest
UR, KE, CH, SHJK, IF, and EAZ are employees of Affimed Therapeutics AG.
ME, FM, and FLG are former employees of Affimed Therapeutics AG.
Supplemental_Material.pdf
Download PDF (343.2 KB)Acknowledgments
We thank G. Moldenhauer (Department of Molecular Immunology, German Cancer Research Center (DKFZ) Heidelberg, Germany) for providing cell lines. We thank B. Stockmeyer (University of Erlangen, Germany) for providing pcDNA3.1(+) plasmid DNA encoding human CD19. We also thank R. Morse for assistance in preparing the manuscript and helpful discussions. The technical assistance of Frank Malischewsky, Sonja Hennemann, Sarah Flößer, Carolin Böhm, Jürgen Weik, Ute Schniegler-Mattox, Thomas Müller, Andreas Müller, Raphael Bleiler, Alexandra Stolarek, Stefanie Wolff, and Tatjana Zingraf (all Affimed Therapeutics AG, Heidelberg, Germany) was gratefully appreciated.
Supplementary Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Grillo-López AJ, White CA, Dallaire BK, Varns CL, Shen CD, Wei A, Leonard JE, McClure A, Weaver R, Cairelli S, et al. Rituximab: the first monoclonal antibody approved for the treatment of lymphoma. Curr Pharm Biotechnol 2000; 1:1-9; PMID:11467356; http://dx.doi.org/10.2174/1389201003379059
- Alduaij W, Illidge TM. The future of anti-CD20 monoclonal antibodies: are we making progress?. Blood 2011; 117:2993-3001; PMID:21209380; http://dx.doi.org/10.1182/blood-2010-07-298356
- Illidge T, Cheadle EJ, Donaghy C, Honeychurch J. Update on obinutuzumab in the treatment of B-cell malignancies. Expert Opin Biol Ther 2014; 14:1507-17; PMID:25190612; http://dx.doi.org/10.1517/14712598.2014.948414
- Remer M, Al-Shamkhani A, Glennie M, Johnson P. Mogamulizumab and the treatment of ccr4-positive t-cell lymphomas. Immunotherapy 2014; 6:1187-206; PMID:25496334; http://dx.doi.org/10.2217/imt.14.94
- Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, Richards JO, Vostiar I, Joyce PF, Repp R, et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res 2008; 68:8049-57; PMID:18829563; http://dx.doi.org/10.1158/0008-5472.CAN-08-2268
- Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: cars take the front seat for hematologic malignancies. Blood 2014; 123:2625-35; PMID:24578504; http://dx.doi.org/10.1182/blood-2013-11-492231
- Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs 2010; 2:129-36; PMID:20190561; http://dx.doi.org/10.4161/mabs.2.2.11221
- Buhmann R, Michael S, Juergen H, Horst L, Peschel C, Kolb H. Immunotherapy with FBTA05 (Bi20), a trifunctional bispecific anti-CD3 x anti-CD20 antibody and donor lymphocyte infusion (DLI) in relapsed or refractory B-cell lymphoma after allogeneic stem cell transplantation: study protocol of an investigator-driven. J Transl Med 2013; 11:160; PMID:23815981; http://dx.doi.org/10.1186/1479-5876-11-160
- Topp MS, N Goekbuget N, Stein AS, Bargou RC, Dombret H, Fielding AK, Ribera JM, Foà F, Zugmaier G, Holland C, et al., Confirmatory openlabel, singlearm, multicenter phase 2 study of the BiTE antibody blinatumomab in patients (pts) with relapsed/refractory Bprecursor acute lymphoblastic leukemia (r/r ALL). J Clin Oncol 2014; 32:5s ( suppl; abstr 7005); PMID:24190110; http://dx.doi.org/10.1200/JCO.2014.56.3247
- Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res 2011; 317:1255-60; PMID:21419116; http://dx.doi.org/10.1016/j.yexcr.2011.03.010
- Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gökbuget N, Neumann S, Goebeler M, Viardot A, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012; 119:6226-33; PMID:22592608; http://dx.doi.org/10.1182/blood-2012-01-400515
- Portell CA, Wenzell CM, Advani AS. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin Pharmacol 2013; 5:5-11; PMID:23671399; http://dx.doi.org/10.2147/CPAA.S42689
- Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother 2010; 59:1197-209; PMID:20309546; http://dx.doi.org/10.1007/s00262-010-0844-y
- Beverley PC, Callard RE. Distinctive functional characteristics of human “T” lymphocytes defined by e rosetting or a monoclonal anti-T cell antibody. Eur J Immunol 1981; 11:329-34; PMID:6788570; http://dx.doi.org/10.1002/eji.1830110412
- Pezzutto A, Dörken B, Rabinovitch PS, Ledbetter JA, Moldenhauer G, Clark EA. CD19 monoclonal antibody HD37 inhibits anti-immunoglobulin-induced b cell activation and proliferation. J Immunol 1987; 138:2793-9; PMID:2437199
- Schwarz M, Röttgen P, Takada Y, Le Gall F, Knackmuss S, Bassler N, Büttner C, Little M, Bode C, Peter K. Single-chain antibodies for the conformation-specific blockade of activated platelet integrin alphaiibbeta3 designed by subtractive selection from naive human phage libraries. FASEB J 2004; 18:1704-6; PMID:15522915
- Honegger A, Plückthun A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol 2001; 309:657-70; PMID:11397087; http://dx.doi.org/10.1006/jmbi.2001.4662
- Padlan EA, Abergel C, Tipper JP. Identification of specificity-determining residues in antibodies. FASEB J 1995; 9:133-9; PMID:7821752
- Kufer P, Lutterbüse R, Kohleisen B, Zeman S, Bäuerle P, inventors; Micromet AG, assignee. Pharmaceutical compositions comprising bispecific anti-CD3, anti-CD19 antibody constructs for the treatment of B-cell related disorders. World patent WO2004/106381. 2004 Dec 9. English
- Traunecker A, Lanzavecchia A, Karjalainen K. Bispecific single chain molecules (janusins) target cytotoxic lymphocytes on HIV infected cells. EMBO J 1991; 10:3655-9; PMID:1834458
- Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011; 117:4542-51; PMID:21300981; http://dx.doi.org/10.1182/blood-2010-09-306449
- Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, Kufer P, Riethmuller G, Bargou R, Baeuerle PA. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer 2002; 100:690-7; PMID:12209608; http://dx.doi.org/10.1002/ijc.10557
- Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther 2012; 136:334-42; PMID:22940266; http://dx.doi.org/10.1016/j.pharmthera.2012.07.013
- Document: Blinatumomab (AMG 103) Background information for the pediatric subcommittee of the oncologic drugs advisory committee meeting 04 December 2012 [Internet]. Silver Spring (MD): U. S. Food and Drug Administration (US); 2012. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM330210.pdf
- Hoffmann P, Hofmeister R, Brischwein K, Brandl C, Crommer S, Bargou R, Itin C, Prang N, Baeuerle PA. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer 2005; 115:98-104; PMID:15688411; http://dx.doi.org/10.1002/ijc.20908
- Tax WJ, Hermes FF, Willems RW, Capel PJ, Koene RA. Fc receptors for mouse igg1 on human monocytes: polymorphism and role in antibody-induced t cell proliferation. J Immunol 1984; 133:1185-9; PMID:6235283
- Ceuppens JL, Bloemmen FJ, Van Wauwe JP. T cell unresponsiveness to the mitogenic activity of okt3 antibody results from a deficiency of monocyte fc gamma receptors for murine igg2a and inability to cross-link the t3-ti complex. J Immunol 1985; 135:3882-6; PMID:2933454
- Smith KG, Austyn JM, Hariri G, Beverley PC, Morris PJ. T cell activation by anti-t3 antibodies: comparison of igg1 and igg2b switch variants and direct evidence for accessory function of macrophage fc receptors. Eur J Immunol 1986; 16:478-86; PMID:2938973; http://dx.doi.org/10.1002/eji.1830160503
- Verwilghen J, Baroja ML, Van Vaeck F, Van Damme J, Ceuppens JL. Differences in the stimulating capacity of immobilized anti-cd3 monoclonal antibodies: variable dependence on interleukin-1 as a helper signal for t-cell activation. Immunology 1991; 72:269-76; PMID:1826671
- Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, Knackmuss SHJ, Kiprijanov S, Little M, Zhukovsky EA. A novel tetravalent bispecific tandab (CD30/CD16a) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 2014; 6:728-39; PMID:24670809
- Rothe A, Topp MS, Eichenauer DA, von Tresckow B, Hummel H, Ravic M, Marschner J, Engert A. Updated phase I data of afm13: a bispecific tandem antibody (tandab) in relapsed/refractory (r/r) hodgkin lymphoma (hl). J Clin Oncol 2014; 32:( suppl; abstr 3025ˆ)
- Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, Horst H, Raff T, Viardot A, Schmid M, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol 2011; 29:2493-8; PMID:21576633; http://dx.doi.org/10.1200/JCO.2010.32.7270
- Dreier T, Baeuerle PA, Fichtner I, Grün M, Schlereth B, Lorenczewski G, Kufer P, Lutterbüse R, Riethmüller G, Gjorstrup P, et al. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J Immunol 2003; 170:4397-402; PMID:12682277; http://dx.doi.org/10.4049/jimmunol.170.8.4397
- Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol 2012; 33:579-89; PMID:22926201; http://dx.doi.org/10.1016/j.it.2012.07.004
- Feng S, Huang Y, Chen Z. Does VEGF secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight-junction proteins in central nervous system leukemia?. Med Hypotheses 2011; 76:618-21; PMID:21398042; http://dx.doi.org/10.1016/j.mehy.2010.12.001
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013; 41: W34-40; PMID:23671333; http://dx.doi.org/10.1093/nar/gkt382
- Mack M, Riethmüller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A 1995; 92:7021-5; PMID:7624362; http://dx.doi.org/10.1073/pnas.92.15.7021
- Kjer-Nielsen L, Dunstone MA, Kostenko L, Ely LK, Beddoe T, Mifsud NA, Purcell AW, Brooks AG, McCluskey J, Rossjohn J. Crystal structure of the human t cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mab OKT3. Proc Natl Acad Sci U S A 2004; 101:7675-80; PMID:15136729; http://dx.doi.org/10.1073/pnas.0402295101
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In John M. Walker, ed., The Proteomics Protocols Handbook. Taylor & Francis; 2005; 571-607.
- Kontula K, Paavonen T, Vuopio P, Andersson LC. Glucocorticoid receptors in hairy-cell leukemia. Int J Cancer 1982; 30:423-6; PMID:7141737; http://dx.doi.org/10.1002/ijc.2910300407
- Carlsson G, Gullberg B, Hafström L. Estimation of liver tumor volume using different formulas - an experimental study in rats. J Cancer Res Clin Oncol 1983; 105:20-3; PMID:6833336; http://dx.doi.org/10.1007/BF00391826
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993; 10:1093-5; PMID:8378254; http://dx.doi.org/10.1023/A:1018943613122