
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The epidermal growth factor receptor (EGFR) and the insulin-like growth factor-1 receptor (IGF-1R) play critical roles in tumor growth, providing a strong rationale for the combined inhibition of IGF-1R and EGFR signaling in cancer therapy. We describe the design, affinity maturation, in vitro and in vivo characterization of the bispecific anti-IGF-1R/EGFR antibody XGFR*. XGFR* is based on the bispecific IgG antibody XGFR, which enabled heterodimerization of an IGF-1R binding scFab heavy chain with an EGFR-binding light and heavy chain by the “knobs-into-holes” technology. XGFR* is optimized for monovalent binding of human EGFR and IGF-1R with increased binding affinity for IGF-1R due to affinity maturation and highly improved protein stability to oxidative and thermal stress. It bears an afucosylated Fc-portion for optimal induction of antibody-dependent cell-mediated cytotoxicity (ADCC). Stable Chinese hamster ovary cell clones with production yields of 2–3 g/L were generated, allowing for large scale production of the bispecific antibody. XGFR* potently inhibits EGFR- and IGF-1R-dependent receptor phosphorylation, reduces tumor cell proliferation in cells with heterogeneous levels of IGF-1R and EGFR receptor expression and induces strong ADCC in vitro. A comparison of pancreatic and colorectal cancer lines demonstrated superior responsiveness to XGFR*-mediated signaling and tumor growth inhibition in pancreatic cancers that frequently show a high degree of IGF-1R/EGFR co-expression. XGFR* showed potent anti-tumoral efficacy in the orthotopic MiaPaCa-2 pancreatic xenograft model, resulting in nearly complete tumor growth inhibition with significant number of tumor remissions. In summary, the bispecific anti-IGF-1R/EGFR antibody XGFR* combines potent signaling and tumor growth inhibition with enhanced ADCC induction and represents a clinical development candidate for the treatment of pancreatic cancer.
Introduction
The epidermal growth factor receptor (EGFR) and the insulin-like growth factor-1 receptor (IGF-1R) are frequently over-expressed receptor tyrosine kinases that show enhanced activation in a variety of human tumors. EGFR and IGF-1R contribute to tumor development and progression by enhancing cell proliferation, inhibiting apoptosis and inducing angiogenesis.Citation1,2 Both receptor tyrosine kinases mediate tumor growth via the PI3K-AKT and RAS-RAF-MAPK pathway, and cross-talk between EGFR and IGF-1R signaling was observed on receptor and downstream signaling levels. Interestingly, several preclinical and clinical studies demonstrated that IGF-1R signaling may induce resistance to EGFR inhibitorsCitation3,4 and EGFR-dependent signaling can confer resistance to IGF-1R inhibitors.Citation5-8 Hence, targeting EGFR and IGF-1R simultaneously is a promising strategy to achieve enhanced tumor growth inhibition.
The first-generation EGFR kinase inhibitors erlotinib or gefitinib are routinely used in the clinic for treatment of tumor malignancies.Citation9 More recently, novel EGFR kinase inhibitors have been approved for cancer therapy, namely osimertinib (AZD9291) for patients with EGFR T790M mutation-positive metastatic non-small cell lung cancer (NSCLC),Citation10 and afatinib, a covalent EGFR-HER2 multikinase inhibitor for treatment of (EGFR mutation positive) NSCLC.Citation11,12 Other (irreversible) EGFR kinase inhibitors are in late-stage clinical development, e.g., dacomitinib (PF-00299804), BI 1482694 (HM61713), rociletinib (CO-1686) (reviewed in ref.Citation13,14). In addition, monoclonal antibodies such as cetuximab and panitumumab, which block the binding of intrinsic receptor ligands (e.g., EGF) to EGFR and inhibit uncontrolled growth of tumor cells are applied in clinical practice.Citation15,16 Recently, the novel IgG1 isotype EGFR antibody necitumumabCitation17-19 was approved for the treatment of metastatic NSCLC. In order to enhance the immune effector function, glycoengineered EGFR antibodies such as imgatuzumab (GA201)Citation20-23 and CetuGEX, a glycoengineered version of cetuximab, have been developed and have been or are in clinical trials. Imgatuzumab is not in active clinical development based on the negative outcome of a Phase 2 trial where it was compared to cetuximab in combination with FOLFIRI in metastatic colorectal cancer.Citation24 As an alternative approach, a synergistic combination of EGFR antibodies, SYM004, is currently in clinical development.Citation25,26
Several monoclonal antibodies targeting IGF-1R, such as R1507 (teprotumumab),Citation27,28 figitumumab (CP-751871),Citation29-32 ganitumab (AMG479),Citation33-35 dalotuzumab (MK-0646)Citation36-38 or cixutumumab (IMC-A12)Citation39-41 entered late-stage clinical development in combination with different chemotherapeutic drugs, (EGFR) kinase inhibitors or antibodies, but are no longer in development due to limited/lack of efficacy in these clinical trials (reviewed in ref. Citation42-44). Particularly, clinical development of R1507 is not being pursued based on its limited clinical efficacy in sarcomaCitation45 and a Phase 2 study in NSCLC in combination with erlotinib.Citation46
Based on the clinical results with monospecific receptor tyrosine kinase (RTK) antibodies such as trastuzumab or cetuximab, the generation of bispecific RTK antibodies has attracted high interest. Several bispecific antibodies recently entered, or are about to enter, clinical trials, including: 1) MM-141, a bispecific antibody against IGF-1R and HER3, is currently in clinical development;Citation47 2) a bispecific, dual action Fab (DAF)-based IgG antibody recognizing EGFR and HER3 simultaneously has been studied in clinical trials;Citation48-50 and 3) bispecific c-Met-EGFR antibodies.Citation51,52 In addition, we and others have described novel tri- and tetraspecific antibodies targeting oncogenic RTKs.Citation53,54
Taken together, clinical testing of IGF-1R and EGFR-targeted therapeutic antibody combinations focused on the treatment of lung or colorectal cancer resulted in discouraging results.Citation36,40 Heterogeneity with regard to mutational status and signaling pathways, e.g., in colorectal cancer, requires a clear hypothesis for patient selection to detect the clinical efficacy of combined EGFR and IGF-1R inhibition. Despite the negative outcome of these clinical studies investigating IGF-1R/EGFR combination therapy, innovative drug design such as combining potent signaling inhibition and enhanced engagement of immune effector functions through glycoengineering of the Fc region in a bispecific IGF-1R/EGFR antibody may help to overcome primary and secondary resistance mechanisms, and holds the potential for successful tumor therapy. Afucosylation or glycoengineering of therapeutic antibodies leads to an approximately 100-fold increase in the affinity to FcγRIIIa receptors and subsequently to an increase in cell death via antibody-dependent cell-mediated cytotoxicity (ADCC).Citation20,21,55,56
We previously described the glycoengineered bispecific antibody XGFR, which was generated using the “knobs-into-holes” (KiH) technology for heterodimerization of an EGFR binding heavy and light chain and an IGF-1R binding scFab heavy chain.Citation57 Here, we describe the highly improved molecule XGFR* derived from the parental molecules anti-EGFR imgatuzumab (GA201)Citation20-23 and an affinity-matured version of anti- IGF-1R R1507.Citation27,28 XGFR* was optimized for maximal monovalent IGF-1R/EGFR signaling inhibition in cells with heterogeneous receptor expression and increased stability to thermal and oxidative stress. It was produced in a stable Chinese hamster ovary (CHO) production cell line at high yields and characterized for product quality. Our results show that XGFR* allowed effective inhibition of key signaling pathways combined with optimal induction of ADCC through Fc receptor-mediated effector functions in vitro, resulting in potent anti-tumoral efficacy in the orthotopic MiaPaCa-2 pancreatic xenograft model.
Results
Design of “one armed scFab” bispecific antibodies XGFR and XGFR* targeting EGFR and IGF-1R simultaneously
XGFR () is a bispecific antibody with an EGFR binding arm composed of the imgatuzumab (GA201) light and heavy chain and a second IGF-1R binding arm based on a single chain Fab-fragment (scFab) of R1507 with the light chain attached to the N-terminus of the VH domain by a 32 amino acid glycine serine linker to form the second heavy chain.Citation57 Heterodimerization of the 2 heavy chains in the molecule was achieved by application of the KiH technology.Citation58 The “knob” mutation (T366W) was introduced into the CH3 domain of the imgatuzumab heavy chain, and 3 “hole” mutations (T366S, L368A, and Y407V) were introduced into the CH3 domain of the scFab heavy chain of R1507. In addition, 2 cysteine residues were introduced (S354C on the “knob” and Y349C on the “hole” side) to form a stabilizing disulfide bridge. In the complementarity-determining regions (CDR) of XGFR*, the CDR2 and CDR3 domains of the VL of the single chain Fab were exchanged by affinity maturation of R1507 to F13B5. The F13B5 light chain variable region contains a W94Y mutation in the CDR3 domain and an aspartic acid residue in the CDR2 domain was deleted (). The CDR1 domain of the variable light chain and all CDR domains of the variable heavy chain of R1507 and F13B5 are identical.
Figure 1. Design of XGFR and XGFR* bispecific antibodies. Schematic diagram of the IGF-1R/EGFR bispecific antibodies XGFR and XGFR*. The bispecific antibodies consist of a human IgG1 heavy and light chain with specificity for EGFR based on the parental antibody GA201 (red and yellow) and a single chain Fab with specificity for IGF-1R derived from the antibody R1507 (blue and light blue) for XGFR and an affinity-matured R1507 (F13B5) for XGFR* (blue and pink). The R1507 or F13B5 light chains were fused by a 32 amino acid (G4S)6GG linker (green) to the N-terminus of the R1507 VH domain. Dimerization of the 2 different heavy chains in the XGFR antibody was facilitated by the knob-into-hole mutations (light gray) in the CH3 domain and an additional disulfide bond.
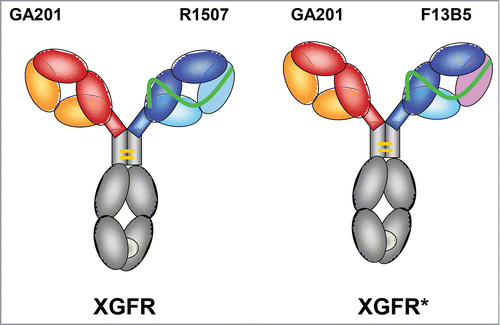
Table 1. Sequence analysis of light chain variable domain complementarity-determining regions.
Affinity maturation of anti IGF-1R antibody R1507
Affinity maturation libraries were constructed on the basis of the R1507 Fab-fragment with randomized positions in CDR1 (residues 30, 31, 32, 34 according to Kabat numbering scheme) and CDR2 (residues 50, 51, 52, 53) of the light chain variable domain (R1507 VL library) and randomized positions in CDR1 (residues 31, 32, 33, 34, 35) and CDR2 (residues 50, 52, 53, 54, 56, 58) of the heavy chain variable domain (R1507 VH library). Phagemid vectors carrying the randomized Fab libraries were transformed into competent E. coli TG1 cells to obtain final library sizes of 1.4 × 1010 for the R1507 VL library and 8.7 × 109 for the R1507 VH library with 65.3% and 73% functional clones.
Selections against the extracellular domains of human or cynomolgus IGF-1R were carried out using a pool of purified R1507 VL and VH library phages. Three different selection strategies were used: 1) decrease of antigen concentration over subsequent rounds of bio-panning (ranging from 10 nM in the first selection round down to 0.8 nM in the third to fifth selection round); 2) competitive selection by addition of parental IgG R1507 at 10-fold antigen concentration or by addition of 1 µM non-biotinylated human IGF-1R to the binding reactions (only in rounds where biotinylated cynomolgus IGF-1R was used as target); or 3) off-rate selections by allowing dissociation of phage antibody antigen complexes for either 3 hours or 3 d. Selections were carried out by either using only human or only cynomolgus IGF-1R during subsequent selection rounds or alternating between these 2 species to avoid affinity-maturation toward one species only. Selection outputs from bio-panning rounds 2 – 5 were screened by surface plasmon resonance (SPR) to identify clones with superior kinetic rate constants and affinity compared to the parental antibody R1507.
The affinity maturation of the IGF-1R antigen binding site and subsequent selection resulted in 5 clones F13B5, L37F7, L39D7, L31D11 and L31D7 with KD values between 1.47 × 10−9 and 2.69 × 10−10 M (). The binding affinity to human IGF-1R of CDR-modified Fab fragments in comparison with the parental Fab fragment R1507 (KD = 1.83 × 10−8 M) was increased 12 – 68 fold by affinity maturation (, ). The affinity-matured Fab fragments showed an approximately 10-fold increased dissociation rate constant (kd), which leads to prolonged binding to the human extracellular IGF-1R domain (). All identified clones were cross reactive to cynomolgus IGF-1R. Based on the results of an in silico oxidation hot spot analysis, the F13B5 Fab fragment was selected for construction of the XGFR* molecule. The W94Y mutation in the CDR3 domain of F13B5 leads to removal of the tryptophan amino acid in the parental antibody R1507 in this position, which was identified as an oxidation hotspot and is still present in the affinity-matured clones L31D7 and L31D11.
Figure 2. Surface plasmon resonance analysis of R1507 affinity maturation. Kinetic rate constants ka and kd as well as affinity (KD) of affinity-matured Fab fragments were measured by SPR using a ProteOn XPR36 (BioRad) instrument at 25°C. An anti-Fab capture antibody was immobilized on a GLM chip to capture purified Fab fragments of affinity-matured clones and a R1507 control Fab. In a one-shot kinetic assay set-up , human IGF-1R was injected as analyte with an association time of 200s and a dissociation time of 600s in a 3-fold dilution series ranging from 33–0.4 nM. Association and dissociation rates were calculated using a simple 1:1 Langmuir binding model.
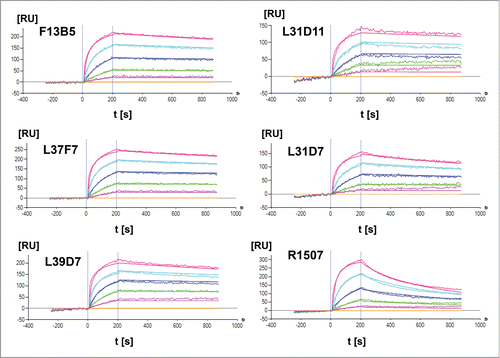
Table 2. Surface plasmon resonance analysis of R1507 affinity maturation. Kinetic rate constants ka and kd as well as affinity (KD) of affinity-matured Fab fragments were measured by SPR.
Expression and purification of XGFR*
For initial experiments, the bispecific antibodies XGFR* and XGFR were produced by transient expression in HEK293 cells. XGFR* was purified to homogeneity by protein A and hydroxyapatite chromatography from cell culture supernatants and subjected to CE-SDS and SDS-PAGE analysis under non-reducing and reducing conditions (). Reduced CE-SDS analysis of XGFR* showed 92.3 kDa F13B5 scFab heavy chain (hole), 59.2 kDa GA201 heavy chain and 26.3 kDa light chain peaks (). Under non-reducing conditions, a molecular weight of 157.7 kDa was measured by CE-SDS, indicating glycosylation of XGFR* in HEK293 cells. Upon transient expression, purification yields of the bispecific antibody XGFR* (37.7 mg/L) were increased 1.8-fold in comparison with XGFR, which gave a final yield of 20.6 mg/L after Protein A and SEC purification (). The product quality of purified XGFR* was further analyzed by analytical size exclusion chromatography and mass spectroscopy (MS) analysis. Analytical size exclusion chromatography showed XGFR* purity of >98% with low levels of high molecular weight aggregates (). The high product quality of purified XGFR* was confirmed by MS analysis and no KiH antibody byproducts such as hole-hole and knob-knob heavy chain homodimers or half antibodies lacking a knob or hole heavy chain were detectable.
Figure 3. Biochemical and biophysical analysis of purified XGFR*. (A) Purity, antibody integrity and molecular weight of XGFR* was characterized by CE-SDS and (B) SDS-PAGE under reducing and non-reducing conditions. (C) Analytical size exclusion chromatography (SEC) was used to estimate the presence of aggregates in the one arm scFab XGFR* molecule after protein A and Hydroxyapatite purification. The chromatogram represents a 20 µg injection.
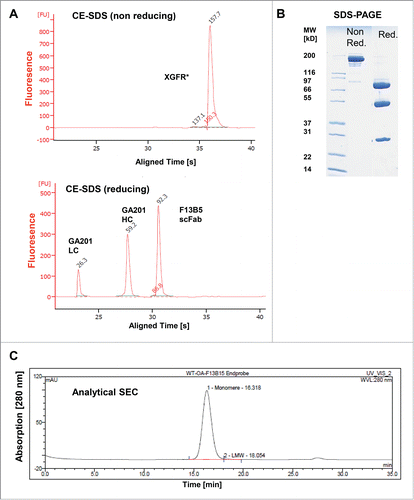
Table 3. Protein purification yield of XGFR* and XGFR.
Generation of stable XGFR* expressing CHO cell lines
For further clinical development, the manufacturing scalability of XGFR* was evaluated in CHO cells. GA201 knob heavy and light chain and the F13B5 OAscFAb hole heavy chain, as well as recombinant β-1,4-N-acetyl-glucosaminyltransferase III and Golgi α-mannosidase II, were over expressed in CHO K1 cells using a glutamine synthetase expression system. Several stable production CHO clones with yields of 2–3 g/L and afucosylation levels above 70% were identified, allowing for large-scale production of the XGFR* bispecific antibody.
SPR analysis of the XGFR* binding affinities to IGF-1R and EGFR
SPR analysis was used to determine the association (ka) and dissociation rate constants (kd), as well as equilibrium constants (KD), of the bispecific antibodies at 37°C. XGFR* or XGFR were captured by an anti-human IgG-Fc antibody on the chip surface and increasing concentrations of either EGFR or IGF-1R receptor extracellular domains were injected. As expected, XGFR and XGFR* bispecific molecules bound to EGFR with an identical low nanomolar KD value of 6.4 nM (). The parental antibody GA201 showed a KD value of 3.0 nM in the control experiment. Binding to the IGF-1R extracellular domain was measured on a C1-Chip with low ligand density to achieve monovalent binding of the IGF1R dimer. Under these experimental conditions, the binding affinities of the R1507 (XGFR) and F13B5 (XGFR*) scFabs were determined as 3.9 and 0.18 nM, respectively (). A KD value of 5.0 nM was determined for the parental control antibody R1507. The half-life (t1/2) of the IGF-1R/antibody complex increased from 2.4 minutes for XGFR to 28.4 minutes for XGFR* and clearly demonstrates the superior binding properties of the affinity-matured molecule. In addition, XGFR lost approximately 50% IGF-1R binding activity in SPR analysis upon 10 d incubation at 40°C under oxidative stress compared to a sample stored at −80°C. In contrast, XGFR* did not show any loss of IGF-1R binding levels when incubated for 10 d at 40°C under oxidative stress, due to the removal of the oxidation hotspot W94Y in the CDR3 domain of the F13B5 scFab.
Table 4. Surface plasmon resonance analysis of bispecific antibody binding affinity to human EGFR.
Table 5. Surface plasmon resonance analysis of bispecific antibody binding affinity to human IGF-1R.
Competitive fluorescence-activated cell sorting analysis of the XGFR* binding to tumor cells
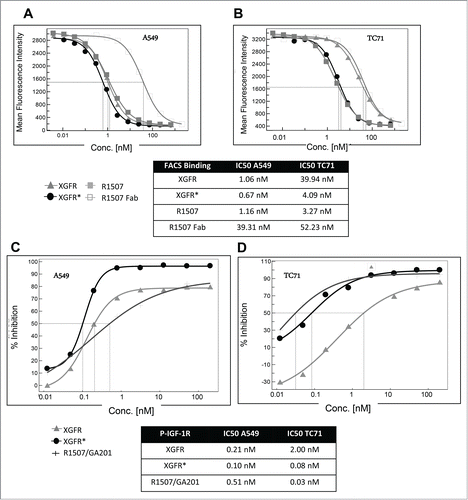
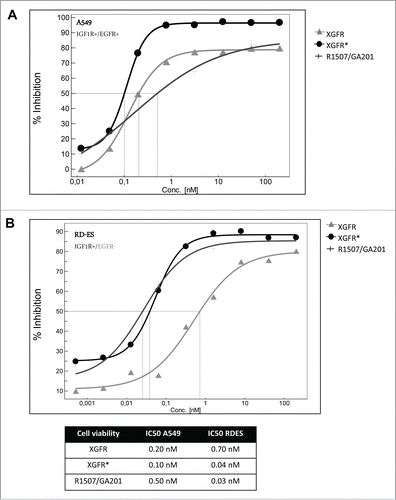
For competitive fluorescence-activated cell sorting (FACS) analysis, tumor cells were incubated with a mixture of phycoerythrin (PE)-labeled monospecific IGF-1R antibody R1507 at 1 μg/ml and varying concentrations of unlabeled IGF-1R/EGFR bispecific antibodies, monospecific R1507 antibody or Fab fragment. Bivalent antibody binding to human adenocarcinoma alveolar basal epithelial A549 cells, which express EGFR and IGF-1R receptors, was similar for XGFR, XGFR* and R1507, with inhibition of PE- labeled R1507 antibody binding at IC50 values of approximately 1.0 nM in competitive FACS analysis (). Here, binding of the EGFR binding arm with a relatively slow dissociation rate compensates the fast off-rate of the IGF-1R binding arm in the XGFR molecule, whereas monovalent binding of the R1507 Fab molecule was approximately 34 fold reduced compared to bivalent R1507 binding to IGF-1R (). A similar effect was found, when binding to cells expressing IGF-1R in absence of EGFR such as the Ewing´s sarcoma cell line TC-71 was analyzed. XGFR and the R1507 Fab fragment exhibited approximately 10-fold reduced binding to TC71cells compared to the bivalent antibody R1507 (). In contrast, the affinity-matured molecule XGFR* with a F13B5 scFab as the IGF-1R binding component showed strongly improved binding to cells expressing IGF-1R in the absence of EGFR and an IC50 value in the low nanomolar range similar to the R1507 bivalent antibody ().
Figure 4. Tumor cell binding and IGF-1R phosphorylation inhibition of XGFR*. (A) Competitive FACS analysis of bispecific constructs XGFR and XGFR*, bivalent IGF-1R antibody R1507 and monovalent R1507 Fab fragment on A549 cells with surface expression of IGF-1R and EGFR and (B) TC-71 cells with surface expression of IGF-1R in absence of EGFR. (C) Inhibition of IGF-1R phosphorylation of XGFR, XGFR* and parental control antibody mixture R1507/GA201 in A549 and (D) TC-71 cells. Titration curves in the indicated concentration range and IC50 values are shown.
XGFR* inhibits IGF-1R/EGFR dependent signaling and tumor cell proliferation
XGFR* effectively inhibited IGF-1R phosphorylation in A549 cells expressing similar levels of IGF-1R and EGFR with an IC50 value of 0.10 nM and a maximal inhibition level of approximately 95% (). The parental antibody mixture R1507/GA201 and XGFR showed lower maximal inhibition and IC50 values of 0.51 nM and 0.21 nM, respectively (). Inhibition of EGFR phosphorylation in A549 cells mediated by XGFR* was comparable to XGFR with a maximal inhibition of 100% and IC50 values of 0.2 nM (data not shown). IGF-1R phosphorylation inhibition of XGFR* (IC50: 0.08 nM) was improved 25 fold compared to XGFR (IC50: 2 nM) in TC71 cells, which express only IGF-1R in the absence of EGFR (). The affinity-matured XGFR* molecule induced IGF-1R phosphorylation inhibition in TC71 cells at a similar level as the R1507/GA201 antibody mixture (). Tumor growth inhibition of XGFR* was analyzed in a 3D cell viability assay and IC50 values of 0.10 nM for inhibition of A549 cells and 0.04 nM for inhibition of Ewing´s sarcoma cell line RD-ES growth were found (). XGFR* mediated tumor growth inhibition of RD-ES cells with expression of IGF-1R without detectable EGFR levels was 17.5-fold improved over XGFR and similar to the R1507/GA201 antibody mixture (). In summary, improved binding of XGFR* resulted in increased inhibition of IGF-1R signaling and growth inhibition of tumor cells with expression of IGF-1R in the absence of EGFR receptors.
Figure 5. XGFR* mediated inhibition of A549 and RD-ES cell proliferation. (A) A549 and (B) RD-ES tumor cells were seeded in poly-HEMA coated plates with increasing concentrations of bispecific antibodies XGFR and XGFR* or parental control antibody mixture R1507/GA201 and incubated for 7 d. Cell viability was measured by luminescence after addition of CellTiter Glo reagent. Titration curves in the indicated concentration range and IC50 values are depicted in the table. At least 2 independent data sets with comparable results were obtained.
ADCC activity mediated by XGFR*
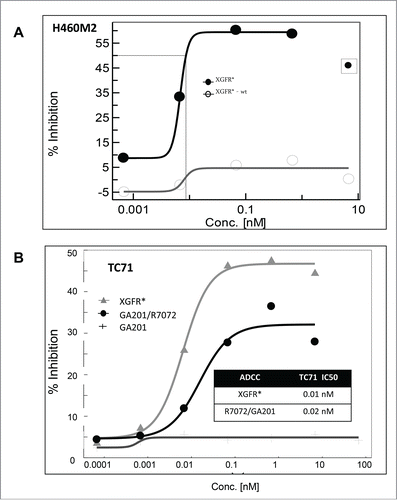
We previously demonstrated that the bivalent IGF-1R antibody R1507 induces IGF-1R internalization and subsequent receptor degradation in the endosomal-lysosomal cell compartment.Citation57 IGF-1R levels in lysates of A549 cells treated with XGFR* were approximately 80% of untreated controls, whereas treatment with the parental IGF-1R antibody R1507 induced receptor degradation to levels of 13.0% after 24 hours (data not shown). Reduced IGF-1R internalization and degradation was found as a unique property of the heterodimeric monovalent IGF-1R/EGFR antibody format, which allows prolonged accessibility of the target antigen and the therapeutic antibody on the cell surface. Hence, the Fc portion of the IgG1 backbone of XGFR* was glycoengineered to enhance Fc receptor-mediated, antibody-dependent effector functions. The resulting afucosylated XGFR* bispecific antibody showed strongly increased cytotoxicity on human lung cancer H460M2 cells compared to the non-glycoengineered wildtype version XGFR*-wt using primary human peripheral blood mononuclear cells (PBMC) as effector cells at an E:T ratio of 25:1 (). ADCC activity relative to the afucosylated parental antibodies R7072 (glycoengineered version of R1507) and GA201 combination was also analyzed with TC71 target cells and human PBMC effector cells. XGFR* showed a highly potent IC50 of 10 pM and a maximal killing efficiency of 45%, which was slightly superior to the combination of the parental antibodies ().
Figure 6. Antibody-dependent cell-meditated cytotoxicity mediated by XGFR*. (A) Induction of in vitro ADCC on H460M2 cells by glycoengineered affinity matured antibody XGFR*. A non-glycoengineered XGFR* antibody was included as control. (B) Induction of in vitro ADCC on TC-71 cells by glycoengineered bispecific antibody XGFR* and parental antibody control mixture R7072/GA201. Tumor cells were seeded in 96-well plates with various antibody concentrations and human PBMC were added at an effector to target ratio E:T of 25:1. Plates were incubated for 4 hours at 37°C, 5% CO2. Cytotoxicity was measured using the LDH Cytotoxicity Detection Kit or the xCelligence system. At least 2 independent data sets with comparable results were obtained. Data points surrounded by square were excluded for curve fitting.
Pancreatic cancer cell lines frequently co-express IGF-1R and EGFR and are sensitive to XGFR* inhibition in vitro
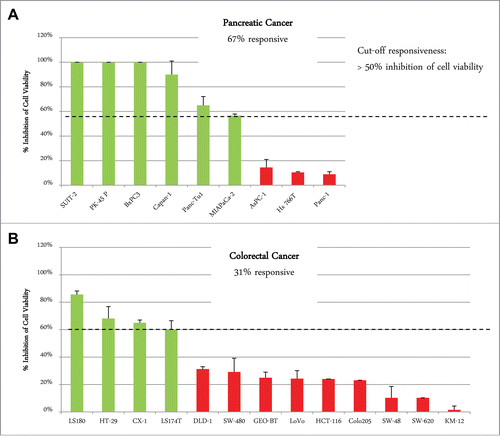
A panel of 8 pancreatic carcinoma and 13 colorectal carcinoma cell lines was analyzed by flow cytometry for surface expression of EGFR and IGF-1R. Co-expression of both receptors on the cell surface was found in all pancreatic cancer cell lines and in 12 of 13 colorectal cancer cell lines, with SW620 cells showing only IGF-1R expression as an exception (). In general, the level of IGF-1R and EGFR expression was higher in pancreatic compared to colorectal carcinoma cell lines (). To determine whether differences in tumor types have an influence on the response to XGFR treatment, 9 pancreatic cancer (PAC) and 13 colorectal cancer (CRC) cell lines were analyzed using an in vitro 3D cell viability assay. Cell lines exhibiting at least 50% inhibition of cell viability by XGFR at 200 nM compared to vehicle control after 5 to 7 d were defined as XGFR-sensitive. While only 31% of CRC cell lines (4 of 13) were responsive to XGFR, 67% of PAC cell lines (6 of 9) responded, indicating higher sensitivity to XGFR treatment in pancreatic carcinoma compared to colorectal carcinoma in the cell lines tested ().
Figure 7. FACS Analysis of IGF-1R and EGFR expression on pancreatic and colorectal carcinoma cell lines. Human pancreatic and colorectal carcinoma tumor cells were incubated with 10µg/ml of phycoerythrin (PE)-conjugated EGFR or IGF-1R antibodies and subjected to FACS analysis using a FACS Canto II. The number of antibodies bound per cell was determined using a Phycoerythrin Fluorescence Quantification Kit and receptor density per cell was calculated.
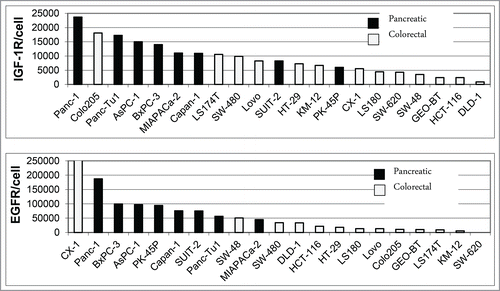
Figure 8. Responsiveness of pancreatic cancer and colorectal cancer cell lines to XGFR* viability inhibition in 3D cellular proliferation assay. Maximal inhibition of cell viability by XGFR* at a concentration of 200 nM in a 3D cellular proliferation assay assessing tumor cell growth after 5–7 d of incubation with a panel of colorectal and pancreatic carcinoma cell lines. Responsiveness was defined as >50% inhibition of cell viability compared to control for each cell line. Mean values of at least 3 independent data sets are shown.
In vivo evaluation in pancreatic cancer xenograft model
As expected for an IgG-like bispecific antibody, XGFR* showed IgG-like pharmacokinetics in the mouse (data not shown). Based on the in vitro data described above, the in vivo anti-tumor efficacy of XGFR* was assessed in the orthotopic MiaPaCa2 pancreatic cancer xenograft model in SCID mice, which enables assessment of the tri-functional mechanism of action: ADCC, EGFR and IGF-1R inhibition in vivo. MiaPaCa2-derived pancreatic tumors were found to have high levels of EGFR and IGF-1R expression (). We chose to compare XGFR* to XGFR, the identical non-glycoengineered wildtype version of XGFR*, as well as cetuximab, an approved anti-EGFR antibody, and AMG479*, an in-house generated version of anti-IGF-1R ganitumab, and the combination therof. Intriguingly, growth and tumor weights at termination of orthotopic MiaPaCa2 xenografts was almost completely inhibited at study termination by XGFR*, with 99% tumor growth inhibition and a significant number of complete tumor remissions observed compared to vehicle control. Notably, no pancreatic tumor was detectable in 10 of 15 XGFR* treated animals (, ). Treatment with XGFR*-wt, a non-glycoengineered wildtype version of XGFR*, also resulted in strong tumor growth inhibition compared to vehicle control (90% tumor growth inhibition); however, in 13 of 15 animals treated with XGFR*-wt tumor tissue was still detectable, indicating that Fc-receptor mediated immune effector functions may have contributed to in vivo efficacy. In contrast to this, the combination of cetuximab and AMG479*, the re-synthesized version of ganitumab, dosed at equimolar concentration, was significantly less efficacious, with only 77% tumor growth inhibition and all mice (15/15) still bearing tumors at study termination. Monotherapy with either cetuximab or AMG479* resulted only in 62% and 41% tumor growth inhibition, respectively, in comparison to the vehicle control group. Thus, each monotherapy alone was less efficacious compared to EGFR/IGF-1R-targeting combination therapy, indicating that both EGFR and IGF-1R inhibition contribute to efficacy in the MiaPaCa2 model. Interestingly, several human colorectal carcinoma models (KM12, Colo205, DLD-1) showed no or minimal sensitivity to IGF-1R/EGFR targeting antibodies, confirming the non-responsiveness of colorectal cancer cell lines observed in the in vitro screening assay (data not shown).
Figure 9. XGFR* in vivo efficacy in human orthotopic MiaPaCa-2 pancreatic carcinoma mouse model. A, Representative example of IGF-1R and EGFR expression in human MiaPaCa-2 pancreatic tumor xenografts grown in SCID mice as detected by immuno-histological staining. B, Tumor weight at termination after treatment with XGFR* and XGFR in comparison to conventional EGFR (cetuximab) and IGF-1R antibodies (AMG479*) and a combination of both antibodies in the orthotopic MiaPaCa-2 pancreatic cancer model in SCID mice. Weekly intraperitoneal treatment with XGFR* or XGFR (20 mg/kg), equimolar EGFR or IGF-1R antibodies (10 mg/kg each) or vehicle control was started 10 d after orthotopic implantation of MiaPaCa-2 tumors. Pancreatic tumor weights and per cent of tumor bearing animals were determined on study day 31 after 3 weeks of treatment (n = 15). Note: Tumor growth could not be monitored during this time due to intra-pancreatic localization of the tumors.
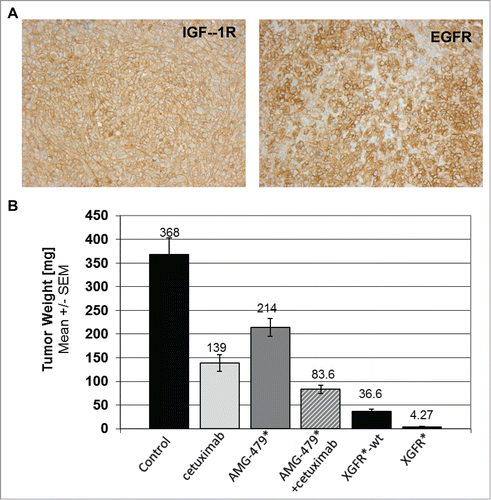
Table 6. Results from orthotopic pancreatic MiaPaCa-2 xenograft model.
Discussion
A wide range of preclinical and clinical data indicate an important role for IGF-1R and EGFR in the progression of cancer, and provide a strong rationale for the combined inhibition of IGF-1R and EGFR signaling.Citation1-3,5-7,59-61 Here, we describe the generation of the novel bispecific antibody XGFR*, which was optimized for inhibition of IGF-1R- and EGFR-dependent signaling in cells with heterogeneous receptor expression and enhanced induction of antibody dependent immune effector functions by glycoengineering of the Fc region. XGFR*is an improved molecule over the precursor antibody XGFR, which is based on the GA201 (EGFR) and R1507 (IGF-1R) parental antibodies.
We previously demonstrated that XGFR has potent anti-tumor efficacy in multiple mouse xenograft tumor models with a complete growth inhibition of AsPC1 human pancreatic tumors and improved survival of SCID beige mice carrying A549 human lung tumors compared to treatment with antibodies targeting either IGF-1R or EGFR alone.Citation57 Anti-tumor activity of XGFR was slightly improved in vitro and similar to the efficacy of combined parental R7202/GA201 antibody treatment in mouse xenograft models.Citation57 The combination of targeting both receptors in a single molecule was clearly more potent than dosing of an individual monospecific IGF-1R or EGFR antibody in vitro and vivoCitation57 XGFR was initially designed to effectively bind EGFR and IGF-1R simultaneously and inhibit downstream signaling in tumor cells with similar expression levels of both receptors. However, due to the fast off rate of R1507 in monovalent binding to IGF-1R, XGFR exhibits only weak binding to cells expressing IGF-1R in the absence of EGFR. Interestingly, monovalent binding of the GA201arm to EGFR was very efficient, with a slow dissociation rate in SPR analysis, and strong binding to cells expressing EGFR with low levels of IGF-1R expression was detectable. Hence, we performed an affinity maturation of the R1507 IGF-1R binding component by introduction of a W94Y mutation in the CDR3 domain and deletion of an aspartic acid residue in the CDR2 domain of the light chain resulting in the F13B5 scFab. The potent binding of XGFR* to cells expressing either one or both of the targeted receptors was demonstrated. Taking the expression pattern in colorectal and pancreatic cancer cell lines observed in this study into account, the superior binding capacity of XGFR* might be crucial to achieve the full efficacy in tumors with heterogeneous receptor expression.
Numerous approaches to generate EGFR/IGF-1R bispecific antibodies have been described before, but the antibody formats had flaws such as low production yields, inherent stability problems, lack of important anti-tumor effector mechanisms, e.g., ADCC, or inferior tumor cell proliferation inhibition compared to the parental antibodies.Citation6,57,62 Our main focus in the development of XGFR* was the generation of a robust molecule that tolerates thermal and oxidative stress, and the scalability of production to generate large amounts of antibody material for clinical development. Due to the removal of the oxidation hotspot W94Y in the CDR3 domain of the F13B5 scFab, we could significantly improve the stability of the molecule. No loss of IGF-1R binding upon incubation for 10 d at 40°C under oxidative stress was observed, whereas XGFR lost approximately 50% IGF-1R binding activity under these conditions. In summary, XGFR* combines an affinity matured, stabilized scFab with KiH heterodimerization of 2 distinct heavy chains to generate a novel bispecific antibody with excellent production yields of 2–3 g/L in CHO cells, thermal and oxidative stability, as well as a very clean side product profile after protein purification.
Treatment of cancer patients with targeted therapeutics often shows that inhibition of key signaling pathways can lead to an initial slow-down in tumor progression. However, a switch to alternative signaling pathways frequently occurs during treatment, resulting in drug resistance.Citation63 In order to achieve long-term treatment responses, it might be important to combine signaling inhibition with an immunotherapeutic activation of the host immune response against the tumor. A strategy to achieve activation of the host immune response against the tumor is the enhanced recruitment of immune effector cells by Fc-engineered therapeutic antibodies that exhibit Fc parts with high affinities for the Fc-receptors on host cells. As has been described previously, afucosylation of the Fc region can lead to enhanced ADCC by natural killer cells and activation of anti-tumor macrophages, which in turn may induce an adaptive immune response against the tumor.Citation64 In contrast to hematological tumors/leukemias, where the role of ADCC for the mechanism of action of CD20 antibodies is believed to be important and afucosylated antibodies such as the glycoengineered Type II CD20 antibody obinutuzumabCitation65-,67 or the afucosylated CCR4 antibody mogamulizumabCitation68,69 have been approved, the role of ADCC in solid tumors such as pancreatic or colorectal cancer still needs to be established, but several glycoengineered/Fc-engineered RTK antibodies are currently in clinical trials. One may speculate that even if afucosylation of XGFR* may have a limited effect at the primary tumor site, it may help to eliminate circulating tumor cells.
In this study, the afucosylated bispecific antibody XGFR* showed potent ADCC activity in vitro, which was strongly enhanced over a non-glycoengineered analog. Another unique property of XGFR* is the reduced receptor internalization upon binding to IGF-1R , which can contribute to prolonged exposure of the XGFR* Fc on the cell surface of the tumor cell, which is crucial for recognition by Fc-receptor bearing immune cells and may lead to increased antibody-dependent immune effector functions in the clinical setting. In the orthotopic MiaPaCa-2 pancreatic xenograft model, the afucosylated bispecific antibody XGFR* showed superior in vivo efficacy over a non-glycoengineered version, as well as the combination of cetuximab, the only approved ADCC-competent anti-EGFR antibody, and a re-synthesized version of AMG479, which was the most advanced anti-IGF-1R antibody at the time.
Pancreatic cancer is a highly aggressive malignancy and shows poor responses to currently available therapeutic agents, and new treatment options for pancreatic cancer are urgently needed. Recently, the IGF-1R-targeted antibody AMG479 in combination with gemcitabine was shown to improve survival of pancreatic tumor patients compared to gemcitabine mono therapy in a Phase 2 trial, but failed to show this effect in a Phase 3 study.Citation33 When combined with EGFR inhibition, pancreatic cancer could be an interesting target indication for XGFR* therapy. The high frequency of IGF-1R and EGFR co-expression observed in human pancreatic cancer cell lines in our study further strengthens a combined treatment concept. In addition, our data indicate that a high percentage of pancreatic cancer cell lines (67%) are sensitive to IGF-1R/EGFR inhibition in a 3D cell viability screening assay, whereas only a small proportion of colorectal cancer cell lines (33%) responded.
In summary, we have demonstrated that XGFR* is a novel bispecific antibody, combining potent IGF-1R and EGFR signaling blockade as well as enhanced induction of ADCC immune effector functions in cells with heterogeneous receptor expression levels. Based on our preclinical data showing EGFR and IGF-1R co-expression and responsiveness of pancreatic carcinoma lines to XGFR* treatment as well as enhanced ADCC induction, XGFR* may represent a promising clinical development candidate for the treatment of pancreatic carcinoma.
Materials and methods
Generation of antibodies
All antibody sequences were generated by gene syntheses and cloned by unique restriction sites into pUC expression vectors. Bispecific and control antibodies were expressed by transient transfection of human embryonic kidney (HEK) cells grown in suspension. Glycoengineered antibodies were produced by co-transfection of the cells with plasmids coding for the carbohydrate modifying enzymes β-1,4-N-acetyl-glucosaminyltransferase III and Golgi α-mannosidase II. HEK293 cell culture supernatants were harvested 7 d after transfection and purified in 2 steps by affinity chromatography using Protein A-Sepharose™ (GE Healthcare) and Superdex 200 size exclusion or hydroxyapatite chromatography. Fractions containing purified bispecific antibodies with less than 5% high molecular weight aggregates were pooled and stored in 6.0 mg/ml aliquots at −80°C. Cetuximab (Erbitux®) was purchased from a local pharmacy. AMG479 (ganitumab) was cloned and produced based on published sequences, and denoted as AMG479*.
Biochemical and biophysical analysis of purified recombinant proteins
The protein concentration of purified protein samples was determined by optical density (OD) measurement at 280 nm, using the molar extinction coefficient calculated on the basis of the amino acid sequence. Purity, antibody integrity and molecular weight of bispecific and control antibodies were analyzed by CE-SDS using microfluidic Labchip technology (Caliper Life Science, USA) and SDS PAGE (Invitrogen). Aggregation of bispecific antibody samples was analyzed by high-performance size exclusion chromatography using a Superdex 200 analytical size-exclusion column (GE Healthcare) in 200 mM KH2PO4, 250 mM KCl, pH 7.0 at 25°C. The integrity of the amino acid backbone and the molecular weight of reduced bispecific antibody light and heavy chains was verified by Electrospray Q-TOF mass spectrometry after removal of N-glycans by enzymatic treatment with Peptide-N-Glycosidase F (Roche).
Oligosaccharide analysis
Oligosaccharides were enzymatically released from the antibodies by Peptide-N-Glycosidase F (Roche). A fraction of the PNGase F-treated sample was subsequently digested with Endoglycosidase H (Roche). The released oligosaccharides were incubated in 150 mM acetic acid prior to purification through a cation exchange resin and analyzed using an Autoflex MALDI/TOF (Bruker Daltonics, Switzerland) in positive ion mode.
Affinity maturation of anti-IGF-1R antibody R1507
Affinity maturation libraries were constructed on the basis of the Fab-fragment of R1507, which was cloned into a phagemid vector enabling subsequent phage display selections. Two sub-libraries were constructed with randomized positions in CDR1 and CDR2 of the variable light or variable heavy chain, respectively. The religated phagemids containing the randomized Fab libraries were used for 60 transformations per sublibrary into electrocompetent E. coli TG1 cells. Phagemid particles displaying the Fab libraries were rescued and purified by PEG/NaCl precipitation to be used for selections. Selections against the extracellular domains of human and/or cynomolgus IGF-1R were carried out in subsequent rounds of bio-panning using R1507 VL and VH library phages. Selection outputs from bio-panning were screened by SPR using ProteOn XPR36 (BioRad) to identify clones with superior kinetic rate constants and affinities compared to parental R1507.
Surface plasmon resonance
SPR experiments were performed using a Biacore T200 instrument with phosphate-buffered saline (PBS)/0.05% Tween20 (v/v). Standard amine coupling to ECD/NHS activated chip surfaces was performed as recommended by the provider (GE Healthcare). Antibodies were captured via anti-human IgG-Fc antibody. A CM5 chip was used for detection of EGFR binding with a ligand density of capture molecule of ˜1000 RU and capture levels ˜40–46 RU of the tested antibodies. A C1-Chip with low ligand density (˜200 RU, capture levels of antibody ˜6–10 RU) was used to achieve monovalent binding of the IGF1R dimer. Five increasing concentrations of the receptors were injected at a flow rate of 50 µL/min for 180 seconds association time and a dissociation time of 1200 s for EGFR and 1600 s for IGF-1R at 37°C. Final regeneration was performed after each cycle using 10 mM glycine pH 1.75, contact time 60 secs and a flow rate of 50 µL/min. Kinetic constants were evaluated by fitting the association and dissociation phase of the analyzed interaction with a Langmuir 1:1 binding model (RI = 0) using usual double referencing (FC1 reference surface with capture molecule and c = 0 nM) by Biacore evaluation Software.
Cell lines
Human lung carcinoma, Ewing's sarcoma, pancreatic and colon carcinoma cell lines A549, TC-71, RD-ES, PK-45P, Capan-1, MiaPaCa-2, AsPC-1, Hs766T, Panc-1, LS180, HT-29, CX-1, LS174T, DLD-1, SW48, SW-480, Lovo, HCT116, Colo205, SW-620 and KM-12 were obtained from ATCC. PancTu-1 was provided by F. Alves and H. Kalthoff, SUIT-2 by Chugai, Japan and GEO-BT by M. Brattain, USA. Cells were routinely cultured in ATCC recommended medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine at 37°C and 5% CO2.
Quantification of receptor levels at the cell surface by FACS analysis
Human tumor cells were seeded into poly Hema-coated 6-well tissue culture plates to prevent adhesion of cells to the plastic surface for 3D-cultures. Coating was performed using 4% poly-HEMA (Polysciences, Warrington, PA) in EtOH and subsequently drying for at least 5 d at 37°C. After 4 hours of cell seeding, Matrigel (BD) was added and spheroids were collected 24h later, washed and trypsinized to generate single cell suspensions. Subsequently, cells were washed twice with ice-cold medium and stained with 10 µg/ml of PE-conjugated EGFR- or IGF-1R-targeted antibodies (BD) or matching isotype controls (BD). Acquisition of data was performed using FACS Canto II and analyzed by FlowJo software. The number of antibodies bound per cell was determined using a Phycoerythrin Fluorescence Quantification Kit (BD) and receptors density per cell was calculated.
Competitive FACS binding assay
Tumor cells were diluted in ice-cold PBS supplemented with 2% fetal calf serum (Gibco) and added to a mixture of PE-labeled monospecific anti-IGF-1R antibody R1507 (final 1 μg/ml) and varying concentrations of unlabeled IGF-1R/EGFR-targeted bispecific antibodies, unlabeled monospecific antibodies or Fab fragments in 96-well microtiter plates (final titration range of 100 to 0.002 μg/ml). After 45 minutes, cells were fixed in the presence of 7-AAD (BD) and fluorescent signal was analyzed by FACS Canto Analyzer (BD Dickinson). Finally, IC50 values were calculated using ExcelFit software.
IGF-1R/EGFR phosphorylation assay
Tumor cells (3 × 104) were seeded in 96-well plates and starved in serum-free medium (1 mg/ml RSA, 10 mM HEPES, 1% Pen/Strep) for 2 hours. To assess phosphorylation, cells were first incubated with either bispecific or control antibodies for 30 min, followed by addition of 5nM IGF-1 (PreProtech) or 10nM EGF (PreProtech) for 10 min and final lysis in 100 µl/well lysis buffer (Cell Lysis Kit, BioRad). Samples were analyzed for EGFR or IGF-1R phosphorylation using the P-EGFR (Tyr) bead kit (Millipore) or P-IGF-1R (Tyr1131) bead kit (BioRad) combined with a phosphoprotein detection reagent (BioRad) by the Luminex detection system.
3D cell viability assay
Tumor cells (density dependent on individual growth kinetics) were seeded in 96-well poly-HEMA coated plates together with increasing concentrations of bispecific or control antibodies and incubated for 7 d. Cell viability was determined using CellTiterGlo® (Promega) according to the manufacturer's instructions.
IGF-1R levels in cell lysates
Tumor cells were cultivated in the presence of 50 nM concentrations of bispecific or control antibodies in standard culture medium for 24 hours at 37°C and 5% CO2. Cells were then lysed with ice-cold cell signaling lysis buffer (Millipore) supplemented with protease inhibitors (Roche). Plates were stored at −20°C until further analysis. IGF-1R levels were detected using an IGF-1R sandwich ELISA using a biotinylated <IGF-1R>hu-1a-IgG (Roche) capture antibody and an <IGF-1Rβ> rabbit (Santa Cruz Biotechnology) detection antibody.
Antibody-dependent cell-mediated cytotoxicity
TC-71 Ewing Sarcoma target cells were collected, washed and resuspended in AIM V® medium (Life Technologies) in round-bottom 96-well plates (30 000 cells/well). The respective amounts of antibody were added and incubated for 10 minutes before addition of human PBMC as effector cells at an effector to target ratio E:T of 25:1. Plates were incubated for 4 hours at 37°C, 5% CO2. Lactate dehydrogenase (LDH) release was measured using the LDH Cytotoxicity Detection Kit (Roche Applied Science). The percentage of ADCC was calculated using the following formula:
Spontaneous release, corresponding to target cells incubated with effector cells without antibody, was defined as 0% cytotoxicity. Maximal release corresponding to target cells lysed with 1% Triton X-100 was defined as 100% cytotoxicity. The average percentage of ADCC and standard deviations of triplicates for each experiment were calculated.
For H460M2 NSCLC, xCelligence system (Roche Applied Science) was used to determine ADCC with comparable conditions as in the LDH release assay. ADCC was determined after 3 hrs and calculated as follows:
Immunohistochemistry analyses of IGF-1R and EGFR expression
Immuno-histological staining procedures for IGF-1R and EGFR were applied using formalin-fixed and paraffin-embedded pancreatic carcinoma specimens. For IGF-1R, a primary rabbit monoclonal antibody (CONFIRM IGF-1R, clone G11, Ventana, cat # 790–4346) was applied on Ventana Benchmark autostainer. Antigen retrieval was done with CC1. Primary antibody provided in a RTU-dispenser at a concentration on 1.7 µg/ml was incubated for 16 min at 37°C. Ultra View universal HRP Multimer (Ventana) was used for detection. For EGFR, a mouse monoclonal antibody (clone 3C6, Ventana cat. #790–2988) was applied on the Ventana Benchmark. Antigen retrieval was done with Protease 1. Primary antibody provided in a RTU-dispenser at a concentration on 1 µg/ml was incubated for 1 hour at 37°C. After A/B block, iView DAB Detection Kit (Ventana) was used for detection. Parameters were semi-quantitatively assessed for: 1) staining intensity of IGF-1R or EGFR expression as negative, weak (+), moderate (++) or strong (+++) staining; 2) the % of IGF-1R or EGFR positive tumor cells; 3) localization (staining pattern) of IGF-1R or EGFR in the tumor cells (membrane, cytoplasm, completeness of membrane staining); and 4) co-localization of the EGFR and IGF-1R in % of tumor cells positive for both markers.
In vivo studies
For assessment of anti-tumor efficacy in the orthotopic MiaPaCa-2 human pancreatic cancer model 1×106 MiaPaCa2 cells were injected into the duodenal lobe of the pancreas of female SCID mice (Charles River). Treatment with bispecific antibodies, control antibodies or vehicle (1x per week, intraperitoneal administration) at indicated doses started 10 d after tumor implantation when a solid tumor was established in the pancreatic tissue. Animals were sacrificed after 3 weeks of treatment and pancreatic tumors were excised and weighed. Tumor growth inhibition was calculated as the ratio treated versus control tumor weight.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sabine Bertl, Michael Antony, Andreas Hinz, Silke Kirchner, Marion Lichtenauer, Ulrike Thomas, Alexandra Baumgartner, Barbara Threm, Sandra Bunte, Sandra Pompiati and Ute Jucknischke for excellent technical assistance and/or help. This work was partially supported by a grant from the Bavarian Ministry for Science and Education.
References
- Capdevila J, Elez E, Macarulla T, Ramos FJ, Ruiz-Echarri M, Tabernero J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat Rev 2009; 35:354-63; PMID:19269105; http://dx.doi.org/10.1016/j.ctrv.2009.02.001
- Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer 2006; 13 Suppl 1:S33-43; PMID:17259557; http://dx.doi.org/10.1677/erc.1.01280
- Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res 2002; 62:200-7; PMID:11782378
- Jones HE, Goddard L, Gee JM, Hiscox S, Rubini M, Barrow D, Knowlden JM, Williams S, Wakeling AE. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 2004; 11:793-814; PMID:15613453; http://dx.doi.org/10.1677/erc.1.00799
- Goetsch L, Gonzalez A, Leger O, Beck A, Pauwels PJ, Haeuw JF, Corvaia N. A recombinant humanized anti-insulin-like growth factor receptor type I antibody (h7C10) enhances the antitumor activity of vinorelbine and anti-epidermal growth factor receptor therapy against human cancer xenografts. Int J Cancer 2005; 113:316-28; PMID:15386423; http://dx.doi.org/10.1002/ijc.20543
- Lu D, Zhang H, Koo H, Tonra J, Balderes P, Prewett M, Corcoran E, Mangalampalli V, Bassi R, Anselma D, et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J Biol Chem 2005; 280:19665-72; PMID:15757893; http://dx.doi.org/10.1074/jbc.M500815200
- Lu D, Zhang H, Ludwig D, Persaud A, Jimenez X, Burtrum D, Balderes P, Liu M, Bohlen P, Witte L, et al. Simultaneous blockade of both the epidermal growth factor receptor and the insulin-like growth factor receptor signaling pathways in cancer cells with a fully human recombinant bispecific antibody. J Biol Chem 2004; 279:2856-65; PMID:14576153; http://dx.doi.org/10.1074/jbc.M310132200
- Haluska P, Carboni JM, TenEyck C, Attar RM, Hou X, Yu C, Sagar M, Wong TW, Gottardis MM, Erlichman C. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther 2008; 7:2589-98; PMID:18765823; http://dx.doi.org/10.1158/1535-7163.MCT-08-0493
- Cataldo VD, Gibbons DL, Perez-Soler R, Quintas-Cardama A. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N Engl J Med 2011; 364:947-55; PMID:21388312; http://dx.doi.org/10.1056/NEJMct0807960
- Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015; 372:1689-99; PMID:25923549; http://dx.doi.org/10.1056/NEJMoa1411817
- Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, Yamamoto N, Yu CJ, Ou SH, Zhou C, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 2015; 16:830-8; PMID:26051236; http://dx.doi.org/10.1016/S1470-2045(15)00026-1
- Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012; 13:528-38; PMID:22452896; http://dx.doi.org/10.1016/S1470-2045(12)70087-6
- Liao BC, Lin CC, Yang JC. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin Oncol 2015; 27:94-101; PMID:25611025; http://dx.doi.org/10.1097/CCO.0000000000000164
- Politi K, Ayeni D, Lynch T. The Next Wave of EGFR Tyrosine Kinase Inhibitors Enter the Clinic. Cancer Cell 2015; 27:751-3; PMID:26058074; http://dx.doi.org/10.1016/j.ccell.2015.05.012
- Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 2011; 29:2011-9; PMID:21502544; http://dx.doi.org/10.1200/JCO.2010.33.5091
- Vermorken JB, Herbst RS, Leon X, Amellal N, Baselga J. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer 2008; 112:2710-9; PMID:18481809; http://dx.doi.org/10.1002/cncr.23442
- Kuenen B, Witteveen PO, Ruijter R, Giaccone G, Dontabhaktuni A, Fox F, Katz T, Youssoufian H, Zhu J, Rowinsky EK, et al. A phase I pharmacologic study of necitumumab (IMC-11F8), a fully human IgG1 monoclonal antibody directed against EGFR in patients with advanced solid malignancies. Clin Cancer Res 2010; 16:1915-23; PMID:20197484; http://dx.doi.org/10.1158/1078-0432.CCR-09-2425
- Thatcher N, Hirsch FR, Luft AV, Szczesna A, Ciuleanu TE, Dediu M, Ramlau R, Galiulin RK, Bálint B, Losonczy G, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol 2015; 16:763-74; PMID:26045340; http://dx.doi.org/10.1016/S1470-2045(15)00021-2
- Paz-Ares L, Mezger J, Ciuleanu TE, Fischer JR, von Pawel J, Provencio M, Kazarnowicz A, Losonczy G, de Castro G Jr, Szczesna A, Crino L, et al. Necitumumab plus pemetrexed and cisplatin as first-line therapy in patients with stage IV non-squamous non-small-cell lung cancer (INSPIRE): an open-label, randomised, controlled phase 3 study. Lancet Oncol 2015; 16:328-37; PMID:25701171; http://dx.doi.org/10.1016/S1470-2045(15)70046-X
- Oppenheim DE, Spreafico R, Etuk A, Malone D, Amofah E, Peña-Murillo C, Murray T, McLaughlin L, Choi BS, Allan S, et al. Glyco-engineered anti-EGFR mAb elicits ADCC by NK cells from colorectal cancer patients irrespective of chemotherapy. Br J Cancer 2014; 110:1221-7; PMID:24496456; http://dx.doi.org/10.1038/bjc.2014.35
- Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, Moessner E, Freytag O, Friess T, Ries CH, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res 2013; 19:1126-38; PMID:23209031; http://dx.doi.org/10.1158/1078-0432.CCR-12-0989
- Paz-Ares LG, Gomez-Roca C, Delord JP, Cervantes A, Markman B, Corral J, Soria JC, Bergé Y, Roda D, Russell-Yarde F, et al. Phase I pharmacokinetic and pharmacodynamic dose-escalation study of RG7160 (GA201), the first glycoengineered monoclonal antibody against the epidermal growth factor receptor, in patients with advanced solid tumors. J Clin Oncol 2011; 29:3783-90; PMID:21900113; http://dx.doi.org/10.1200/JCO.2011.34.8888
- Delord JP, Tabernero J, Garcia-Carbonero R, Cervantes A, Gomez-Roca C, Bergé Y, Capdevila J, Paz-Ares L, Roda D, Delmar P, et al. Open-label, multicentre expansion cohort to evaluate imgatuzumab in pre-treated patients with KRAS-mutant advanced colorectal carcinoma. Eur J Cancer 2014; 50:496-505; PMID:24262587; http://dx.doi.org/10.1016/j.ejca.2013.10.015
- Bridgewater JA, Cervantes A, Markman B, et al. GAIN-(C): Efficacy and safety analysis of imgatuzumab (GA201), a novel dual-acting monoclonal antibody (mAb) designed to enhance antibody-dependent cellular cytotoxicity (ADCC), in combination with FOLFIRI compared to cetuximab plus FOLFIRI in second-line KRAS exon 2 wild type (e2WT) or with FOLFIRI alone in mutated (e2MT) metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2015; 33:2405-6; PMID:26033821
- Dienstmann R, Patnaik A, Garcia-Carbonero R, Cervantes A, Benavent M, Roselló S, Tops BB, van der Post RS, Argilés G, Skartved NJ, et al. Safety and Activity of the First-in-Class Sym004 Anti-EGFR Antibody Mixture in Patients with Refractory Colorectal Cancer. Cancer Discov 2015; 5:598-609; PMID:25962717; http://dx.doi.org/10.1158/2159-8290.CD-14-1432
- Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, Kragh M. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res 2010; 70:588-97; PMID:20068188; http://dx.doi.org/10.1158/0008-5472.CAN-09-1417
- Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, Toner GC, Maki RG, Meyers PA, Chugh R, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol 2011; 29:4541-7; PMID:22025149; http://dx.doi.org/10.1200/JCO.2010.34.0000
- Kurzrock R, Patnaik A, Aisner J, Warren T, Leong S, Benjamin R, Eckhardt SG, Eid JE, Greig G, Habben K, et al. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin Cancer Res 2010; 16:2458-65; PMID:20371689; http://dx.doi.org/10.1158/1078-0432.CCR-09-3220
- Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, Benner RJ, Scranton JR, Olszanski AJ, Jassem J. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J Clin Oncol 2014; 32:2059-66; PMID:24888810; http://dx.doi.org/10.1200/JCO.2013.54.4932
- Karp DD, Paz-Ares LG, Novello S, Haluska P, Garland L, Cardenal F, Blakely LJ, Eisenberg PD, Langer CJ, Blumenschein G Jr, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol 2009; 27:2516-22; PMID:19380445; http://dx.doi.org/10.1200/JCO.2008.19.9331
- Scagliotti GV, Bondarenko I, Blackhall F, Barlesi F, Hsia TC, Jassem J, Milanowski J, Popat S, Sanchez-Torres JM, Novello S, et al. Randomized, phase III trial of figitumumab in combination with erlotinib versus erlotinib alone in patients with nonadenocarcinoma nonsmall-cell lung cancer. Ann Oncol 2015; 26:497-504; PMID:25395283; http://dx.doi.org/10.1093/annonc/mdu517
- Cohen BD, Baker DA, Soderstrom C, Tkalcevic G, Rossi AM, Miller PE, Tengowski MW, Wang F, Gualberto A, Beebe JS, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res 2005; 11:2063-73; PMID:15756033; http://dx.doi.org/10.1158/1078-0432.CCR-04-1070
- Kindler HL, Richards DA, Garbo LE, Garon EB, Stephenson JJ Jr, Rocha-Lima CM, Safran H, Chan D, Kocs DM, Galimi F, et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann Oncol 2012; 23:2834-42; PMID:22700995; http://dx.doi.org/10.1093/annonc/mds142
- Rosen LS, Puzanov I, Friberg G, Chan E, Hwang YC, Deng H, Gilbert J, Mahalingam D, McCaffery I, Michael SA, et al. Safety and pharmacokinetics of ganitumab (AMG 479) combined with sorafenib, panitumumab, erlotinib, or gemcitabine in patients with advanced solid tumors. Clin Cancer Res 2012; 18:3414-27; PMID:22510349; http://dx.doi.org/10.1158/1078-0432.CCR-11-3369
- Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, Benedetto PW, Friberg G, Deng H, McCaffery I, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 2012; 30:1849-56; PMID:22508822; http://dx.doi.org/10.1200/JCO.2011.37.2359
- Sclafani F, Kim TY, Cunningham D, Kim TW, Tabernero J, Schmoll HJ, Roh JK, Kim SY, Park YS, Guren TK, et al. A Randomized Phase II/III Study of Dalotuzumab in Combination With Cetuximab and Irinotecan in Chemorefractory, KRAS Wild-Type, Metastatic Colorectal Cancer. J Natl Cancer Inst 2015; 107:djv258; PMID:26405092; http://dx.doi.org/10.1093/jnci/djv258
- Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodríguez-Braun E, Domingo A, Guijarro J, Gamez C, Rodon J, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res 2011; 17:6304-12; PMID:21810918; http://dx.doi.org/10.1158/1078-0432.CCR-10-3336
- Scartozzi M, Bianconi M, Maccaroni E, Giampieri R, Berardi R, Cascinu S. Dalotuzumab, a recombinant humanized mAb targeted against IGFR1 for the treatment of cancer. Curr Opin Mol Ther 2010; 12:361-71; PMID:20521225
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, Bassi R, Abdullah R, Hooper AT, Koo H, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res 2003; 63:8912-21; PMID:14695208
- Reidy DL, Vakiani E, Fakih MG, Saif MW, Hecht JR, Goodman-Davis N, Hollywood E, Shia J, Schwartz J, Chandrawansa K, et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol 2010; 28:4240-6; PMID:20713879; http://dx.doi.org/10.1200/JCO.2010.30.4154
- Weickhardt A, Doebele R, Oton A, Lettieri J, Maxson D, Reynolds M, Brown A, Jackson MK, Dy G, Adjei A, Fetterly G, Lu X, et al. A phase I/II study of erlotinib in combination with the anti-insulin-like growth factor-1 receptor monoclonal antibody IMC-A12 (cixutumumab) in patients with advanced non-small cell lung cancer. J Thorac Oncol 2012; 7:419-26; PMID:22237261; http://dx.doi.org/10.1097/JTO.0b013e31823c5b11
- Scagliotti GV, Novello S. The role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumors. Cancer Treat Rev 2012; 38:292-302; PMID:21907495; http://dx.doi.org/10.1016/j.ctrv.2011.07.008
- Iams WT, Lovly CM. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin Cancer Res 2015; 21:4270-7; PMID:26429980; http://dx.doi.org/10.1158/1078-0432.CCR-14-2518
- Allison M. Clinical setbacks reduce IGF-1 inhibitors to cocktail mixers. Nat Biotechnol 2012; 30:906-7; PMID:23051797; http://dx.doi.org/10.1038/nbt1012-906c
- Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, Ladanyi M, Grippo JF, Dall G, Staddon AP, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer 2014; 120:2448-56; PMID:24797726; http://dx.doi.org/10.1002/cncr.28728
- Ramalingam SS, Spigel DR, Chen D, Steins MB, Engelman JA, Schneider CP, Novello S, Eberhardt WE, Crino L, Habben K, et al. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J Clin Oncol 2011; 29:4574-80; PMID:22025157; http://dx.doi.org/10.1200/JCO.2011.36.6799
- Fitzgerald JB, Johnson BW, Baum J, Adams S, Iadevaia S, Tang J, Rimkunas V, Xu L, Kohli N, Rennard R, et al. MM-141, an IGF-IR- and ErbB3-directed bispecific antibody, overcomes network adaptations that limit activity of IGF-IR inhibitors. Mol Cancer Ther 2014; 13:410-25; PMID:24282274; http://dx.doi.org/10.1158/1535-7163.MCT-13-0255
- Li C, Huang S, Armstrong EA, Francis DM, Werner LR, Sliwkowski MX, van der Kogel A, Harari PM. Antitumor Effects of MEHD7945A, a Dual-Specific Antibody against EGFR and HER3, in Combination with Radiation in Lung and Head and Neck Cancers. Mol Cancer Ther 2015; 14:2049-59; PMID:26141946; http://dx.doi.org/10.1158/1535-7163.MCT-15-0155
- Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, Sliwkowski MX, Harari PM. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res 2013; 73:824-33; PMID:23172311; http://dx.doi.org/10.1158/0008-5472.CAN-12-1611
- Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, Totpal K, Wong A, Lee CV, Stawicki S, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011; 20:472-86; PMID:22014573; http://dx.doi.org/10.1016/j.ccr.2011.09.003
- Castoldi R, Ecker V, Wiehle L, Majety M, Busl-Schuller R, Asmussen M, Nopora A, Jucknischke U, Osl F, Kobold S, et al. A novel bispecific EGFR/Met antibody blocks tumor-promoting phenotypic effects induced by resistance to EGFR inhibition and has potent antitumor activity. Oncogene 2013; 32:5593-601; PMID:23812422; http://dx.doi.org/10.1038/onc.2013.245
- Zheng S, Moores S, Jarantow S, Pardinas J, Chiu M, Zhou H, Wang W. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. MAbs 2016; 0:1-11; PMID:26761634; http://dx.doi.org/10.1080/19420862.2015.1136762
- Castoldi R, Jucknischke U, Pradel LP, Arnold E, Klein C, Scheiblich S, Niederfellner G, Sustmann C. Molecular characterization of novel trispecific ErbB-cMet-IGF1R antibodies and their antigen-binding properties. Protein Eng Des Sel 2012; 25:551-9; PMID:22936109; http://dx.doi.org/10.1093/protein/gzs048
- Hu S, Fu W, Xu W, Yang Y, Cruz M, Berezov SD, Jorissen D, Takeda H, Zhu W. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Res 2015; 75:159-70; PMID:25371409; http://dx.doi.org/10.1158/0008-5472.CAN-14-1670
- Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 1999; 17:176-80; PMID:10052355; http://dx.doi.org/10.1038/6179
- Ferrara C, Grau S, Jager C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A 2011; 108:12669-74; PMID:21768335; http://dx.doi.org/10.1073/pnas.1108455108
- Schanzer JM, Wartha K, Croasdale R, Moser S, Künkele KP, Ries C, Scheuer W, Duerr H, Pompiati S, Pollman J, et al. A novel glycoengineered bispecific antibody format for targeted inhibition of epidermal growth factor receptor (EGFR) and insulin-like growth factor receptor type I (IGF-1R) demonstrating unique molecular properties. J Biol Chem 2014; 289:18693-706; PMID:24841203; http://dx.doi.org/10.1074/jbc.M113.528109
- Ridgway JB, Presta LG, Carter P. 'Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996; 9:617-21; PMID:8844834; http://dx.doi.org/10.1093/protein/9.7.617
- Buck E, Eyzaguirre A, Rosenfeld-Franklin M, Thomson S, Mulvihill M, Barr S, Brown E, O'Connor M, Yao Y, Pachter J, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res 2008; 68:8322-32; PMID:18922904; http://dx.doi.org/10.1158/0008-5472.CAN-07-6720
- Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 2008; 118:2609-19; PMID:18568074
- Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoel MJ, Fartoux L, Venot C, Bladt F, Housset C, Rosmorduc O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res 2009; 15:5445-56; PMID:19706799; http://dx.doi.org/10.1158/1078-0432.CCR-08-2980
- Croasdale R, Wartha K, Schanzer JM, Kuenkele KP, Ries C, Mayer K, Gassner C, Wagner M, Dimoudis N, Herter S, et al. Development of tetravalent IgG1 dual targeting IGF-1R-EGFR antibodies with potent tumor inhibition. Arch Biochem Biophys 2012; 526:206-18; PMID:22464987; http://dx.doi.org/10.1016/j.abb.2012.03.016
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008; 27:3944-56; PMID:18297114; http://dx.doi.org/10.1038/onc.2008.19
- Lazar GA, Dang W, Karki S, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005-10; PMID:16537476; http://dx.doi.org/10.1073/pnas.0508123103
- Mossner E, Brunker P, Moser S, Püntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010; 115:4393-402; PMID:20194898; http://dx.doi.org/10.1182/blood-2009-06-225979
- Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, Chagorova T, de la Serna J, Dilhuydy MS, Illmer T, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014; 370:1101-10; PMID:24401022; http://dx.doi.org/10.1056/NEJMoa1313984
- Lee HZ, Miller BW, Kwitkowski VE, Ricci S, DelValle P, Saber H, Grillo J, Bullock J, Florian J, Mehrotra N, et al. U.S. Food and drug administration approval: obinutuzumab in combination with chlorambucil for the treatment of previously untreated chronic lymphocytic leukemia. Clin Cancer Res 2014; 20:3902-7; PMID:24824310
- Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, Fujimoto K, Yamamoto K, Miyamoto T, Uike N, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol 2014; 32:1157-63; PMID:24616310; http://dx.doi.org/10.1200/JCO.2013.52.0924
- Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs 2012; 4:419-25; PMID:22699226; http://dx.doi.org/10.4161/mabs.20996