
ABSTRACT
Bispecific immunoglobulins (Igs) typically contain at least two distinct variable domains (Fv) that bind to two different target proteins. They are conceived to facilitate clinical development of biotherapeutic agents for diseases where improved clinical outcome is obtained or expected by combination therapy compared to treatment by single agents. Almost all existing formats are linear in their concept and differ widely in drug-like and manufacture-related properties. To overcome their major limitations, we designed cross-over dual variable Ig-like proteins (CODV-Ig). Their design is akin to the design of circularly closed repeat architectures. Indeed, initial results showed that the traditional approach of utilizing (G4S)x linkers for biotherapeutics design does not identify functional CODV-Igs. Therefore, we applied an unprecedented molecular modeling strategy for linker design that consistently results in CODV-Igs with excellent biochemical and biophysical properties. CODV architecture results in a circular self-contained structure functioning as a self-supporting truss that maintains the parental antibody affinities for both antigens without positional effects. The format is universally suitable for therapeutic applications targeting both circulating and membrane-localized proteins. Due to the full functionality of the Fc domains, serum half-life extension as well as antibody- or complement-dependent cytotoxicity may support biological efficiency of CODV-Igs. We show that judicious choice in combination of epitopes and paratope orientations of bispecific biotherapeutics is anticipated to be critical for clinical outcome. Uniting the major advantages of alternative bispecific biotherapeutics, CODV-Igs are applicable in a wide range of disease areas for fast-track multi-parametric drug optimization.
Introduction
Bispecific antibodies may target soluble ligands or membrane receptors to modulate signaling, or they may bind cell surface proteins, such as in effector cell retargeting strategies.Citation1,2 Two bispecific antibody therapeutics, catumaxomab (Removab®) and blinatumomab (Blincyto®), have been approved, and over ten bispecific antibodies are currently in clinical development for treatment of inflammatory diseases, hemophilia A, or cancers.Citation3,4
The drug-like properties of bispecific biotherapeutics described in the literature vary widely. Molecular weights range from ∼55 kDa to over 300 kDa, their valences for antigen binding are from two to six, and serum half-lives are reported to be between ∼0.5 hours and ∼2 weeks.Citation1,2,5-7 Each format presents a particular strength in physicochemical properties, manufacturing, formulation, pharmacokinetics, or pharmacodynamics while being limited in the other evaluation criteria.Citation1 Sustained structural and functional homogeneity throughout manufacture, storage, and in vivo exposure is critical to their successful therapeutic application, but recognized in many bispecific formats as limiting factor. For example, any format isolating a variable fragment (Fv) by replacing its adjacent constant fragment (Fc) with a peptide linking heavy or light chain Fv domains is associated with decreased Fv stability.Citation2,8 Introduction of disulfide bridges or improved interface interactions stabilize Fv dimerization in specific cases; however, such strategies remain associated with loss of antigen affinity or increased aggregation propensity.Citation9-12 Asymmetric bispecific antibodies including Fc domains give rise to improperly paired side-products even when optimized.Citation2 Thus, they generally require further engineering, extensive purification, or special production systems imposing specific limitations.Citation8,13-22 Furthermore, their avidity per antigen is reduced compared to the parental antibodies.
Symmetric Fc containing bispecific immunoglobulins (Igs) overcome limitations of asymmetric constructs. Improperly paired side-products are avoided and avidity per antigen is preserved at the parental level. The dual-variable-domain IgG (DVD-Ig) format overcomes limitations of Fv instability by N-terminally extending both heavy and light chains of a natural antibody with another Fv fragment ().Citation23 However, some DVD-Igs show a positional effect; thus, they maintain antigen-affinity at the level of the parental antibody only at the outer, N-terminal Fv (Fv1). Antigen affinity at the inner Fv (Fv2) is significantly reduced.Citation24,25 Structural studies suggest that Fv1 may hinder antigen access by swinging “like a bucket” over the antigen-binding region of Fv2.Citation26,27
Biotherapeutic agents require a proper balance of target tissue penetration and maintenance of therapeutic concentrations at the site of action. Low molecular weight bispecific Igs devoid of Fc domains aim at superior pharmacodynamics by increased target tissue penetration. They potentially require continuous intravenous infusion over weeks to maintain therapeutic serum concentrations.Citation28,29 The high molecular weight of bispecific agents including Fc domains may limit tissue penetration. Nevertheless, such constructs preserve the potential to bind to Ig receptors (FcRs). As a consequence, they may have extended serum half-lifes by endocytic salvage via neonatal FcR (FcRn), which may compensate for slow tissue penetration.Citation30-35 Furthermore, they may elicit antibody-dependent cell-mediated cytotoxicity or phagocytosis (ADCC or ADCP, respectively) via Fcγ receptors (FcγRs), which is important for cancer biotherapeutics.Citation3,36-39 Fully functional Fc domains may also confer the potential to trigger the classical pathway of complement-dependent humoral response, and thereby elicit complement-dependent cytotoxicity (CDC).Citation40,41 To overcome major limitations of existing bispecific biotherapeutics and to create a universally applicable format that can be fine-tuned in multi-parametric drug optimization, we designed the CODV format to serve as a versatile platform for the development of bispecific agents.
Results
Inspired by the structures of natural antibodies, different types of CODV biotherapeutics may be engineered. Both heavy and light chains of a first parent IgG (IgGantigen1) are N-terminally extended by an additional Fv fragment taken from a second parent IgG (Fvantigen2, IgGantigen2). Two variants are achieved by employing either heavy or light chain as template (), and two types of each variant may be engineered. In variant 1, the heavy chain is the template and the variable domain of the second parent antibody (VHantigen2) extends the heavy chain of IgGantigen1 N-terminally. However, the light chain is crossed-over by inserting variable domain of IgGantigen2 (VLantigen2) between VLantigen1 and the constant domain of the light chains (CL; : Type 1). The variable and constant domains of the light chains are connected by linkers L1 and L2, respectively. The variable domains and the first constant domain of the heavy chains are connected by linkers L3 and L4, respectively. CODV-Igs are formed by oligomerization of two light and two heavy chains.
Figure 1. CODV format gives rise to four distinct three-dimensional arrangements of Fvs and Fc. Each sketch represents half of a CODV-Ig that dimerizes by heavy chain pairing, resulting in tetramers of HC2LC2 stoichiometry. The two-dimensional sketches translate the different three-dimensional arrangements of the immunoglobulin domains. We adopt the following nomenclature for CODV biotherapeutics: in CODV-Igantigen1 x antigen2 and CODV-Fabantigen1 x antigen2 Fvantigen1 is N-terminal on the heavy chain and consequently located between Fvantigen2 and the first constant domain (Fc1) on the light chain no matter which engineering variant and linker lengths are concerned. We apply optimized all-glycine linkers of 7, 5, 1, and 2 residues for L1 through L4 in this work unless indicated otherwise.
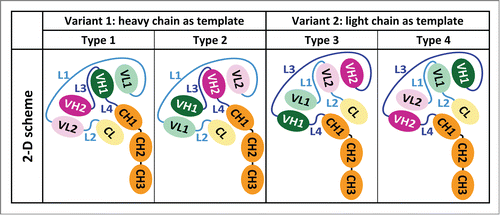
Swapping the Fv domains on both, heavy and light chains while maintaining the linker lengths results in type 2 (). Such constructs are distinct in three dimensions from the variant 2 constructs, which employ the light chain as template. Type 4 is obtained by N-terminally extending the light chains at Fvantigen1 by VLantigen2 and inserting VHantigen2 between VHantigen1 and the first constant domain of the heavy chains (CH1; ). Type 3 is derived from type 4 by swapping the Fv domains on both, heavy and light chains ().
We developed CODV prototypes using parental monoclonal antibodies directed against interleukins IL4 and IL13. Initial results indicated that identification of appropriate lengths of four linking peptide stretches L1 to L4 requires significant design (Supplements). Use of all-glycine linkers limited to a maximum of 20 residues, and with no consideration of the order of the two Fv domains, resulted in a search space of 21Citation4 constructs. To rationally limit the exploration of this search space, we employed a three-dimensional (3D) modeling strategy that used homology modeling of FvIL4 and protein-protein docking of the homology model of FvIL4 and the crystallographic models of both IL4 or FvIL13 (Fig. S1) at key steps. We identified four different spatial arrangements of FvIL4 and FvIL13 with suitable connectivity, and for which we expected no steric interference at the antigen binding sites either by the other Fv or the constant domains (). These arrangements suggest distinct sets of linker lengths of three to 15 residues, and give rise to either variant 1 or 2 (). They also suggest that linker insertion at L4 or L2 is not obligatory in variant 1 or 2, respectively.
Figure 2. CODV format generates a self-supporting architecture that profoundly differs from DVD format. One- and two-dimensional schemes and crystal structures or three-dimensional models are depicted for the complexes CODV-FabIL4 x IL13/IL4, CODV-FabIL13 x IL4/IL4/IL13, CODV-IgIL4 x IL13/IL4/IL13, and DVD-FabIL12 x IL18/IL18. CODV-FabIL4 x IL13 of the crystal structures is a type 1 construct, CODV-FabIL13 x IL4 of the depicted initial models a type 4 construct.
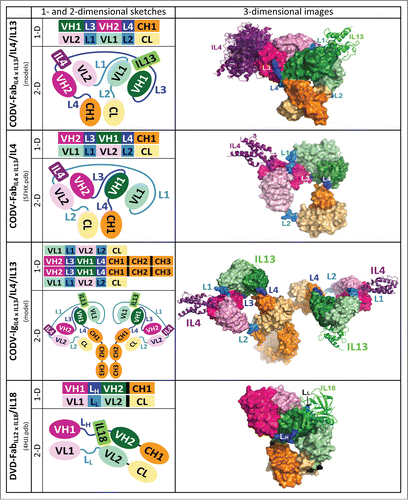
We confirmed these results experimentally by producing 150 CODV constructs using all-glycine linkers. We obtained type 1 and type 4 constructs with good yield and low aggregation propensity (, Table S1). Their binding affinity for both antigens is very well preserved. The median KD values of 6 pM and 60 pM for IL4 and IL13 binding, respectively, are as good as the KD values of the parental antibodies of 8 and 80 pM, respectively (, Table S1). Constructs of types 2 and 3 evince that inversion of the Fv domains results in equally well-behaved CODV-Ig. The optimal CODV-IgIL4 x IL13 prototype with linker lengths of 7, 5, 1, and 2 residues for L1 through L4, which was produced at 15 mg/L culture medium with 5% aggregation, binds IL4 and IL13 with a KD of 2.7 and 24.5 pM, respectively. We improved yield and aggregation propensity further by maintaining linker lengths and replacing the all-glycine stretches by sequences of arbitrary peptide composition or derived from natural CH1-CH2 domain transitions or VL-CL transitions of both k or λ light chains of IgGs. These constructs are extremely well-behaved, with excellent yields, aggregation propensities, and KD values ().
Table 1. The CODV format is universally applicable by maintaining antigen affinities at the level of the parental antibodies without positional effect. The table indicates protein format, targeted antigens, CODV format type, and linker sequences. LL and LH of DVD-Ig correspond to L1 and L3 of CODV-Ig, respectively; Fv2-Fc transitions of DVD-Ig do not require linker insertions that would correspond to L2 and L4 of CODV-Ig. Data on protein production yield, aggregation propensity measured by SEC, and antigen affinities determined by SPR as well as activities in cellular assays are reported for selected constructs of all four CODV types explored during the optimization of linker lengths and compositions. The CODV format is compared to the DVD-Ig format targeting various antigens. Additional data are reported in Table S1 and 2. Lines with background shaded in white or gray differentiate constructs by order of targeted antigens. Abbreviations: na = not applicable, nm = not measured, np = not produced, nr = not reported.
The presence of either Fv does not affect the binding properties of the other Fv. Furthermore, antigen binding to FvIL4 and FvIL13 is independent and tetravalent (Fig. S2). Notably, we observe no positional, cooperative, or allosteric effects for CODV-IgIL4 x IL13 in binding studies (Fig. S2).
A self-supporting architecture for universal use
The variation of linker lengths and sequences influences yield and aggregation propensity, but the vast majority of CODV constructs maintains antigen affinity at the level of the parental antibodies (, Tables S1 and 2). This suggests that almost all explored linkers permit proper Fv and Fc pairing, although to various extends. Once Fv and Fc domains are correctly paired, the CODV assemblies are stable.
We support this hypothesis by thermal stability studies and the determination of CODV-Fab crystal structures. We characterized thermal stability of CODV-Igs and CODV-Fabs directed against IL4, IL13, tumor necrosis factor (TNF), IL12/23, IL1β, insulin-like growth factor 1 receptor (IGF1R), and human epidermal growth factor receptor 2 (HER2) by differential scanning fluorimetry. Melting temperatures of the CODV-Igs and CODV-Fabs are in the typical range observed for natural IgGs (Tables S4A and B). Melting temperatures of CODV constructs directed against IL4 and IL13 are slightly reduced compared to the parental antibodies. Antigen order and linker composition do not affect melting temperatures significantly, however linker lengths affects melting temperatures slightly in the selected examples. Notably, only one melting temperature is observed in CODV-Igs, suggesting that melting of Fab- and Fc domains occurs concomitantly. We consider this observation as a hallmark of the circular self-contained architecture that functions as a self-supporting truss. Thermal stability of some CODV-constructs may be slightly reduced. We conclude that CODV architecture is overall as stable as natural Igs due to proper Fv and Fc pairing.
We crystallized CODV-FabIL4 x IL13 both in apo-form and complexed with IL4, and determined the structures at 2.7 and 2.6 Å resolution, respectively (, Table S4, Fig. 6). Both structures reveal a compact assembly formed by heavy and light chains. Both chains are properly dimerized as expected from the initial design and evinced by in vitro characterization. CODV-FabIL4 x IL13 is stabilized by interactions at the dimer interfaces of both Fvs and Fc1. Linkers L1 to L4 are well defined in the electron density maps. Longer linkers are indeed required in the light chain to surround the heavy chain template for proper dimerization (). The structure of the complex reveals IL4 bound to FvIL4. The IL4 epitope of CODV-IgIL4 x IL13 involves residues at the C- and N-terminal ends of helices H1 and H3, respectively, and part of loop H2-H3 (Fig. S4). Thus, IL4 interacts only with complementarity-determining region 3 (CDR3) of VLIL4 and CDR2 and CDR3 of VHIL4.
Systematic maintenance of antigen affinities by both Fv domains at the level of the parental antibodies is a marked difference between CODV- and DVD-Igs, which we consider the most closely related bispecific Igs ().Citation26,27 The crystal structures of CODV-FabIL4 x IL13 evince reasons for maintenance of antigen affinities and independence of antigen binding in CODV format. The binding sites of FvIL4 and FvIL13 are about 65 Å apart, oriented in opposing directions, and very few interactions are observed between these domains or with Fc1 (, Fig. S5). Cooperative effects in antigen binding by Igs are proposed to be mediated by domain interactions between constant and variable domains.Citation42 Residues of the second framework loop on the light chain may play a particular role.Citation42-44 This loop corresponds to residues LysL157 to LysL163 in FvIL4 and LysL43 to LysL49 in FvIL13. These critical residues are not involved in direct domain contacts. Thus, the absence of cooperative or allosteric effects in surface plasmon resonance (SPR) measurements of antigen binding is coherent with the structural data.
No antigen binding site is obstructed by the presence of the other Fv or Fc1. Instead, the crossover architecture provides self-supporting constructs that allow for little flexibility in the relative placements of the Fv and Fc domains. Comparison of the crystal structures of IL4-bound and antigen-free CODV-FabIL4 x IL13 suggests that the linkers maintain the relative placement of FvIL4 and FvIL13 tightly and allow for limited reorientation of Fc1 (Fig. S6). Access to the antigen binding sites is also unobstructed in the antigen-free structure. The comparison does not raise any concern on their accessibility even for large antigens. The well-maintained binding affinities for IL4 and IL13 of CODV-Igs with various linker lengths suggest that the self-supporting property is inherent to CODV architecture. Therefore, we conclude that the CODV format may be suitable for a wide variety of uses.
We evince universal use of the CODV-Ig format by constructs arbitrarily combining Fv domains from clinically applied monoclonal antibodies of different IgG germ lines. They bind antigens of various biological functions and sizes. They are directed against clinically validated targets, including TNF, IL12/23, IL1β, IGF1R, HER2, and EGFR. All constructs are well behaved in terms of yield, aggregation propensity, and antigen binding compared to corresponding DVD-Igs (, Table S2).
CODV proteins are fully functional in terms of antigen binding. We also show efficient inhibition of cell signaling proteins by inhibition of IL4, IL13, or TNF signaling in cellular assays: IC50 values range from 33 to 54 pM, 0.9 to 4.5 nM, and 95 to 785 nM for IL4, IL13, or TNF signaling, respectively (, Table S2). Thus, CODV-Igs have the potential to satisfy therapeutic aims in clinical applications.
Paratope orientations affect function
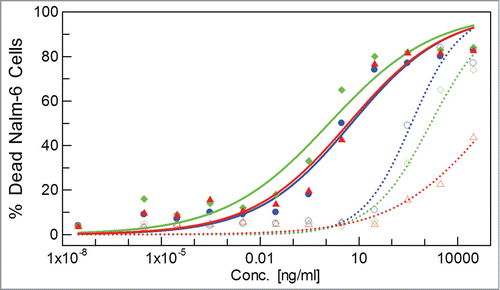
We further proof the concept of their biological functionality by a T-cell dependent cytotoxicity assay probing CODV-Fabs directed against CD3ε or αβ-TCR expressed on primary human T-cells and CD19 expressed on NALM-6 cells (). This assay can only translate the biological efficacy of such “T-cell engager” molecules if both antigens on either T-cell or target cell are simultaneously engaged.Citation45-48 DVD-FabCD3ε x CD19 and DVD-FabCD19 x CD3ε are generated with identical primary sequences for comparison. We observe highest and comparable cytotoxicity for CODV-FabCD3e x CD19, DVD-FabCD3ε x CD19, and CODV-Fabαβ-TCR x CD19 with EC50 values of about 4 ng/ml. We note a positional effect in both formats when inverting Fv order: EC50 values are higher by 2 to 3 orders of magnitude for CODV-Fabs. The cytotoxicity of DVD-FabCD19 x CD3ε is lower by more than 5 orders of magnitude. In DVD-Fabs this positional effect could be explained by the positional effect on antigen affinity. However, the CODV format is not prone to a positional effect in antigen binding, yet we observe it in biological efficacy. Thus, we demonstrate the ability of these bispecific molecules to bind two membrane-bound antigens simultaneously, and show that the relative orientation in 3 dimensions of the antigen binding sites with respect to their epitopes in the biological environment is a critical property of multifunctional biotherapeutic proteins.
Figure 3. Relative three-dimensional arrangements of Fvs in bispecific biotherapeutics are a critical parameter in in cellulo – and by extension in in vivo – efficacy beyond in vitro antigen affinities. Quantification of dead Nalm-6 cells in function of concentration of bispecific immunoglobulins: CODV-FabCD3 x CD19 and CODV-FabCD19 x CD3 (solid and dashed blue lines, filled and open circles, respectively), CODV-Fabαβ x CD19 and CODV-FabCD19 x αβ (solid and dashed green lines, filled and open squares, respectively), DVD-FabCD3 x CD19 and DVD-FabCD19 x CD3 (solid and dashed red lines, filled and open triangles, respectively). DVD-Fabs are constructed with (G4S)2 linkers on heavy and light chains.
CODV-Igs retain intrinsic IgG functions
IgGs exert biological activities by interactions of their Fcs with Fc receptors and complement proteins such as C1q.Citation36,40,49-53 To determine the effects of CODV architecture on Fc functionality, we quantified the affinity of CODV-IgIL4 x IL13 for FcRn and FcγR1 by SPR. The affinity for FcγR1 is very similar to isotype control IgG1IL4 while the affinities for FcRn receptors are slightly decreased by ∼2.5-fold (Table S3). We conclude that Fc functionality is a priori conserved in CODV-Igs.
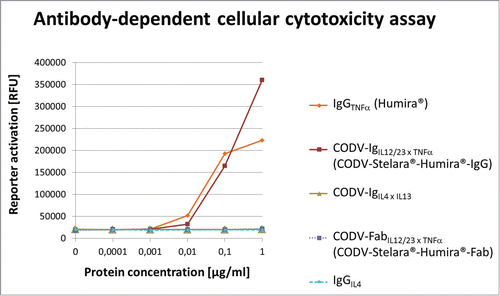
FcγRs bind to natural IgGs at the transition of Fc1 to Fc2.Citation50,51 Superposition and comparison of the crystal structure of IgG1 or the model of CODV-IgIL4 x IL13/IL4/IL13 with the crystal structure of the FcγR3/Fc complex indicate that the binding mode of FcγRs is unchanged (Fig. S8). Therefore, we assessed the ADCC functionality of CODV-IgIL12/23 x TNFα on Chinese hamster ovary (CHO) cells as target cells that express membrane-bound TNF by using Jurkat cells as effector cells that express FcγIIIR. Antibody binding to FcγIIIR with concurrent target cell engagement activates a NFAT driven luciferase reporter. Thereby, the assay reveals ADCC of the antibody. CODV-IgIL12/23 x TNFα and positive control IgGTNFa induce ADCC (). Soluble TNF successfully competes with target cell engagement and inhibits ADCC (Fig. S10). CODV-IgIL4 x IL13, IgGIL4, and CODV-FabIL12/23 x TNFα do not show ADCC due to lack of either target or effector cell engagement, respectively (). Thus, we evidence the potential of CODV architecture to achieve ADCC in vivo by tethering effector cells to target cells.
Figure 4. CODV architecture retains intrinsic functionalities of natural IgGs such as ADCC and CDC. Immunoglobulins are tested on CHO cells as target cells that express membrane-bound TNF; effector cells are Jurkat cells that express FcγIIIR and activate an NFAT driven luciferase reporter upon antibody-binding and cross-linking due to target cell engagement. Thus, ADCC activity of IgGTNFα, CODV-IgIL12/23 x TNFα, CODV-IgIL4 x IL13, CODV-FabIL12/23 x TNFα, and IgGIL4 is measured by activation of the Jurkat luciferase reporter translating ADCC (orange line and tilted square, redline and square, green line and triangle, blue dotted line and square, cyan dashed line and circle, respectively). Further data are reported in Fig. S10.
IgG antibodies elicit CDC by activating the classical pathway of the complement cascade via binding to C1 complex. Formation of ordered IgG hexamers as observed in the crystal packing of the structure of IgGgp120 increases affinity to C1 and thereby potentiates complement activation when viral or cellular surface targets are recognized.Citation40 Superposition of the CODV-IgIL4 x IL13/IL4/IL13 model on the immune-competent IgGgp120 hexamer reveals that no steric hindrance for hexamerization by Fc2-Fc3 is introduced by the CODV architecture. Furthermore, the CDR regions remain accessible to antigens (Fig. S9). Thus, we assessed CDC activity of CODV-IgIL12/23 x TNFα on CHO cells expressing membrane-bound TNF. Complement-dependent cytolysis was detected by measurement of LDH release into the supernatant. CODV-IgIL12/23 x TNFα and positive control IgGTNFa induce CDC. Negative controls CODV-IgIL4 x IL13 and CODV-FabIL12/23 x TNFα do not elicit CDC due to lack of either target cell or complement binding, respectively (Fig. S10). Thereby, we show that the novel bispecific Ig format potentially increases therapeutic benefit by antigen-promoted CDC in specific indications.
CODV-Ig binding to membrane receptors
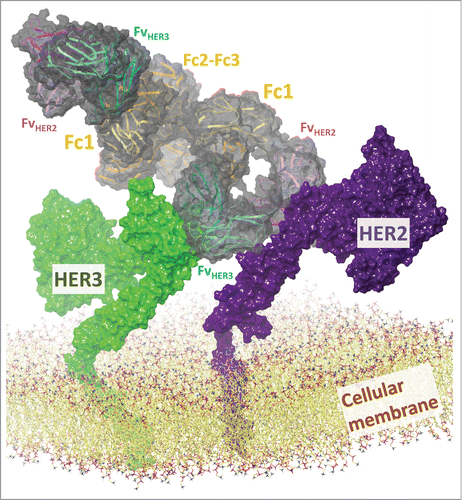
Finally, we asked how CODV-Igs could engage in cell-surface antigen binding, as in the dual receptor-targeting of HER2 and HER3 for which Phase 2 clinical studies are reported.Citation1,54 Formation of HER2/HER3 heterodimers is a potent mitogenic signal that strongly depends on correct association of their transmembrane helices.Citation55 Dual HER2/HER3 targeting aims at inhibiting their signaling, which can be achieved by simultaneously blocking HER2/HER3 dimerization and preventing their engagement in other heterodimeric complexes.Citation1,54 We constructed homology models of CODV-IgHER2 x HER3/HER2/HER3 complexes including the transmembrane helices of HER2 and HER3. The models evince the possibility of concomitant binding of large antigens pairs at cell surfaces (, Supplements). They suggest that dimer association of transmembrane helices required for signaling may be blocked by suitably chosen paratopes of which the required orientation is maintained by the self-supporting CODV architecture (Supplements).
Figure 5. CODV-Ig format is compatible with concomitant binding of large antigens at cellular surfaces and may inhibit signaling competent receptor dimerization. The models of CODV-IgHER2 x HER3/HER2/HER3 integrate the Fv domains of a “tryptophan-rich antibody” targeting HER2 and of antibody DL11 directed against HER3 and EGFR, as well as the N-terminal extracellular domains and the transmembrane helices of HER2 and HER3. The bispecific antibody is depicted by a ribbon diagram colored in magenta, green, orange, and blue for FvHER2, FvHER3, Fc regions, and linkers, respectively, and its solvent-accessible surface in transparent dark gray. Dark and light shades of the ribbon diagram differentiate heavy and light chains. HER2 and HER3 are depicted by solvent accessible surfaces in dark violet and green, respectively. The lipid bilayer modeled as 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) is shown by an all-atom representation that excludes hydrogen atoms while indicating carbon, oxygen, nitrogen, and phosphor atoms in yellow, red, blue, and violet, respectively.
Discussion
The design of the CODV format is an example of applied research in drug discovery, yet it implicates innovation and its realization is based on atypical solutions from the point of modeling. Protein engineering usually involves site-directed mutagenesis, loop grafting, or creation of fusion proteins by connecting several domains by simple linkers. Intellectually and technically, these tasks are well mastered, in particular when experimentally derived 3D models support design. The design of fusion proteins also generally faces unperturbed folding of the individual domains as the only difficulty. More challenging designs are pursued in the fields of molecular evolution, protein folding and engineering. Here, protein design may aim at the creation of novel domain interfaces possibly with enzymatic function or adaptation of interfaces of oligomerization or repeating unit assembly that are perturbed when steric restraints are not properly taken care of in the design.Citation56-62 Immunoglobulin domains and Fv fragments are versatile modules of proven value in biotherapeutics engineering.Citation1 Our CODV design differs from all previously reported designs because we do not N- or C-terminally attach VL and VH domains to create linearly extended domain strings as in, for example, diabody (Db), single-chain variable fragment (scFv), or DVD-Ig formats.Citation1 Instead, our cross-over design creates a circular self-contained architecture that gives rise to a self-supporting truss composed of Fv1, Fv2, and Fc1. The CODV design is thus akin to the design of circularly closed repeat architectures such as β-propeller and β-trefoil folds.Citation56,63-65 Steric constraints must be matched to permit correct VH/VL pairing of Fv1 and Fv2, as in engineering of circularly closed repeat architectures. Inappropriate linker lengths may result in VH/VL mismatch, leading to dimerization of DVD-Ig type with loss of target affinity as suggested by our initial constructs (Supplements). To rationally identify appropriate linker lengths and to limit the exploration of the search space of the 21Citation4 all-glycine linker mutants, we applied protein-protein docking of experimental and homology models of FvIL13, IL4, and FvIL4. Protein-protein docking is revealed as a sufficiently robust method to identify suitable domain arrangements for protein engineering, even if it is not reliable enough to routinely and unambiguously predict protein complexes or domain configurations as they are experimentally observed.Citation66,67 Indeed, the modeling approach proved surprisingly efficient as we experimentally explored only ∼0.08% of the search space and identified almost exclusively constructs with expected molecular weight that maintain parental target affinities.
The design of a robust, universally applicable bispecific biotherapeutic platform has to satisfy various parameters with regard to target antigen binding. Well-controlled affinity is one of these.Citation39,68 Moreover, the proper balance of target affinity and avidity is critical to ascertain high efficacy with reduced undesired side-effects.Citation68 As the CODV design guarantees unobstructed paratopes and stable VH/VL dimerization, it fulfils these requirements. Nevertheless, target affinity and avidity are not the sole parameters that drive efficacy. The 3D orientation and distance of the paratopes of bispecific agents can determine the biological effect. For instance, N- or C-BsAbsHer2 x Her2 in which the heavy chain of one IgGHER2 is N- or C-terminal extended by a second Fv-containing unit directed against another HER2 epitope have distinct cellular activities.Citation21 Also, IgG-scFv and IgG-Fab built with the two previously mentioned FvHER2 significantly differ in cellular assays.Citation22 Similarly, bispecific tandem scFv and tandem Fabs comprising the same FvEGFR and FvCD3 also show distinct outcome in cellular evaluations.Citation22 The circular self-contained architecture of the CODV format imposes defined paratope orientations by the lengths of linkers L1 to L4 that act like reins to adjust the antigen binding site directions. This possibility is unique among bispecific biotherapeutic formats because alternative architectures intrinsically allow for more flexible hinge-like movements of the Fv joining linkers. Thus, our results in the T-cell engager assays that systematically compare target antigen order in CODV- and DVD-Fabs clearly evince the importance of 3D paratope orientation at an even more subtle level compared to the cited examples when target engagement aims at spatially tethering molecular or cellular units.
A universally applicable bispecific biotherapeutics platform should also provide uncompromised functionality of Fc domains to permit tailoring of pharmacokinetic properties and mechanism of drug action. Engineering interaction with FcRn gives control over the pharmacokinetic/pharmacodynamic profiles of biotherapeutic drugs, whether linear or non-linear pharmacokinetics will determine clearance.Citation29,33,34,69,70 Thereby administration and dosing strategy can be tailored in function of disease requirements, which may differ widely in chronic and ad-hoc treatments. Systematic pharmacokinetic studies show that increased FcRn affinity leads to increased elimination half-life of IgGs; however, modulation of in vitro affinity does not result in predictive modification of the in vivo elimination profile. Affinity differences as much as 100-fold do not necessarily translate into statistically significant modification of elimination half-life, while the same level of in vitro affinity may give rise to significantly different elimination kinetics.Citation71 FcRn contribute to IgG homeostasis by binding a histidine-rich site overlapping Fc2 and Fc3 of IgG.Citation53 Superposition of the crystal structure of the FcRn/Fc complex and the model of CODV-IgIL4 x IL13/IL4/IL13 suggests that the binding mode of FcRn should be preserved in the CODV-Ig format (Fig. S7). No steric interference due to the presence of the Fv domains is obvious and binding of soluble antigens is likely to not interfere with FcRn-binding. As in vitro FcRn affinity is only minimally affected compared to isotype control, we anticipate that IgG-derived CODV-Igs are subjected to FcRn-dependent homeostasis. CODV-Fabs show the expected serum half-life of four to six hours in preclinical pharmacokinetic studies in mice.
Interaction with FcγRs or complement may be desired as part of the mechanism of drug action as in the trifunctional biotherapeutics catumaxomab and ertumaxomab or monoclonal antibodies daratumumab, 2F2, and 7D8.Citation3,38,40,72-75 The circular self-contained CODV architecture with its voluminous Fab region could raise the concern that binding of FcRs or hexamerization which potentiates complement activation might be perturbed.Citation40,51,76 Also, both, binding of surface antigens on target cells with concomitant FcγR engagement on immune effector cells and assembly of the complement components on antigen-bound CODV-Ig are subjected to 3D constraints that one might imagine incompatible with CODV architecture. These concerns have no general applicability for two reasons: 1) conservation of FcRn binding at the level of reference IgG evinces the potential of fine-tuning of pharmacokinetics of CODV-Igs when intermittent dosing schedules are desired; and 2) the results in our ADCC and CDC assays evidence that CODV-Igs are also applicable in complex mechanisms of drug action. Site-directed mutagenesis studies show how modulated binding of IgGs to FcRs or C1q can alter their biological effect.Citation5,32,40,70,77-79 Because our characterization of the CODV format documents the excellent conservation of the properties of natural IgG, we anticipate that these mutations equivalently affect CODV-Ig functionalities, and that multi-parametric drug optimizations benefit from such mutations.
The close similarity of CODV proteins to natural IgG also facilitates exploration of the format in early stages of biotherapeutic drug discovery. Our results reveal that CODV architecture generates a structurally autonomous Fab equivalent with expected characteristics that can be employed accordingly. Furthermore, well-established techniques of IgG production and purification by affinity chromatography on protein A columns are applicable to CODV-Igs (Supplements). Protein A binds to the Fc of most human IgG at the transition of CH2 to CH3 and to Fv regions built with HC of sub-group III.Citation80-84 Binding of protein A to IgG is concentration dependent and can be described by a bivalent binding mechanism.Citation85-87 Superposition of crystal structures of Fc or Fab domains in complex with sub-domains of protein A on CODV-Ig models constructed with the type 1 crystal structure of CODV-FabIL4 x IL13 show that these binding sites are unobstructed in CODV-Ig except for steric interferences observed at the site of Fv1.Citation81,88-90 Superposition of the Fab/protein A complexes on the initial type 4 model suggests unobstructed protein A binding sites on both Fv1 and Fv2. Thus, choice of linker lengths may influence purification of CODV-Igs, in particular when Fv domains with HC of sub-group III are employed. Nevertheless, our experience in working with both CODV and DVD formats shows that the characteristics of the parental IgG of which the Fv domains are derived determine ease of production and purification in both bispecific formats. CODV and DVD proteins built with Fv domains of easily obtained Ig are well produced and purified, whereas bispecific proteins constructed with Fv domains of parental Ig that are performing worse regularly are more difficult to obtain in good quality and quantity. Indeed, the properties of employed Fvs dominate over the effect that Ig formats may have in production and purification of bispecific proteins. Therefore, we recommend systematic exploration of a large choice of parental Igs to choose the best Fv pair for the construction of bispecific Ig.
In summary, CODV architecture is universally suitable for the development of bispecific multifunctional therapeutic proteins simultaneously targeting circulating or cell-surface antigens to elicit a curative effect. The format allows modification of the protein sequence to optimize properties such as antigen binding, solubility, or stability as it is state-of-the-art in monoclonal antibody design. It allows control of antigenicity by optimization of the framework regions and ensures efficient generation of structurally and functionally homogeneous protein. It is universally applicable with respect to targeting strategy, targeted antigens, integrated Fvs, isotype of Fc framework, and disease areas. The format unites the major advantages of alternative bispecific biotherapeutics to serve as a versatile starting point for fast-track biotherapeutic drug discovery. We currently apply it in disease areas as diverse as oncology, genetic, or infectious diseases, and expect major contributions to satisfy unmet medical needs.
Methods
Modeling and experimental work were carried out by state-of-the-art procedures under conventional conditions. In Supplements, we report details and references on: design and analysis by molecular modeling; expression plasmid generation, protein production, and purification by affinity chromatography; biochemical characterization and size-exclusion chromatography; kinetic analysis of antigen- and FcR-binding by SPR; IC50 determination in cellular assays of IL4/IL13 or TNF activity; thermostability studies by DSF; protein crystallography; T-cell dependent cytotoxicity, ADCC, and CDC assays.
Initial design of CODV proteins
Homology models, CODV-Fab, and CODV-Ig models were constructed using programs of Schrödinger software package unless indicated otherwise by state-of-the-art procedures described in detail in Supplements.Citation91-95 Initial CODV models are designed by the following 3D-modeling strategy (Fig. S1). Protein-protein docking of FvIL13 and FvIL4 was carried out with program ZDock employing standard settings in software package Discovery Studio.Citation96-98 2000 unrefined poses with 99 cluster centers and 478 singletons of the first screening round were exported for analysis in Maestro Package. The 99 cluster centers and the 478 singletons were visually inspected. Complexes of FabIL13/IL13 and FvIL4/IL4 were superimposed on each pose to reveal the possibility of proper N/C-terminal connectivity between Fvs and Fc, absence of steric interference at the paratopes potentially leading to loss in antigen binding, and to estimate linker lengths. Selected poses were merged with the structure of FabIL13/IL13 to construct homology models of CODV-FabIL4 x IL13 with minimal, intermediate, and maximal linker lengths. Selected models of CODV-FabIL4 x IL13 with minimal and intermediate linker lengths were checked for structural stability by molecular dynamics simulations at 298.15 K, 2 fs time steps, 100 or 500 ps trajectory, Surface Generalized Born implicit solvent model, and standard settings in program Impact. Neither structural instability nor any other structural problems were revealed by the molecular dynamics simulations. Detailed descriptions of construction and structural analysis of the final model of CODV-IgIL4 x IL13/IL4/IL13 and homology models of CODV-IgHER2 x HER3/HER2/HER3 are provided in Supplements.
The crystallographic coordinates are deposited in the Protein Data Bank (5FHX.pdb, 5HCG.pdb).
Disclosure of potential conflicts of interest
All authors are employees of Sanofi R&D.
Supplementary_Data.docx
Download MS Word (7.4 MB)Acknowledgments
A. S. acknowledges generous support by Drs J-Chr. Mozziconacci, D. Rinaldo, and J. Weiser. We also thank Dr D. Usener (Sanofi) for contribution of the TNF-expressing CHO cell line.
References
- Kontermann R. Dual targeting strategies with bispecific antibodies. Mabs 2012; 4:182-97; PMID:22453100; http://dx.doi.org/10.4161/mabs.4.2.19000
- Marvin JS, Zhu Z. Recombinant approaches to IgG-like bispecific antibodies. Acta Pharmacol Sin 2005; 26:649-58; PMID:15916729; http://dx.doi.org/10.1111/j.1745-7254.2005.00119.x
- Bokemeyer C. Catumaxomab - trifunctional anti-EpCAM antibody used to treat malignant ascites. Expert Opin Biol Ther 2010; 10:1259-69; PMID:20624115; http://dx.doi.org/10.1517/14712598.2010.504706
- Dhimolea E, Reichert JM. World Bispecific Antibody Summit, September 27-28, 2011, Boston, MA. Mabs 2012; 4:4-13; PMID:22327426; http://dx.doi.org/10.4161/mabs.4.1.18821
- Unverdorben F, Färber-Schwarz A, Richter F, Hutt M, Kontermann RE. Half-life extension of a single-chain diabody by fusion to domain B of staphylococcal protein A. Protein Eng Des Sel 2012; 25:81-88; PMID:22238430; http://dx.doi.org/10.1093/protein/gzr061
- Müller D, Karle A, Meissburger B, Höfig I, Stork R, Kontermann RE. Improved Pharmacokinetics of Recombinant Bispecific Antibody Molecules by Fusion to Human Serum Albumin. J Biol Chem 2007; 282:12650-60; PMID:17347147; http://dx.doi.org/10.1074/jbc.M700820200
- Holliger P, Wing M, Pound JD, Bohlen H, Winter G. Retargeting serum immunoglobulin with bispecific diabodies. Nat Biotechnol 1997; 15:632-36; PMID:9219264; http://dx.doi.org/10.1038/nbt0797-632
- Von Kreudenstein TS, Escobar-Carbrera E, Lario PI, D'Angelo I, Brault K, Kelly JF, Durocher Y, Baardsnes J, Woods RJ, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability. mAbs 2013; 5:646-54; PMID:23924797; http://dx.doi.org/10.4161/mabs.25632
- Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: Bisulfide-stabilized Fv fragments. Nat Biotechnol 1996; 14:1239-45; PMID:9631086; http://dx.doi.org/10.1038/nbt1096-1239
- Raag R, Whitlow M. Single-chain Fvs. FASEB J 1995; 9:73-80; PMID:7821762
- Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol 2005; 347:773-89; PMID:15769469; http://dx.doi.org/10.1016/j.jmb.2005.01.053
- Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001; 305:989-1010; PMID:11162109; http://dx.doi.org/10.1006/jmbi.2000.4265
- Fenn S, Schiller CB, Griese JJ, Duerr H, Imhof-Jung S, Gassner C, Moelleken J, Regula JT, Schaefer W, Thomas M, et al. Crystal structure of an anti-Ang2 CrossFab demonstrates complete structural and functional integrity of the variable domain. PloS One 2013; 8:e61953; PMID:23613981; http://dx.doi.org/10.1371/journal.pone.0061953
- Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs 2015; 7:377-89; PMID:25621507; http://dx.doi.org/10.1080/19420862.2015.1007816
- Carter P. Bispecific human IgG by design. J Immunol Methods 2001; 248:7-15; PMID:11223065; http://dx.doi.org/10.1016/S0022-1759(00)00339-2
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel 2010; 23:195-202; PMID:20299542; http://dx.doi.org/10.1093/protein/gzp094
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J BiolChem 2010; 285:19637-46; PMID:20400508; http://dx.doi.org/10.1074/jbc.M110.117382
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 2011; 108:11187-92; PMID:21690412; http://dx.doi.org/10.1073/pnas.1019002108
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotech 2014; 32:191-8; PMID:24463572; http://dx.doi.org/10.1038/nbt.2797
- Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotech 2013; 31:753-8; PMID:23831709; http://dx.doi.org/10.1038/nbt.2621
- Wu X, Sereno AJ, Huang F, Zhang K, Batt M, Fitchett JR, He D, Rick HL, Conner EM, Demarest SJ. Protein design of IgG/TCR chimeras for the co-expression of Fab-like moieties within bispecific antibodies. Mabs 2015; 7:364-76; PMID:25611120; http://dx.doi.org/10.1080/19420862.2015.1007826
- Wu X, Sereno AJ, Huang F, Lewis SM, Lieu RL, Weldon C, Torres C, Fine C, Batt MA, Fitchett JR, et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. Mabs 2015; 7:470-82; PMID:25774965; http://dx.doi.org/10.1080/19420862.2015.1007826
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotech 2007; 25:1290-7; PMID:17934452; http://dx.doi.org/10.1038/nbt1345
- DiGiammarino EL, Harlan JE, Walter KA, Ladror US, Edalji RP, Hutchins CW, Lake MR, Greischar AJ, Liu J, Ghayur T, et al. Ligand association rates to the inner-variable-domain of a dual-variable-domain immunoglobulin are significantly impacted by linker design. Mabs 2001; 3:487-94; PMID:21814039; http://dx.doi.org/10.4161/mabs.3.5.16326
- Rao E, Mikol V, Li D, Kruip J, Davison M. Humanized antibodies that bind interleukin-4 and/or interleukin-13 WO 2009052081 A2
- Correia I, Sung J, Burton R, Jakob CG, Carragher B, Ghayur T, Radziejewski C. The structure of dual-variable-domain immunoglobulin molecules alone and bound to antigen. Mabs 2013; 5:364-72; PMID:23572180; http://dx.doi.org/10.4161/mabs.24258
- Jakob CG, Edalji R, Judge RA, DiGiammarino E, Li Y, Gu J, Ghayur T. Structure reveals function of the dual variable domain immunoglobulin (DVD-Ig) molecule. Mabs 2013; 5:358-63; PMID:23549062; http://dx.doi.org/10.4161/mabs.23977
- Wu C. Diabodies: Molecular engineering and therapeutic applications. Drug News Perspect 2009; 22:453-8; PMID:20016854
- Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, Jin L, Gorlatov S, Ciccarone V, Chen F, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: Preclinical activity and safety in nonhuman primates. Sci Transl Med 2015; 7:289ra82; PMID:26019218; http://dx.doi.org/10.1126/scitranslmed.aaa5693
- Bumbaca D, Boswell CA, Fielder PJ, Khawli LA. Physiochemical and Biochemical Factors Influencing the Pharmacokinetics of Antibody Therapeutics. AAPS J 2012; 14:554-8; PMID:22610647; http://dx.doi.org/10.1208/s12248-012-9369-y
- Kamei DT, Lao BJ, Ricci MS, Deshpande R, Xu H, Tidor B, Lauffenburger DA. Quantitative methods for developing Fc mutants with extended half-lives. Biotechnol Bioeng 2005; 92:748-60; PMID:16136591; http://dx.doi.org/10.1002/bit.20624
- Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer W, Blumberg RS. Neonatal Fc Receptor: From Immunity to Therapeutics. J Clin Immunol 2010; 30:777-89; PMID:20886282; http://dx.doi.org/10.1007/s10875-010-9468-4
- Roopenian DC, Sun VZ. Clinical Ramifications of the MHC Family Fc Receptor FcRn. J Clin Immunol 2010; 30:790-7; PMID:20848168; http://dx.doi.org/10.1007/s10875-010-9458-6
- Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E, et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med 2015; 287ra70; PMID:25972002; http://dx.doi.org/10.1126/scitranslmed.aaa4802
- Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs 2015; 7:331-43; PMID:25658443; http://dx.doi.org/10.1080/19420862.2015.1008353
- Dornan D, Spleiss O, Yeh RF, Duchateau-Nguyen G, Dufour A, Zhi J, Robak T, Moiseev SI, Dmoszynska A, Solal-Celigny P, et al. Effect of FCGR2A and FCGR3A variants on CLL outcome. Blood 2010; 116:4212-22; PMID:20705761; http://dx.doi.org/10.1182/blood-2010-03-272765
- Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008; 7:2517-27; PMID:18723496; http://dx.doi.org/10.1158/1535-7163.MCT-08-0201
- Ströhlein MA, Heiss MM. The trifunctional antibody catumaxomab in treatment of malignant ascites and peritoneal carcinomatosis. Future Oncol 2010; 6:1387-94; PMID:20919824; http://dx.doi.org/10.2217/fon.10.111
- Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, Wang L, Weiskopf K, Majeti R. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. Mabs 2015; 7:946-56; PMID:26083076; http://dx.doi.org/10.1080/19420862.2015.1062192
- Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJR, et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Sci 2014; 343:1260-3; PMID:24626930; http://dx.doi.org/10.1126/science.1248943
- Wang W, Wang E, Balthasar J. Monoclonal Antibody Pharmacokinetics and Pharmacodynamics. Clin Pharmacol Therapeutics 2008; 84:548-58; http://dx.doi.org/10.1038/clpt.2008.170
- Sela-Culang I, Kunik V, Ofran Y. The structural basis of antibody-antigen recognition. Front Immunol 2013; 4:302; PMID:24115948; http://dx.doi.org/10.3389/fimmu.2013.00302
- Sela-Culang I, Alon S, Ofran Y. A systematic comparison of free and bound antibodies reveals binding-related conformational changes. J Immunol 2012; 189:4890-9; PMID:23066154; http://dx.doi.org/10.4049/jimmunol.1201493
- Masuda K, Sakamoto K, Kojima M, Aburatani T, Ueda T, Ueda H. The role of interface framework residues in determining antibody VH/VL interaction strength and antigen-binding affinity. FEBS J 2006; 273:2184-94; PMID:16649995; http://dx.doi.org/10.1111/j.1742-4658.2006.05232.x
- Birnbaum ME, Berry R, Hsiao YS, Chen Z, Shingu-Vazquez MA, Yu X, Waghray D, Fischer S, McCluskey J, Rossjohn J, et al. Molecular architecture of the ab T cell receptor-CD3 complex. PNAS USA 2014; 111:17576-81; PMID:25422432; http://dx.doi.org/10.1073/pnas.1420936111
- Haagen IA, van de Griend R, Clark M, Geerars A, Bast B, de Gast B. Killing of human leukaemia/lymphoma B cells by activated cytotoxic T lymphocytes in the presence of a bispecific monoclonal antibody (aCD3/aCD19). Clin Exp Immunol 1992; 90:368-75; PMID:1281055; http://dx.doi.org/10.1111/j.1365-2249.1992.tb05853.x
- Mack M, Riethmüller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. PNAS USA 1995; 92:7021-5; PMID:7624362; http://dx.doi.org/10.1073/pnas.92.15.7021
- Kim CH, Axup JY, Lawson BR, Yun H, Tardif V, Choi SH, Zhou Q, Dubrovska A, Biroc SL, Marsden R, et al. Bispecific small molecule-antibody conjugate targeting prostate cancer. PNAS USA 2013; 110:17796-801; PMID:24127589; http://dx.doi.org/10.1073/pnas.1316026110
- Hirvinen M, Heiskanen R, Oksanen M, Pesonen S, Liikanen I, Joensuu T, Kanerva A, Cerullo V, Hemminki A. Fc-gamma receptor polymorphisms as predictive and prognostic factors in patients receiving oncolytic adenovirus treatment. J Transl Med 2013; 11:1-12; PMID:23281771; http://dx.doi.org/10.1186/1479-5876-11-193
- Sondermann P, Huber R, Jacob U. Crystal structure of the soluble form of the human fcgamma-receptor IIb: a new member of the immunoglobulin superfamily at 1.7 Å resolution. EMBO J 1999; 18:1095-103; PMID:10064577; http://dx.doi.org/10.1093/emboj/18.5.1095
- Sondermann P, Kaiser J, Jacob U. Molecular basis for immune complex recognition: A comparison of Fc-receptor structures. J Mol Biol 2001; 309:737-49; PMID:11397093; http://dx.doi.org/10.1006/jmbi.2001.4670
- Raghavan M, Bjorkman PJ. Fc receptors and their interactions with immunoglobulins. Annu Rev Cell Dev Biol 1996; 12:181-220; PMID:8970726; http://dx.doi.org/10.1146/annurev.cellbio.12.1.181
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nat 1994; 372:379-83; PMID:7969498; http://dx.doi.org/10.1038/372379a0
- Merrimack. MM-111 Phase 2 trial. http://www.merrimackpharma.com/pipeline/mm-111/mm-111-phase-2-trial (Access Date February 3, 2015) (2015)
- Warren CM, Landgraf R. Signaling through ERBB receptors: Multiple layers of diversity and control. Cell Signal 2006; 18:923-33; PMID:16460914; http://dx.doi.org/10.1016/j.cellsig.2005.12.007
- Blaber M, Lee J. Designing proteins from simple motifs: opportunities in Top-Down Symmetric Deconstruction. Curr Opin Struct Biol 2012; 22:442-50; PMID:22726756; http://dx.doi.org/10.1016/j.sbi.2012.05.008
- Urvoas A, Valerio-Lepiniec M, Minard P. Artificial proteins from combinatorial approaches. Trends Biotechnol 2012; 30:512-20; PMID:22795485; http://dx.doi.org/10.1016/j.tibtech.2012.06.001
- Sawyer N, Chen J, Regan L. All repeats are not equal: A module-based approach to guide repeat protein design. J Mol Biol 2013; 425:1826-38; PMID:23434848; http://dx.doi.org/10.1016/j.jmb.2013.02.013
- Sawyer N, Speltz EB, Regan L. NextGen protein design. Biochem Soc Trans 2013; 41:1131-6; PMID:24059497; http://dx.doi.org/10.1042/BST20130112
- Merkx M, Golynskiy MV, Lindenburg LH, Vinkenborg JL. Rational design of FRET sensor proteins based on mutually exclusive domain interactions. Biochem Soc Trans 2013; 41:1201-5; PMID:24059509; http://dx.doi.org/10.1042/BST20130128
- Huang J, Koide A, Makabe K, Koide S. Design of protein function leaps by directed domain interface evolution. PNAS USA 2008; 105:6578-83; PMID:18445649; http://dx.doi.org/10.1073/pnas.0801097105
- Zhou X, Wang H, Zhang Y, Gao L, Feng Y. Alteration of substrate specificities of thermophilic α/β hydrolases through domain swapping and domain interface optimization. Acta Biochim Biophys Sin (Shanghai) 2012; 44:965-73; PMID:23099882; http://dx.doi.org/10.1093/abbs/gms086
- Broom A, Doxey AC, Lobsanov YD, Berthin LG, Rose DR, Howell PL, McConkey BJ, Meiering EM. Modular evolution and the origins of symmetry: Reconstruction of a 3-fold symmetric globular protein. Structure 2012; 20:161-71; PMID:22178248; http://dx.doi.org/10.1016/j.str.2011.10.021
- Yadid I, Tawfik DS. Functional β-propeller lectins by tandem duplications of repetitive units. Protein Eng Des Sel 2011; 24:185-95; PMID:20713410; http://dx.doi.org/10.1093/protein/gzq053
- Voet ARD, Noguchi H, Addy C, Simoncini D, Terada D, Unzai S, Park SY, Zhang KYJ, Tame JRH. Computational design of a self-assembling symmetrical β-propeller protein. PNAS USA 2014; 111:15102-7; PMID:25288768; http://dx.doi.org/10.1073/pnas.1412768111
- Lensink MF, Wodak SJ. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013; 81:2082-95; PMID:24115211; http://dx.doi.org/10.1002/prot.24428
- Moal IH, Torchala M, Bates PA, Fernández-Recio J. The scoring of poses in protein-protein docking: current capabilities and future directions. BMC Bioinformatics 2013; 14:1-15; PMID:23323762; http://dx.doi.org/10.1186/1471-2105-14-286
- Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall'Acqua WF. Insights into the molecular basis of a bispecific antibody's target selectivity. Mabs 2015; 7:461-9; PMID:25730144; http://dx.doi.org/10.1080/19420862.2015.1022695
- Wang J, Iyer S, Fielder PJ, Davis JD, Deng R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: comparative evaluation of prediction approaches in early drug development. Biopharm Drug Dispos 2015
- Ying T, Wang Y, Feng Y, Prabakaran P, Gong R, Wang L, Crowder K, Dimitrov DS. Engineered antibody domains with significantly increased transcytosis and half-life in macaques mediated by FcRn. Mabs 2015; 7:922-30; PMID:26179052; http://dx.doi.org/10.1080/19420862.2015.1067353
- Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn Affinity-Pharmacokinetic Relationship of Five Human IgG4 Antibodies Engineered for Improved In Vitro FcRn Binding Properties in Cynomolgus Monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; http://dx.doi.org/10.1124/dmd.112.045864
- Jäger M, Schoberth A, Ruf P, Hess JR, Lindhofer H. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor 2. Cancer Res 2009; 69:4270-6; PMID:19435924; http://dx.doi.org/10.1158/0008-5472.CAN-08-2861
- Diermeier-Daucher S, Ortmann O, Buchholz S, Brockhoff G. Trifunctional antibody ertumaxomab: Non-immunological effects on Her2 receptor activity and downstream signaling. Mabs 2012; 4:614-22; PMID:22820509; http://dx.doi.org/10.4161/mabs.21003
- Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H, Chan C, Parren PWHI, Hack CE, Dechant M, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004; 104:1793-800; PMID:15172969; http://dx.doi.org/10.1182/blood-2004-01-0039
- de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, Oomen LA, Peipp M, Valerius T, Slootstra JW, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol 2011; 186:1840-8; PMID:21187443; http://dx.doi.org/10.4049/jimmunol.1003032
- Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-Å crystal structure of the human IgG1 Fc fragment-FcgRIII complex. Nat 2000; 406:267-73; http://dx.doi.org/10.1038/35018508
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. PNAS USA 2006; 103:4005-10; PMID:16537476; http://dx.doi.org/10.1073/pnas.0508123103
- Mimoto F, Kadono S, Katada H, Igawa T, Kamikawa T, Hattori K. Crystal structure of a novel asymmetrically engineered Fc variant with improved affinity for FcgRs. Mol Immunol 2014; 58:132-38; PMID:24334029; http://dx.doi.org/10.1016/j.molimm.2013.11.017
- Mimoto F, Igawa T, Kuramochi T, Katada H, Kadono S, Kamikawa T, Shida-Kawazoe M, Hattori K. Novel asymmetrically engineered antibody Fc variant with superior Fc gamma R binding affinity and specificity compared with afucosylated Fc variant. mAbs 2013; 5:229-36; PMID:23406628; http://dx.doi.org/10.4161/mabs.23452
- Haake DA, Franklin EC, Frangione B. The modification of human immunoglobulin binding to staphylococcal protein A using diethylpyrocarbonate. J Immunol 1982; 129:190-2; PMID:6211482
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-.ANG. resolution. Biochem 1981; 20:2361-70; PMID:7236608; http://dx.doi.org/10.1021/bi00512a001
- Sasano M, Burton DR, Silverman GJ. Molecular selection of human antibodies with an unconventional bacterial B cell antigen. J Immunol 1993; 151:5822-39; PMID:8228264
- Sasso EH, Silverman GJ, Mannik M. Human IgM molecules that bind staphylococcal protein A contain VHIII H chains. J Immunol 1989; 142:2778-83; PMID:2495325
- Sasso EH, Silverman GJ, Mannik M. Human IgA and IgG F(ab')2 that bind to staphylococcal protein A belong to the VHIII subgroup. J Immunol 1991; 147:1877-83; PMID:1909733
- Nelson JT, Kim S, Reuel NF, Salem DP, Bisker G, Landry MP, Kruss S, Barone PW, Kwak S, Strano MS. Mechanism of Immobilized Protein A Binding to Immunoglobulin G on Nanosensor Array Surfaces. Anal Chem 2015; 87:8186-93; PMID:26149633; http://dx.doi.org/10.1021/acs.analchem.5b00843
- Ogi H, Motohisa K, Hatanaka K, Ohmori T, Hirao M, Nishiyama M. Concentration dependence of IgG-protein A affinity studied by wireless-electrodeless QCM. Biosens Bioelectron 2007; 22:3238-42; PMID:17420120; http://dx.doi.org/10.1016/j.bios.2007.03.003
- Saha K, Bender F, Gizeli E. Comparative study of IgG binding to proteins G and A: Nonequilibrium kinetic and binding constant deetermination with the acoustic waveguide device. Anal Chem 2003; 75:835-42; PMID:12622374; http://dx.doi.org/10.1021/ac0204911
- Deis LN, Wu Q, Wang Y, Qi Y, Daniels KG, Zhou P, Oas TG. Suppression of conformational heterogeneity at a protein-protein interface. PNAS USA 2015; 112:9028-33; PMID:26157136; http://dx.doi.org/10.1073/pnas.1424724112
- Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, Silverman GJ. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. PNAS USA 2000; 97:5399-404; PMID:10805799; http://dx.doi.org/10.1073/pnas.97.10.5399
- Donaldson JM, Zer C, Avery KN, Bzymek KP, Horne DA, Williams JC. Identification and grafting of a unique peptide-binding site in the Fab framework of monoclonal antibodies. PNAS USA 2013; 110:17456-61; PMID:24101516; http://dx.doi.org/10.1073/pnas.1307309110
- Madhavi SG, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013; 27:221-34; PMID:23579614; http://dx.doi.org/10.1007/s10822-013-9644-8
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol 2002; 320:597-608; PMID:12096912; http://dx.doi.org/10.1016/S0022-2836(02)00470-9
- Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins 2004; 55:351-67; PMID:15048827; http://dx.doi.org/10.1002/prot.10613
- Guo Z, Mohanty U, Noehre J, Sawyer TK, Sherman W, Krilov G. Probing the a-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem Biol Drug Des 2010; 75:348-59; PMID:20331649; http://dx.doi.org/10.1111/j.1747-0285.2010.00951.x
- Schrödinger. Schrödinger Release 2014-4: Maestro, version 10.0, New York, NY: Schrödinger, LLC (2014) 2015
- Chen R, Weng Z. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins 2002; 47:281-94; PMID:11948782; http://dx.doi.org/10.1002/prot.10092
- Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS One 2011; 6:e24657 EP; PMID:21949741; http://dx.doi.org/10.1371/journal.pone.0024657
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2011, San Diego: Dassault Systèmes (2011)