
ABSTRACT
Undesired solution behaviors such as reversible self-association (RSA), high viscosity, and liquid-liquid phase separation can introduce substantial challenges during development of monoclonal antibody formulations. Although a global mechanistic understanding of RSA (i.e., native and reversible protein-protein interactions) is sufficient to develop robust formulation controls, its mitigation via protein engineering requires knowledge of the sites of protein-protein interactions. In the study reported here, we coupled our previous hydrogen-deuterium exchange mass spectrometry findings with structural modeling and in vitro screening to identify the residues responsible for RSA of a model IgG1 monoclonal antibody (mAb-C), and rationally engineered variants with improved solution properties (i.e., reduced RSA and viscosity). Our data show that mutation of either solvent-exposed aromatic residues within the heavy and light chain variable regions or buried residues within the heavy chain/light chain interface can significantly mitigate RSA and viscosity by reducing the IgG's surface hydrophobicity. The engineering strategy described here highlights the utility of integrating complementary experimental and in silico methods to identify mutations that can improve developability, in particular, high concentration solution properties, of candidate therapeutic antibodies.
Abbreviations
| RSA | = | reversible self-association |
| HDX-MS | = | hydrogen-deuterium exchange mass spectrometry |
| mAb | = | monoclonal antibody |
| Fab | = | fragment antigen-binding |
| Fv | = | variable fragment |
| Fc | = | fragment crystallizable |
| CDR | = | complementarity-determining region |
| IgG | = | immunoglobulin G |
| VH | = | heavy chain variable domain |
| VL | = | light chain variable domain |
| KD | = | binding affinity, i.e., dissociation equilibrium constant |
| SASA | = | solvent accessible surface area |
| AC-SINS | = | affinity-capture self-interaction nanoparticle spectroscopy |
| DLS | = | dynamic light scattering |
| DSC | = | differential scanning calorimetry |
| DSF | = | differential scanning fluorimetry |
| HPSEC | = | high performance size-exclusion chromatography |
| HIC | = | hydrophobic interaction chromatography |
| Cp | = | centipoise |
Introduction
Monoclonal antibodies (mAbs) represent the fastest growing class of biologic drugs in the biopharmaceutical market.Citation1 Increased demand for enhanced patient convenience through lower dosing frequency and subcutaneous self-administration requires development of high concentration liquid formulations, typically greater than 100 mg/mL. In addition to typical physical (e.g., conformational, colloidal), chemical (e.g., oxidation, deamidation), and interfacial (e.g., freeze-thaw, shear) instabilities,Citation2 at high concentrations, undesired solution properties such as reversible self-association (RSA), high viscosity, and liquid-liquid phase separation can introduce substantial challenges.Citation3-7
RSA (i.e., native, non-covalent, and reversible oligomerization of monomeric species) is typically induced in the crowded environments of high concentrations due to the reduced intermolecular distances and increased probability of molecular collisions.Citation8 The self-associated species can pose manufacturing and delivery challenges (e.g., high viscosity, clogging of the lines and filters Citation3,9) or cause pain to patients upon administration.Citation10,11 In certain cases, the self-associated species can affect bioactivity and pharmacokinetic properties of biologic drug candidatesCitation12,13 or, depending on the structural and environmental conditions, transform to irreversible aggregates via further covalent linkages.Citation14
A number of studies have illustrated the diversity of structural mechanisms by which self-association can occur.Citation9,15-22 Antibodies have been found to self-interact through sites within both the variable fragment (Fv) and constant regions of the antigen-binding fragment (Fab) domains, as well as the crystallizable fragment (Fc) domain.Citation9,15,18-20 In addition to the multitude of distinct domain surfaces that can compose the binding interfaces, the nature of the driving forces behind antibody self-association can also be diverse, with both electrostatic and hydrophobic interactions playing key roles.Citation6,7,18,21,23
Potential strategies to mitigate self-association and other undesired high concentration solution properties may include protein engineering approaches,Citation20,24-26 as well as formulation development and optimization efforts.Citation5-7,9,18,19,21,23,27 While the former can be limited due to the degree of knowledge of sites of interactions, the latter can be time and resource intensive and may not always be fully successful to a desired degree for all antibodies. Historically, a number of computational and analytical screening methods have been used to predict aggregationCitation28-30 and high concentration formulation risks.Citation17,31-36 While these approaches can provide valuable information and help screening of potential lead candidates during discovery and early stage development, they do not necessarily provide insight into the underlying molecular mechanisms involved. With regard to high concentration challenges, the availability of methodologies that can probe native intermolecular interactions at the structural and molecular level can effectively guide corresponding protein engineering and formulation efforts designed to mitigate the development risks.
Recently, characterization studies of the RSA of mAb-C, a model human monoclonal IgG1 were presented.Citation15,18 This antibody exhibits self-association even at relatively low concentrations (<10 mg/mL) and becomes highly viscous when formulated at higher concentrations.Citation15,18 Using a novel hydrogen-deuterium exchange mass spectrometry (HDX-MS) technique, structural regions within mAb-C heavy and light chain variable domains (VH and VL, respectively) were identified that exhibited a decreased rate of deuterium exchange as a function of increasing protein concentration.Citation15 This result suggested that the self-interacting interfaces of mAb-C monomers are composed of residues within the identified regions that presumably become more shielded from the surrounding solvent at the interaction interfaces. In this report, we build upon those findings and describe single residue mutations within the HDX-MS-identified VH and VL regions that can potentially prevent mAb-C self-association and reduce viscosity at high concentrations.
Data herein show that the rationally engineered mutations, through either direct or indirect reduction in antibody hydrophobicity, do indeed reduce mAb-C self-association tendencies, as well as its viscosity at high concentrations. The selected mutations were identified by incorporating data from the aforementioned HDX-MS study, in silico structural modeling, and high throughput self-association screening techniques. The combination of these complementary techniques provides a powerful and comprehensive approach that can be applied to any mAb to identify and engineer the structural determinants of RSA and the linked high concentration issues. Finally, the strategy employed here can minimize the more extensive characterization, formulation development and optimization efforts that are required to better understand and mitigate high concentration issues.
Results
Mutagenesis of mAb-C based on homology modeling
Previously, HDX-MS was utilized to reveal two key regions within mAb-C structure (VH amino acids 35–59: HWVRQAPGQGLEWMGWINPHSGGTN and VL amino acids 36–71: YQQKPGKAPKLLIYVASSLQSGVPSRFSGSGSGTDF) that likely encompass the self-interaction interfaces, leading to self-association of this mAb.Citation15 In addition, previous formulation studies assessing the effect of specific anionic and cationic species as well as aromatic amino acids suggested that histidine and hydrophobic/aromatic residues may play key roles in modulating mAb-C self-association.Citation18 To better understand the molecular mechanisms responsible for mAb-C self-association, targeted mutations within the identified HDX-MS interacting interfaces were performed. Two key histidine residues within the VH region (H35 and H53) were each independently mutated to alanine, although studies with H53 mutation did not show any effect on mAb-C self-association (data not shown), and it was therefore not studied further. To guide the choice of hydrophobic/aromatic residues to mutate, we reasoned that only surface-exposed residues would contribute to the observed self-association. In the absence of an x-ray crystal structure, a homology model of mAb-C was generated and side chain solvent accessible surface area (SASA) and surface aggregation prediction analysis of the modeled Fv was performed. The latter analysis identifies surface regions composed of hydrophobic residues that are both adjacent and solvent exposed. The hydrophobic residues within the HDX-MS-identified interfaces that are predicted to form aggregation-prone surfaces include W50 in the VH region and Y49 and L54 in the VL region (). Therefore, in addition to H35, these exposed hydrophobic residues were also mutated to determine their effect on mAb-C self-association ().
Table 1. mAb-C VH and VL residues that impact self-association. The Fv location (Kabat numbering), germline encoded residue for the given position, and introduced substitutions are listed. SASA was measured using BioLuminate and a mAb-C homology model. Aggregation surface indicates whether the particular residue was predicted to contribute to an exposed hydrophobic patch as predicted by BioLuminate aggregation surface analysis.
Affinity-capture self-interaction nanoparticle spectroscopy
We used affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS), which is a high-throughput screening technique, to evaluate the effect of the selected mutations on mAb-C self-association.Citation35,36 The H35 is positioned at the VH/VL interface and is largely buried according to the SASA analysis of the homology model (). Despite the lack of solvent exposure, mutation of this residue to alanine (H35A) resulted in a significant reduction in mAb-C self-association as evident by the significant decrease in plasmon shift signals measured by AC-SINS (). In contrast, mutation to asparagine (H35N), an uncharged polar amino acid, showed no effect (). Other charged residues (K and D) were also introduced at position 35; however, they prevented expression and could not be examined further. To test whether other small, non-polar side chains reduced mAb-C self-association propensity, the H35 was mutated to G and V residues, both of which had an effect comparable to that of alanine (Fig. S1).
Figure 1. Single CDR mutations prevent mAb-C self-association. (A) AC-SINS analysis of self-association of mAb-C variants in PBS, pH 7.4. mAb-C exhibits high self-association in PBS (25 nm plasmon shift). Mutation of histidine 35 of the VH to alanine, but not asparagine, substantially decreases self-association. Mutation of tryptophan 50 of the VH to arginine, and tyrosine 49 and leucine 54 of the VL to aspartate, reduce self-association to varying extents. Plasmon shifts are normalized to the plasmon wavelength of each antibody incubated with control nanoparticles that lack anti-Fc capture antibody. (B) DLS determination of mAb-C variant hydrodynamic size as a function of concentration. The H35A, W50R, Y49D, and L54D mutations prevent mAb-C interaction at up to 10 mg/mL. H35N also decreases hydrodynamic size, but to a much lesser extent. Error bars indicate standard deviation of triplicate measurements.
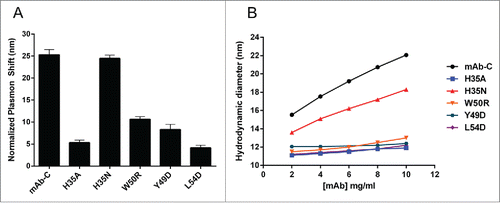
The germline gene encoding the heavy chain of mAb-C (IGHV1-2) exists as multiple alleles, and, depending on the allele, the amino acid at position 50 is either a tryptophan or arginine.Citation37 To determine the effect of this variation between alleles, W50 was mutated to arginine (W50R), which resulted in a significant decrease in mAb-C RSA, although to a smaller extent compared to the H35A mutation (). Within the VL, mutation of Y49 to aspartate (Y49D) significantly decreased the extent of mAb-C self-association. Mutation of L54 to aspartate (L54D) resulted in an even greater degree of reduction in self-association compared to the Y49D substitution (). Collectively, these data indicate that the introduction of single mutations into the previously identified HDX-MS VH or VL regions is sufficient to minimize or prevent mAb-C reversible self-association tendencies.
Dynamic light scattering
The AC-SINS technique indirectly measures antibody self-association by measuring the clustering of gold nanoparticles induced by self-interaction of antibody captured on the particle surface.Citation35 As a complementary technique to AC-SINS and to directly measure the self-association of free antibody in solution, dynamic light scattering (DLS) was utilized. As previously reported, mAb-C exhibits increasing hydrodynamic size (an ∼8 nm increase in hydrodynamic diameter) as a function of increasing protein concentration, indicative of reversible self-association ().Citation17 Analysis of the selected mAb-C mutants revealed that the DLS results were consistent with those of the AC-SINS method. The H35A, Y49D, L54D, and W50R mutations quite effectively inhibited the observed increase in mAb-C hydrodynamic size over the tested concentration range (). In contrast, the H35N mutation yielded only a partial reduction in hydrodynamic size, consistent with the AC-SINS findings, suggesting only a moderate effect on mAb-C self-interactions (). Collectively, the AC-SINS and DLS results reveal that the engineered mutations in either of the HDX-MS-identified VH or VL regions do indeed mitigate, although to different degrees, the self-association tendencies of mAb-C.
Capillary isoelectric focusing and hydrophobic interaction chromatography
To better understand the effect of the engineered mutations on the chemical properties of mAb-C, charge and hydrophobicity were measured. Capillary isoelectric focusing (cIEF) revealed that the mutants can be discriminated by isoelectric point (pI) and charge isoform distribution, but that the absolute effect on net charge was minimal (≤0.2 pI unit difference between major charge isoforms) (). These results suggest that a change in charge, i.e., electrostatic interactions, is unlikely to explain the observed RSA behavior.
Figure 2. Charge and hydrophobicity analysis of mAb-C variants. (A) Capillary isoelectric focusing of mAb-C variants shows only minor differences in pI (indicated above peaks) and charge heterogeneity. (B) Hydrophobic interaction chromatography analysis indicates that the H35A, W50R, Y49D, and L54D mutations substantially reduce surface hydrophobicity of mAb-C.
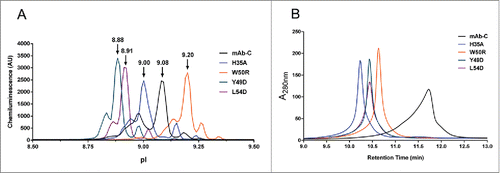
Our previous studies showed that the extent of mAb-C self-association was reduced in the presence of aromatic amino acids (e.g., tryptophan, phenylalanine, tyrosine).Citation18 Further, data showed that the aromatic residues that are more hydrophobic in nature (e.g., tryptophan), more effectively reduced self-association of mAb-C. To test whether the engineered mutations decreased the surface hydrophobicity of mAb-C, hydrophobic interaction chromatography (HIC) was performed. HIC is used to assess and differentiate the intrinsic degree of hydrophobicity by measuring the retention time of proteins on the HIC column. Molecules with high relative hydrophobicity interact with the column more readily, resulting in longer retention times.Citation38,39 HIC analysis showed that mAb-C exhibited the longest retention time (12.0 minutes) and a broad, asymmetric profile (). The H35A mutant, which had a mutation of only the buried histidine residue, had a shorter retention time (10.2 minutes) and a much narrower, symmetric profile (). The W50R mutation also reduced retention time (10.6 minutes) and yielded a more symmetric peak (). The Y49D and L54D mutants displayed almost identical peak profiles with a similar retention time of 10.4 minutes (). These findings indicate that the mutations in the HDX-MS-identified VH and VL domains, particularly H35A, decrease the surface hydrophobicity of mAb-C.
Differential scanning calorimetry and differential scanning fluorimetry
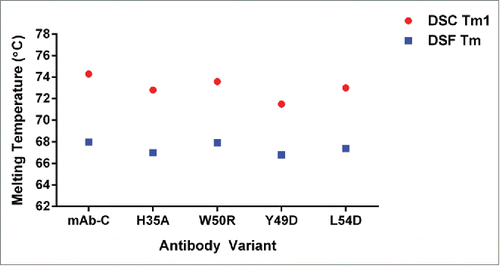
To assess the effect of the engineered mutations on the structural stability of mAb-C, differential scanning calorimetry (DSC) and differential scanning fluorimetry (DSF) were employed. These analyses revealed no significant impact from the mutations on mAb-C thermostability (). The mid-point of thermal transition (Tm) measured by DSC showed values all within 74 ± 2°C, suggesting the Fv mutations had no effect on the global thermodynamic stability of mAb-C (herein, only the first mid-point of thermal transition (Tm1) is reported for purpose of stability comparison). The tertiary structural stability of mAb-C was examined with DSF using the extrinsic fluorescent dye, Sypro Orange. All Tm values fell within the range of 68 ± 2°C, suggesting no significant impact of the mutations on the tertiary structural stability of mAb-C. Collectively, the findings indicate that the Fv mutations do not compromise the global structural stability of mAb-C.
Figure 3. Thermostability analysis of mAb-C variants by differential scanning calorimetry (DSC) and differential scanning fluorimetry (DSF) show that the introduced mutations have a negligible effect on physical stability.
High performance size exclusion chromatography
Recently, it was reported that colloidally unstable monoclonal antibodies can be discriminated by retention time and peak profile derived from high-performance size exclusion chromatography (HPSEC).Citation34 The HPSEC analysis of mAb-C showed a retention time of 8.3 minutes and an asymmetric, tailing peak (Fig. S2). The H35A, W50R, Y49D, and Y54D mutations exhibited longer retention times (8.7, 8.8, 8.7, and 8.6 minutes, respectively) and resulted in narrower and more symmetric peaks (Fig. S2). These findings suggest that the engineered mutations affect mAb-C interaction with the HPSEC column substrate, and support the suggestion that mAbs prone to self-association can be potentially flagged by their aberrant HPSEC profiles.
In addition to evaluation of the structural stability of mAb-C by DSC and DSF, the effects of the engineered mutants on mAb-C aggregation and fragmentation propensities were tested under accelerated storage conditions. Samples were incubated at 45°C for 2 weeks, with control samples incubated at 4°C for a similar time period. The HPSEC results showed no significant differences in degradation rates when comparing mAb-C to its variants under the stressed conditions tested (). These results show that the introduced mutations have no significant effect on mAb-C aggregation and fragmentation rates.
Table 2. MAb-C variants exhibit minor and comparable degradation following thermal stress. Antibodies were incubated at either 4°C or 45°C for two weeks and the percent of monomer (Mon.), aggregate (Agg.), and fragment (Frag.) was determined by HPSEC analysis. The percent change in each species was calculated from the difference between the 45°C and 4°C treated samples.
Viscosity
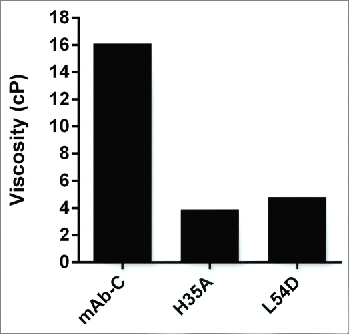
Previous studies have shown that a direct link can exist between reversible self-association and elevated viscosity at high concentrations.Citation3 Such an observation was made for mAb-C, in which conditions (e.g., increasing protein concentration, reducing temperature, presence of anionic salt species) that led to induced self-association also resulted in elevated viscosity at high concentrations.Citation15 To test whether the Fv mutations had any effect on viscosity at high concentrations, the H35A and L54D mutants were selected as representative VH and VL mutants, respectively, with the most impact on RSA. Viscosity measurements were performed at a protein concentration of 70 mg/mL in phosphate-buffered saline (PBS) buffer, pH 7.4, at 4°C. MAb-C showed a viscosity of 16.1 cP, while H35A and L54D resulted in viscosity values of 3.9 and 4.8 cP, respectively (). These viscosity results indicate that the selected mutations not only mitigated mAb-C RSA, but also significantly reduced its viscosity at higher concentrations.
Figure 4. MAb-C VH and VL mutations reduce viscosity. Antibody viscosity was measured at 70 mg/mL in PBS, pH 7.4 at 4°C by an Anton-Paar MCR-301 cone-and-plate rheometer.
Discussion
Herein, we have provided a molecular level understanding of the mechanism of reversible self-association for a model human IgG1, mAb-C. Integrating previous structural analysis (HDX-MS) with in silico modeling has enabled the identification of single amino acid mutations that can significantly mitigate the RSA propensity, as well as reduce high concentration viscosity without compromising structural stability. Notably, our results revealed two approaches to prevent RSA: 1) removal of exposed hydrophobic residues within the VH or VL; and 2) indirect modulation of surface hydrophobicity by mutation of a buried residue positioned at the VH/VL interface.
Previous HDX-MS studies identified self-interacting regions within the VH and VL domain of mAb-C that became less solvent exposed as a function of increasing protein concentration.Citation15 It is evident from the homology model of mAb-C that the HDX-MS-identified interface regions are on the opposing surfaces of the Fv. While one surface is on the VH and is primarily positioned along the VH/VL interface, the other is on the VL, spanning complementarity-determining region (CDR)2 and the adjacent framework residues (). This spatial arrangement suggests that self-association of mAb-C is driven by an asymmetric interaction involving surfaces on both the VH and VL. The location of the two hydrophobic patches identified by in silico aggregation surface analysis shows that the VH patch (W50 and Y33) is positioned at a cleft formed between the VH and VL (). Within this patch, only W50 is located within the HDX-MS interface region, though Y33 is adjacent to W50. We attempted to determine the contribution of Y33 to mAb-C RSA, but mutating this position prevented antibody expression, and, therefore, any further studies. Thus, it is unclear whether Y33 contributes to self-association or not. The second aggregation-prone patch (Y49 and L54) is located on the opposing surface of the VL (). The fact that the W50R, Y49D, and L54D mutations notably reduced RSA supports the conclusion that the two hydrophobic patches drive the mAb-C self-association. This is further highlighted by the HIC analysis where the W50R, Y49D, and L54D mutations reduced surface hydrophobicity, as indicated by their shorter column retention times compared to mAb-C. It is interesting that the different alleles of the VH germline gene of mAb-C (IGHV1-2) encode either a W or an R, since these residues are so chemically dissimilar. The vast majority of human VH germline genes encode a hydrophobic residue at position 50, so it is intriguing to speculate that the R-encoding allele emerged, in part, because of its beneficial effect on stability.Citation37
Figure 5. Cartoon of mAb-C homology model depicting location of mutated VH and VL residues. The HDX-identified binding interface (green) includes VH FR2, a segment of CDR H2, CDR L2, and a segment of VL FR2 and FR3. (A) H35 (magenta) is positioned at the VH/VL interface beneath adjacent aromatic side chains, whereas W50 (orange), Y49 (pink), and L54 (purple) are exposed. Mutation of H35 to a small, non-polar, amino acid (e.g., alanine) may create a cavity that allows W50 or other nearby hydrophobic residues to become more buried into the cleft between the VH/VL interface. VH and VL framework are in blue and gray, respectively. CDRs excluded from the HDX interface are labeled cyan (H1), light blue (H3), yellow (L1), and brown (L3). (B) Aggregation surface analysis of mAb-C Fv. Aggregation prone regions of the surface are depicted in red. A predicted aggregation patch composed of W50 and Y33 is located on the VH (top image, green oval). On the VL, Y49 and L54 compose an aggregation surface (bottom image, green oval).
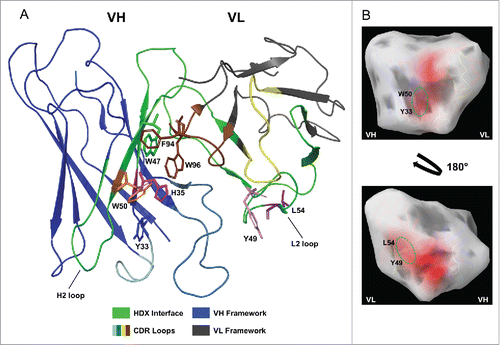
Perhaps the most interesting observation was the drastic effect of H35 mutation on mAb-C RSA and viscosity. In our homology model, this position is largely buried at the VH/VL interface and is therefore not expected to form direct contact between mAb-C monomers. Based on the model, H35 is located within a hydrophobic pocket created by aromatic residues extending from both the VH and VL (W50 and Y33 on the VH, and W47, F94, and W96 on the VL) (). W50 and H35 are adjacent, located on opposing β-strands. The fact that mutation of histidine to alanine, glycine, and valine, but not asparagine, prevented self-association suggests that the size and/or polarity of the side chain mediate the degree of RSA. One possibility is that small, non-charged/polar residues at position 35 allow greater packing of neighboring aromatic residues toward the interior of the VH/VL interface. Such a local structural rearrangement would be consistent with the decreased surface hydrophobicity of the H35A mutant. Indeed, previous studies support this concept. A structural study of a VH single domain antibody revealed that mutation of H35 to glycine or alanine increased solubility, expression, and thermostability.Citation16 The H35 glycine mutation created a cavity into which an adjacent tryptophan at position 95 became buried. This resulted in increased hydrophilicity of the VH surface, which is normally interfaced with the VL.Citation16 A similar result was observed in another study on the VH single domain antibodies HEL4 and DP47d, which are highly and poorly soluble, respectively.Citation40 In contrast to DP47d, HEL4 has a glycine at position 35. The HEL4 crystal structure revealed that tryptophan 47 is positioned within a cavity formed by the glycine 35 substitution. When glycine was exchanged for serine at position 35 in DP47d, solubility was significantly enhanced.Citation40 Thus, in both studies, a glycine at position 35 was found to increase solubility by allowing neighboring aromatic side chains to orient in a manner by which their solvent exposure was reduced.
While the aforementioned studies were designed to improve the solution properties of VH domain antibodies, we found that a similar mutation (histidine to alanine) is also beneficial to the full-length mAb, expanding the utility of introducing a small side chain at VH position 35 beyond single domain antibodies. Even though the hydrophobic residues at the VH/VL of IgG are largely buried, this mutation can still improve the mAb's solution behavior. Recently, substitution of aspartate at VH position 35 was identified as a strategy to prevent heat-induced aggregation of antibodies expressed on phage.Citation41 Thus, the location of VH position 35 within the folded antibody structure appears to significantly affect stability by modulating surface hydrophobicity/hydrophilicity.
In the context of the mAb-C studies reported here, the H35A mutation may force W50 to become more buried. However, the fact that W50R does not exhibit an equivalent extent of reduction in RSA compared to that of H35A suggests that the H35A mutation may affect the structural arrangement of other hydrophobic residues in addition to W50R. Comparison of the crystal structures of mAb-C and the H35A mutant could conclusively determine whether the alanine mutation causes rearrangement of W50 or other side chains into more buried conformations. Alternatively, the H35A mutation may affect the overall pairing geometry between the VH and VL in a manner that decreases exposure of aromatic residues. It will also be valuable to evaluate whether small side-chain substitutions at VH position 35 can be beneficial to antibodies other than mAb-C, derived from similar or distinct germlines. Histidine at position 35 is only present in a subset of germlines, so it is unclear at this point if mutation would have a similar effect across structurally diverse antibodies. Finally, the structure of the VL may also be important. The benefit of mutating H35 might only be observed when specific VH and VL chains are paired.
To ensure that the engineered mutations described here did not affect the structural stability of mAb-C, thermal measurements with DSC and DSF were performed, with data highlighting no significant effect on mAb-C structural stability. In addition, evaluation of aggregation and fragmentation propensities under accelerated storage conditions showed no differences between mAb-C and its mutants. This is in contrast to the HEL4/DP47d study, where the introduction of glycine at position 35 caused a decrease in thermostability.Citation40 Thus, while the presence of a small, non-polar side chain at position 35 appears to benefit the solution properties of different antibody formats, the effect on structural stability might be detrimental in the absence of a stabilizing VL domain.
Rheology measurements showed that the mutations that prevented mAb-C RSA also resulted in significant reduction of its viscosity at high concentrations. The two representative VL and VH mutants, L54D and H35A, resulted in an approximately 70% and 75% reduction in mAb-C viscosity, respectively. A report by Sharma et al. suggested that the difference in predicted net charge (at pH 5.5) of the VH and VL sequence correlates with antibody viscosity. Of the analyzed antibodies, those with a high degree of charge asymmetry between the VH and VL had higher viscosity and vice versa.Citation30 The validity of this analysis is based on the presumption that viscosity is driven by electrostatic interactions between the oppositely charged VH and VL domains. Due to the presence of basic amino acids, both the VH and VL of mAb-C are predicted to have a positive net charge and, by this analysis, they are predicted to have low viscosity. While in general, it could be that electrostatic interactions are the major driver of antibody viscosity, our study describes a case where RSA and viscosity are mainly driven by hydrophobic interactions. This case study highlights the varied nature of antibody self-association, and supports the notion that the accuracy of antibody stability predictions are increased by considering the three-dimensional structure of the antibody, as opposed to only the linear Fv sequence.
While it was not the focus of this work, the effect of the identified mutations on antigen binding was also examined. In brief, while the VH mutations decreased binding affinity (KD) significantly (by ∼200-400 fold), the VL mutations reduced KD only minimally (by ∼12–18 fold) compared to mAb-C, which has a sub-nanomolar KD (data not shown). This offers light chain mutants as potential next-generation candidates with improved developability properties. This study therefore highlights the importance in understanding the regions of the antibody structure that compose the binding interface, as each residue would be expected to have a different effect on antibody activity.
High concentration formulations can introduce substantial hurdles during biopharmaceutical development. Understanding the molecular mechanisms that lead to undesired solution behaviors such as RSA and viscosity can inform our ability to engineer designer mAbs with improved high concentration solution properties. The latter can offer a number of advantages, including greater patient convenience, by allowing more robust drug product dosing and presentation. By pushing the boundaries of high concentrations, the designer mAbs could result in reduced frequency of dosing and more compatible molecular and formulation platforms for use with self-administered devices, such as prefilled syringes and auto-injectors. Finally, these molecules could result in a more robust manufacturing process design space where associated risks with high concentrations, such as clogging and fouling of the filters, membranes, and pumps, can be minimized.
Presently, a major challenge to generate these designer mAbs is to determine which residues within an antibody structure promote native protein-protein interactions. The strategy employed here to optimize mAb-C, namely combining experimental data with modeling analyses, addresses this problem while avoiding the pitfalls of purely computational or mathematical-based predictions. In particular utilization of a high resolution technique such as HDX-MS enabled identification of multiple sequence hot spots responsible for high concentration issues observed. While a number of mutations were shown to mitigate reversible self-association and viscosity, a subsection of those residues also maintained comparable binding affinity, presenting themselves as potentially viable designer mAbs. We anticipate that future studies integrating in silico tools, such as spatial aggregation prediction and spatial charge map analysis with structural approaches (e.g., HDX-MS) and high-throughput screening assays (e.g., AC-SINS), will support a Quality by Design approach to generate antibodies with improved high concentration solution properties.Citation31,41
Materials and methods
Antibody generation
A plasmid encoding mAb-C (human IgG1κ) was generated by ligating synthesized DNA gBlocks (IDT Inc., Coralville, IA) encoding codon-optimized VH and VL sequence into an in-house IgG1 mammalian expression vector. Point mutations (e.g., H35A, L54N) were introduced by site-directed mutagenesis using the QuickChange Multi Lightning mutagenesis kit (Agilent Technologies, Santa Clara, CA). All antibody numbering is based on Kabat numbering.Citation31,41 The resulting plasmids were transiently transfected into suspension-adapted HEK293 cells using 293Fectin transfection reagent (Life Technologies, Carlsbad, CA). Cells were grown in FreeStyle 293-F Expression Medium (Life Technologies). The expressed antibodies were purified from clarified and filtered cell supernatant by affinity chromatography using a HiTrap Protein A column (GE Healthcare Life Sciences, Marlborough, MA). Antibody was eluted using Pierce IgG Elution Buffer (ThermoFischer Scientific, Waltham, MA) and neutralized with 1 M Tris, pH 8.0. Antibodies were dialyzed into PBS, pH 7.4 (Life Technologies) and purity (monomer content) was assessed by high performance size-exclusion chromatography; all antibodies were ≥ 98% monomer (Fig. S1).
Affinity capture self-interaction nanoparticle spectroscopy
AC-SINS was performed as described elsewhere.Citation36 Briefly, citrate-stabilized 20 nm gold nanoparticles (Ted Pella Inc., Redding, CA) were combined at a 10:1 ratio with either 0.4 mg/mL ChromePure goat IgG, whole molecule (Jackson ImmunoResearch, West Grove, PA) to yield non-capture nanoparticles, or a 0.4 mg/mL mixture of goat IgG and AffiniPure goat anti-human IgG Fcγ fragment specific antibody (Jackson ImmunoResearch), such that 20% of the antibody mixture is goat IgG and 80% is anti-human IgG Fcγ, to yield capture nanoparticles. Prior to addition of the nanoparticles, both goat IgG and anti-human IgG Fcγ were dialyzed into 20 mM potassium acetate, pH 4.3. After a 1 h incubation at room temperature, the nanoparticles were blocked by addition of 0.1 μM poly(ethyleneglycol) methyl ether thiol (2000 MW, Sigma-Aldrich, St. Louis, MO). The nanoparticles were concentrated by centrifugation (20K × g for 5 min) in siliconized tubes (VWR, Radnor, PA) and re-suspended in 1/10th of the starting volume. To assess self-association, 5.5 μL of either non-capture or capture nanoparticles were combined with 49.5 μL of 50 μg/mL of antibody in PBS, pH 7.4 and in a 96-well, 0.5 mL round-bottom polypropylene plate (Agilent Technologies). For testing crude antibody preparations (Fig. S1), expressed antibody in clarified, filtered cell supernatant from 293 transfected cells was diluted to 50 μg/mL in PBS, pH 7.4. Samples were incubated for 1 h at room temperature, and then 50 μL was transferred to a Nunc 384-well polystyrene plate (ThermoFisher Scientific). Absorbance was measured on a SPECTROstar Nano UV/Vis plate reader (BMG Labtech, Cary, NC) from 490-700 nM. The wavelength of peak absorbance (i.e., plasmon wavelength) was identified using the Find Maximum calculation tool in the MARS data analysis software (BMG Labtech). For each antibody, the normalized plasmon shifts were calculated by subtracting the plasmon wavelength of the non-capture nanoparticles from the capture particles. Results were graphed using Prism (GraphPad, La Jolla, CA).
Capillary isoelectric focusing
Capillary IEF was performed using a PeggySue instrument (ProteinSimple, San Jose, CA). Briefly, antibody was diluted to 4 μg/mL in Sample Diluent (ProteinSimple) and combined with an ampholyte mixture composed of 50% Premix G2 pH 4-9 (ProteinSimple) and 50% Ampholyte pH 8-10.5 (prepared by combining Pharmalyte 8-10.5 (GE Healthcare Life Sciences) with Pre-mix G2 Ampholyte-free separation gradient (ProteinSimple), plus 0.5% N,N,N′,N′-tetramethylenediamine (Amresco, Solon, OH) and fluorescent pI standards (pI 5.5, 7.3, 8.4, and 9.7; ProteinSimple). Detection was performed using horseradish peroxidase-conjugated mouse anti-human IgG Fc (Jackson ImmunoResearch) and Peroxide XDR/Luminol reagents (ProteinSimple). Data analysis was performed using the Compass software (ProteinSimple) and results were graphed using Prism.
Hydrophobic interaction chromatography
For HIC analysis, 50 μg of each sample was analyzed on an Agilent 1200 Series High Pressure Liquid Chromatography instrument and TSKgel Butyl-NPR 4.6 mm ID × 3.5 cm, 2.5 mm column (Tosoh Bioscience, LLC, King of Prussia, PA). Two mobile phase buffers were used in complementary 0-100% gradients: Buffer A: 25 mM Tris-HCl, 1.5 M ammonium sulfate, pH 7.0; Buffer B: 25 mM Tris-HCl, 5% 2-propanol, pH 7.0. Sample flow rate was 1 mL/min with a 20 min total run time. UV detection was performed at A280 nm with a bandwidth of 16 and reference correction at 360 nm with a 100 bandwidth.
High performance size-exclusion chromatography
For HPSEC, 100 μg of each sample were analyzed on an Agilent 1200 series HPLC and TSKgel G3000SWXL column (Tosoh Bioscience LLC, Montgomeryville, PA). The mobile phase used was 100 mM sodium phosphate, pH 6.8. Sample flow rate was 1 mL/min. UV detection was performed at A280 nm. Sample monomer, aggregate, and fragment percentages were calculated based on curve area integration using the HPLC ChemStation software (Agilent). For accelerated stability testing, samples were diluted to 1 mg/mL in PBS, pH 7.4 (Invitrogen) and incubated for 2 weeks at either 4°C or 45°C. After incubation, samples were analyzed by HPSEC as described above. The change in monomer, fragment, and aggregate content was calculated from the difference in each species at 45°C versus 4°C.
Rheology measurements
Viscosity measurements were conducted using a cone-plate geometry of diameter 19.972 mm, 1° angle. A 35 µL sample volume was tested at 4°C with an MCR-301 torsional rheometer (Anton Paar) that detects torque (T) in the range 0.1 µN.m ≤ T ≤ 200 µN.m. Motor adjustment and tool inertia correction were performed before measurements, and the residual T with air was ≤ ± 0.02 µN.m. All measurements were performed at 103 S−1. A hood covered the cone-plate geometry to mitigate evaporation. Six data points were acquired and results were averaged.
Dynamic light scattering
Each sample was prepared over the concentration range of 2–10 mg/mL in PBS buffer, pH 7.2. The hydrodynamic diameter of samples was determined using a high-throughput 384-well plate DynaPro DLS instrument (Wyatt Technology, Santa Barbara, California) equipped with a 633 nm laser. The scattered light was monitored at 173° to the incident beam and autocorrelation functions were generated using a digital autocorrelator. Thirty µL of sample was loaded to each well and the plate was centrifuged at 2000 rpm for 2 min to remove any air bubbles. Samples were run in triplicates, and data were collected using 10 5 s acquisitions per sample. The hydrodynamic diameter was calculated from the diffusion coefficient based on the Stokes-Einstein equation using the method of cumulants. Temperature was controlled using a Peltier-based controller and the hydrodynamic diameter data were collected upon adequate equilibration at 25°C. Finally, viscosity corrections were applied where appropriate (data not shown).
Differential scanning fluorescence
Samples were prepared at a final protein concentration of 0.5 mg/mL in PBS buffer, pH 7.2. SYPRO® Orange was added immediately before the experiment to achieve a final dilution of 5000X relative to the initial stock concentration. Twenty-five µL of sample was added in triplicate to white-walled PCR plates. A Chromo4 Real Time PCR Detector (Bio-Rad, Hercules, CA) was used as a thermal cycler, and the fluorescence emission was detected using the software's custom dye calibration routine. The PCR plate containing the test samples was subjected to a temperature ramp from 20°C to 90°C in increments of 0.2°C with 10 s pauses after each temperature increment. The total assay time was approximately 3 h. The Tm was calculated by the software using a mathematical second derivative method to calculate the inflection point of the curve. The reported Tm is an average of two measurements.
Differential scanning calorimetry
DSC studies were performed using a VP capillary DSC system (MicroCal, Piscataway, NJ). The samples were analyzed at concentration of 1.0 mg/mL in PBS buffer, pH 7.2. The change in the heat capacity (Cp) was measured as the samples were heated from 20°C to 90°C at a rate of 90°C/h. For all samples, the relevant PBS buffer blank was used as a reference. Normalized Cp data were corrected for buffer baseline. Data analysis was performed using OriginLab software (OriginLab Corporation, Northampton, Massachusetts). The Tm was calculated from raw data fit using OriginTM scientific plotting software and the Levenberg-Marquardt nonlinear least square method.
In silico homology modeling
The homology model of mAb-C variable domain was generated using BioLuminate v.1.9 (Schrödinger, New York, NY). For framework regions (based on Chothia definition), template coordinates from PDB structure 4JDV were chosen based on a sequence homology search.Citation42 The identity between the mAb-C and VH and VL template framework sequence is 99% and 77%, respectively, with similarity of 100% and 94%; similarity is calculated by dividing the number of matching residues according to BLOSUM62 matrix scoring by the total number of residues.Citation43 By Chothia definition H35 and W50 are within the framework region 2, as opposed to CDR1 and CDR2, respectively, if using Kabat numbering, as indicated in . CDRs were modeled using the BioLuminate Basic Loop Model function. The identity and similarity of the CDR loops were as follows (% identity, % similarity): H1 (100, 100), H2 (67, 100), H3 (25, 31), L1 (82, 82), L2 (71, 100), L3 (67, 100). The BioLuminate Aggregation Surface tool was used to identify residues predicted to contribute to aggregation patches; default tool parameters were applied. Solvent accessible surface area of side chains was calculated using the Residue Analysis tool. All antibody structure images were generated using BioLuminate and PyMol (Schrödinger).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Figure_1_2.zip
Download Zip (351.9 KB)Acknowledgment
The authors would like to thank Nancy Craighead for her critical review of this manuscript.
References
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010; 9:767-74; PMID:20811384; http://dx.doi.org/10.1038/nrd3229
- Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci 2007; 96:1-26; PMID:16998873; http://dx.doi.org/10.1002/jps.20727
- Liu J, Nguyen MD, Andya JD, Shire SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci 2005; 94:1928-40; PMID:16052543; http://dx.doi.org/10.1002/jps.20347
- Mason BD, Zhang L, Remmele RL, Jr, Zhang J. Opalescence of an IgG2 monoclonal antibody solution as it relates to liquid-liquid phase separation. J Pharm Sci 2011; 100:4587-96; PMID:21638285; http://dx.doi.org/10.1002/jps.22650
- Nishi H, Miyajima M, Nakagami H, Noda M, Uchiyama S, Fukui K. Phase separation of an IgG1 antibody solution under a low ionic strength condition. Pharm Res 2010; 27:1348-60; PMID:20401522; http://dx.doi.org/10.1007/s11095-010-0125-7
- Yadav S, Liu J, Shire SJ, Kalonia DS. Specific interactions in high concentration antibody solutions resulting in high viscosity. J Pharm Sci 2010; 99:1152-68; PMID:19705420; http://dx.doi.org/10.1002/jps.21898
- Yadav S, Shire SJ, Kalonia DS. Factors affecting the viscosity in high concentration solutions of different monoclonal antibodies. J Pharm Sci 2010; 99:4812-29; PMID:20821382; http://dx.doi.org/10.1002/jps.22190
- Laue T. Proximity energies: a framework for understanding concentrated solutions. J Mol Recognit 2012; 25:165-73; PMID:22407980; http://dx.doi.org/10.1002/jmr.2179
- Kanai S, Liu J, Patapoff TW, Shire SJ. Reversible self-association of a concentrated monoclonal antibody solution mediated by Fab-Fab interaction that impacts solution viscosity. J Pharm Sci 2008; 97:4219-27; PMID:18240303; http://dx.doi.org/10.1002/jps.21322
- Burckbuchler V, Mekhloufi G, Giteau AP, Grossiord JL, Huille S, Agnely F. Rheological and syringeability properties of highly concentrated human polyclonal immunoglobulin solutions. Eur J Pharm Biopharm 2010; 76:351-6; PMID:20719247; http://dx.doi.org/10.1016/j.ejpb.2010.08.002
- Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci 2004; 93:1390-402; PMID:15124199; http://dx.doi.org/10.1002/jps.20079
- Brems DN, Alter LA, Beckage MJ, Chance RE, DiMarchi RD, Green LK, Long HB, Pekar AH, Shields JE, Frank BH. Altering the association properties of insulin by amino acid replacement. Protein Eng 1992; 5:527-33; PMID:1438163; http://dx.doi.org/10.1093/protein/5.6.527
- Clodfelter DK, Pekar AH, Rebhun DM, Destrampe KA, Havel HA, Myers SR, Brader ML. Effects of non-covalent self-association on the subcutaneous absorption of a therapeutic peptide. Pharm Res 1998; 15:254-62; PMID:9523312; http://dx.doi.org/10.1023/A:1011918719017
- Alford JR, Kendrick BS, Carpenter JF, Randolph TW. High concentration formulations of recombinant human interleukin-1 receptor antagonist: II. Aggregation kinetics. J Pharm Sci 2008; 97:3005-21; PMID:17924426; http://dx.doi.org/10.1002/jps.21205
- Arora J, Hickey JM, Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Weis DD, Volkin DB. Hydrogen exchange mass spectrometry reveals protein interfaces and distant dynamic coupling effects during the reversible self-association of an IgG1 monoclonal antibody. MAbs 2015; 7:525-39; PMID:25875351; http://dx.doi.org/10.1080/19420862.2015.1029217
- Barthelemy PA, Raab H, Appleton BA, Bond CJ, Wu P, Wiesmann C, Sidhu SS. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem 2008; 283:3639-54; PMID:18045863; http://dx.doi.org/10.1074/jbc.M708536200
- Esfandiary R, Hayes DB, Parupudi A, Casas-Finet J, Bai S, Samra HS, Shah AU, Sathish HA. A systematic multitechnique approach for detection and characterization of reversible self-association during formulation development of therapeutic antibodies. J Pharm Sci 2013; 102:3089-99; PMID:23794522; http://dx.doi.org/10.1002/jps.23654
- Esfandiary R, Parupudi A, Casas-Finet J, Gadre D, Sathish H. Mechanism of reversible self-association of a monoclonal antibody: role of electrostatic and hydrophobic interactions. J Pharm Sci 2015; 104:577-86; PMID:25407315; http://dx.doi.org/10.1002/jps.24237
- Nishi H, Miyajima M, Wakiyama N, Kubota K, Hasegawa J, Uchiyama S, Fukui K. Fc domain mediated self-association of an IgG1 monoclonal antibody under a low ionic strength condition. J Biosci Bioeng 2011; 112:326-32; PMID:21783411; http://dx.doi.org/10.1016/j.jbiosc.2011.06.017
- Wu SJ, Luo J, O'Neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel 2010; 23:643-51; PMID:20543007; http://dx.doi.org/10.1093/protein/gzq037
- Yadav S, Laue TM, Kalonia DS, Singh SN, Shire SJ. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol Pharm 2012; 9:791-802; PMID:22352470; http://dx.doi.org/10.1021/mp200566k
- Yadav S, Sreedhara A, Kanai S, Liu J, Lien S, Lowman H, Kalonia DS, Shire SJ. Establishing a link between amino acid sequences and self-associating and viscoelastic behavior of two closely related monoclonal antibodies. Pharm Res 2011; 28:1750-64; PMID:21626060; http://dx.doi.org/10.1007/s11095-011-0410-0
- Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic vs. electrostatic inter-molecular interactions. MAbs 2015; 7:212-30; PMID:25559441; http://dx.doi.org/10.4161/19420862.2014.985504
- Bethea D, Wu SJ, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O'Neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel 2012; 25:531-7; PMID:22915597; http://dx.doi.org/10.1093/protein/gzs047
- Teplyakov A, Obmolova G, Wu SJ, Luo J, Kang J, O'Neil K, Gilliland GL. Epitope mapping of anti-interleukin-13 neutralizing antibody CNTO607. J Mol Biol 2009; 389:115-23; PMID:19361524; http://dx.doi.org/10.1016/j.jmb.2009.03.076
- Chow CK, Allan BW, Chai Q, Atwell S, Lu J. Therapeutic Antibody Engineering To Improve Viscosity and Phase Separation Guided by Crystal Structure. Mol Pharm 2016; 13:915-23; PMID:26849155; http://dx.doi.org/10.1021/acs.molpharmaceut.5b00817
- Tomar DS, Kumar S, Singh SK, Goswami S, Li L. Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. MAbs 2016; 8:216-28; PMID:26736022; http://dx.doi.org/10.1080/19420862.2015.1128606
- Chennamsetty N, Helk B, Voynov V, Kayser V, Trout BL. Aggregation-prone motifs in human immunoglobulin G. J Mol Biol 2009; 391:404-13; PMID:19527731; http://dx.doi.org/10.1016/j.jmb.2009.06.028
- Obrezanova O, Arnell A, de la Cuesta RG, Berthelot ME, Gallagher TR, Zurdo J, Stallwood Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. MAbs 2015; 7:352-63; PMID:25760769; http://dx.doi.org/10.1080/19420862.2015.1007828
- Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A 2014; 111:18601-6; PMID:25512516; http://dx.doi.org/10.1073/pnas.1421779112
- Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs 2016; 8:43-8; PMID:26399600; http://dx.doi.org/10.1080/19420862.2015.1099773
- Hasegawa H, Woods CE, Kinderman F, He F, Lim AC. Russell body phenotype is preferentially induced by IgG mAb clones with high intrinsic condensation propensity: relations between the biosynthetic events in the ER and solution behaviors in vitro. MAbs 2014; 6:1518-32; PMID:25484054; http://dx.doi.org/10.4161/mabs.36242
- Jacobs SA, Wu SJ, Feng Y, Bethea D, O'Neil KT. Cross-interaction chromatography: a rapid method to identify highly soluble monoclonal antibody candidates. Pharm Res 2010; 27:65-71; PMID:19911257; http://dx.doi.org/10.1007/s11095-009-0007-z
- Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs 2015; 7:752-8; PMID:25961854; http://dx.doi.org/10.1080/19420862.2015.1048410
- Sule SV, Dickinson CD, Lu J, Chow CK, Tessier PM. Rapid analysis of antibody self-association in complex mixtures using immunogold conjugates. Mol Pharm 2013; 10:1322-31; PMID:23383873; http://dx.doi.org/10.1021/mp300524x
- Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs 2014; 6:483-92; PMID:24492294; http://dx.doi.org/10.4161/mabs.27431
- Giudicelli V, Chaume D, Lefranc MP. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res 2005; 33:D256-61; PMID:15608191; http://dx.doi.org/10.1093/nar/gki010
- Gagnon P, Mayes T, Danielsson A. An adaptation of hydrophobic interaction chromatography for estimation of protein solubility optima. J Pharm Biomed Anal 1997; 16:587-92; PMID:9502154; http://dx.doi.org/10.1016/S0731-7085(97)00153-2
- Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. MAbs 2014; 6:852-8; PMID:24751784; http://dx.doi.org/10.4161/mabs.28693
- Jespers L, Schon O, James LC, Veprintsev D, Winter G. Crystal structure of HEL4, a soluble, refoldable human V(H) single domain with a germ-line scaffold. J Mol Biol 2004; 337:893-903; PMID:15033359; http://dx.doi.org/10.1016/j.jmb.2004.02.013
- Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A 2012; 109:10879-84; PMID:22745168; http://dx.doi.org/10.1073/pnas.1202866109
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; http://dx.doi.org/10.1016/0022-2836(87)90412-8
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 1992; 89:10915-9; PMID:1438297; http://dx.doi.org/10.1073/pnas.89.22.10915