
ABSTRACT
Nivolumab is a therapeutic fully human IgG4 antibody to programmed death 1 (PD-1). In this study, a nivolumab biosimilar, which was produced in our laboratory, was analyzed and characterized. Sequence variants that contain undesired amino acid sequences may cause concern during biosimilar bioprocess development. We found that low levels of sequence variants were detected in the heavy chain of the nivolumab biosimilar by ultra performance liquid chromatography (UPLC) and tandem mass spectrometry. It was further identified with UPLC-MS/MS by IdeS or trypsin digestion. The sequence variant was confirmed through addition of synthetic mutant peptide. Subsequently, the mixing base signal of normal and mutant sequence was detected through DNA sequencing. The relative levels of mutant A424V in the Fc region of the heavy chain have been detected and demonstrated to be 12.25% and 13.54%, via base peak intensity (BPI) and UV chromatography of the tryptic peptide mapping, respectively. A424V variant was also quantified by real-time PCR (RT-PCR) at the DNA and RNA level, which was 19.2% and 16.8%, respectively. The relative content of the mutant was consistent at the DNA, RNA and protein level, indicating that the A424V mutation may have little influence at transcriptional or translational levels. These results demonstrate that orthogonal state-of-the-art techniques such as LC- UV- MS and RT-PCR should be implemented to characterize recombinant proteins and cell lines for development of biosimilars. Our study suggests that it is important to establish an integrated and effective analytical method to monitor and characterize sequence variants during antibody drug development, especially for antibody biosimilar products.
Abbreviations
| PD-1 | = | programmed death 1 |
| UPLC | = | Ultra-performance liquid chromatography |
| Q-Tof | = | quadrupole-time of flight |
| MS | = | mass spectrometry |
| ESI | = | electrospray ionization |
| DTT | = | dithiothreitol |
| FA | = | formic acid |
| PTMs | = | post-translational modifications |
| RT-PCR | = | Real time-Polymerase Chain Reaction |
| SNP | = | single nucleotide polymorphism |
| CID | = | collision induced dissociation |
| BPI | = | Base Peak Intensity |
Introduction
The field of recombinant monoclonal antibody (mAb) therapeutics has developed rapidly. So far, more than 30 therapeutic mAbs have been approved by the US Food and Drug Administration for the treatment many kinds of diseases, such as cancers and immune-mediated disorders.Citation1
The process of translating recombinant DNA into the desired protein is critical to production of human proteins in Escherichia coli, yeast, or mammalian cells, including Chinese hamster ovary (CHO) cells.Citation2 However, in addition to expressing the product of interest, cells may also generate an array of sequence variants, which regarded as “product-related impurities.”Citation3 Sequence variants, which result from unintended amino acid substitutions, are protein isoforms containing undesired amino acid sequences that may cause concern during the production of mAbs and other therapeutic proteins.Citation2-7 Therefore, it is necessary to detect potential sequence variants to confirm the purity and safety of the product during clone selection and bioprocess development. To ensure product safety, efficacy and consistency, manufacturers who produce the therapeutic mAbs are increasingly using advanced analytical methods and technologies during product characterization and development.
The mechanisms for the generation of the sequence variant can be grouped into the 3 broad categories: 1) mutations at the DNA level; 2) mistranslation or improper tRNA acylation by either nonsense read-through or misreading at the level of transcription or translation; and 3) miscleavage during the post-translational processing.Citation7,8 For recombinant proteins, most sequence variants can be observed at the DNA level. For example, 3 variants have been verified by polymerase chain reaction (PCR) analysis to be a genomic nucleotide mutation at the DNA level.Citation6
Although peptide mapping with UV detection is still a simple but powerful method to detect sequence variants in mAbs, the sequence variants can only be detected while the percentage is 1 to 5% or above. Because sequence variants in the therapeutic mAbs are generally very rare, detecting and characterizing sequence variants is a substantial challenge.Citation7 With the improvement of technology and analytical methods, small amounts of sequence variant in the recombinant human mAbs can be detected and characterized. Several sequence variants have been described.Citation2,9 Yang et al developed a method to detect sequence variants that used complementary enzymes to produce multiple peptide mappings. The peptides are separated by reverse phase-ultra performance liquid chromatography (RP-UPLC) coupled with an accurate, high-resolution and sensitivity mass spectrometer. The acquired mass spectra data are imported to a software (Mascot Error Tolerant Search; ETS) to automate searches of a known protein sequence database.Citation7 Due to the sensitivity of the technology, this method has become useful to detect sequence variants, especially when only trace amounts of sequence variants are present. For example, LTQ-Orbitrap can detect sequence variants with levels as low as 0.01%.Citation2 Sequence variants can be detected and characterized by a series of methods. Wade et al. analyzed hemoglobin sequence variants through a high resolution analytical method that included chromatographic and electrophoretic techniques, DNA and MS analysis.Citation10 Dorai et al. used the method of quantitative polymerase chain reaction (QPCR), which can be used to help rapidly identify single nucleotide polymorphism (SNP) and detect DNA sequence variation.Citation11
Sequence variants may affect the physicochemical and biological properties (e.g., activity, stability, safety) of protein therapeutics.Citation12-15 However, compared with the native protein, these sequence variants may contain only one or a few amino acid substitutions, which makes them difficult to separate from the desired product during downstream purification.Citation16 Therefore, sequence variants in mAbs produced for therapeutic use should be strictly monitored and detected through-out the development process.
Nivolumab, a fully human IgG4 programmed death 1 (PD-1) immune-checkpoint–inhibitor antibody, disrupts the negative signal that regulates T-cell activation and proliferation through binding to PD-1 on activated immune cells to selectively block the interaction of the PD-1 receptor with its 2 known programmed death ligands, PD-L1 and PD-L2. The heavy chain constant region of nivolumab is a human IgG4 isotype with an S228P mutation, which replaces a serine residue in the hinge region with the proline residue found at the corresponding position in IgG1.Citation17,18 Administered intravenously, nivolumab was first approved in 2014 for the treatment of unresectable malignant melanoma in Japan.Citation19 Nivolumab is now approved in numerous countries for a variety of cancer types.Citation20 In an open-label, randomized, Phase 3 study involving patients with ipilimumab-refractory melanoma, nivolumab was associated with a higher rate of objective response than chemotherapy (32% vs. Eleven%).Citation21
A nivolumab biosimilar candidate, an IgG4 mAb, was expressed in CHO cells in our laboratory. Here, we report the observation of a Ala to Val sequence variant at position of 424 in the Fc region of the heavy chain of the recombinant antibody. The variant was detected by combining UPLC-UV-MS/MS by IdeS and trypsin digestion. The A424V sequence variant was further confirmed by adding a synthetic mutant peptide that replaced Ala with Val in the native peptide and cell line genomic DNA sequencing. Subsequently, RT-PCR was also used to quantitate the sequence variant at the DNA and RNA level. This study proved that it is necessary to establish an integrated and effective analytical method to detect and analyze sequence variants in the antibody drug development, particularly for biosimilars. These methods include subunit analysis by LC-MS, middle-down mass spectrometry following IdeS digestion and peptide mapping analysis.
Results
Mass spectral analysis of reduced heavy chain
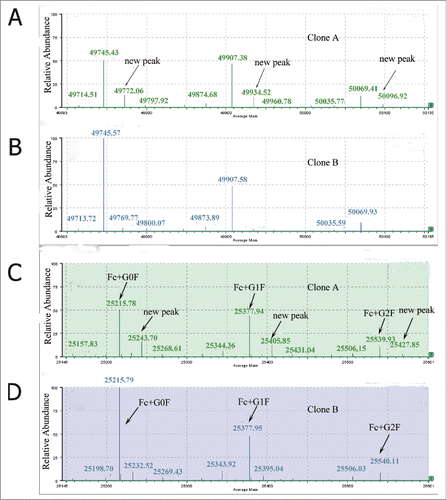
The nivolumab biosimilar antibody was expressed in CHO cells in our laboratory and 2 final seed clones were selected for further developments. The two seed clones were clone A: (23A-C8-Agms-C1-B5) and clone B: (18C-G6-Agms-A1-C3), respectively. The recombinant antibody was purified by affinity chromatography on Protein A-Sepharose (GE Healthcare) and separated into free light and heavy chain by breaking the inter-chain disulfide linkages.Citation22 Two prominent peaks of reduced antibody from the 2 seed clones were observed in the RP-UPLC profile (Fig. S1). There was no obvious difference between the 2 clones on the total ion chromatography (TIC) response. The light chain mass of the 2 clones is consistent and the average mass of the light chain is 23,370 Da. The deconvoluted mass spectra of the heavy chain of the 2 clones are shown in . The reduced protein level of carbohydrate heterogeneity was observed for the mAb where N-linked glycans accounted for the main observed heterogeneity in the deconvoluted heavy chain mass data. The main molecular weight of the heavy chain was 49,745 Da, which included the glycan (G0F) on the heavy chain. The other 2 main molecular weight of the heavy chain were 49,907 Da and 50,069 Da, which correspond to heavy chain with G1F and G2F, respectively. Clone A revealed a new species with a mass shift of approximately +28 Da from the G0F and G1F glycan forms (). The new molecular weight was not observed in clone B (). This suggested that a sequence variant or some new modification may exist in the heavy chain of clone A. Thus, further identification of the new peaks and further research at the molecular level were urgently needed.
Figure 1. Deconvoluted mass spectra of the reduced antibody heavy chain(A and B) and Fc fragment generated by IdeS (C and D).
Identification of the sequence variant by Ides digesting
Ides digesting of the heavy chain of both clone A and clone B were carried out to identify the region (Fd or Fc region) in which the sequence variant or modification occurs. IdeS is that cysteine protease cleaves at the hinge region of all IgG subclasses. Application of IdeS has been demonstrated in many literature reports on IgG1 and IgG2 molecules, as well as Fc fusion proteins.Citation23-28 Three predominant reduced peaks were observed in the chromatography by IdeS digestion, which represent the 3 released domains −Fc/2, LC and Fd, respectively. The molecular weights of the LC and Fd domains of the clone A were consistent with that of clone B. The deconvoluted mass spectra of the Fc/2 domain of both clones were showed in . There were 3 main peaks observed in both clone A and B, respectively. The heterogeneity of Fc/2 domain resulted from the N-glycan forms. There was no obvious difference between the 2 clones in the main 3 peaks, which representing the Fc/2 domain with G0F, G1F and G2F glycan forms. Visual comparison of the profiles showed a new peak with a mass shift of +28 Da in clone A () that was not observed in clone B (). The results were consistent with the change that we found in the reduced heavy chain (). These data demonstrated that the sequence variant or modification was located in the Fc domain of the heavy chain in clone A. Subsequently, additional details on modification or sequence variant were investigated using peptide mapping.
Identification of the Ala to Val variant by tryptic peptide mapping
Tryptic peptide mapping was a useful technique for characterizing the proteins amino acid sequences and their post-translational modifications (PTMs).Citation29 The peptide map method could be used as an alternative, or it could be combined with an oligosaccharide mapping method to acquire site-specific information for proteins with multiple modification sites. Using a data acquisition C18 LC-MS with alternate changeable energy full scanning (LC-MSE) was useful for peptide mapping.Citation30 Unlike LC-UV/MS peptide mapping methods, MSE acquired exclusive fragments data for all precursors. The LC-MSE data of a proteolytic digest are collected throughout the entire LC-MS experiment, preserving the chromatographic profile of all the detected peptides and their associated product ions. Product ion information is obtained from all the isotopes and charge states of any given precursor peptide as they are simultaneously fragmented. LC-MSE can be used for analyzing peptide maps of the protein digest.Citation31 More than 95% sequence coverage and good analytical reproducibility were reached in replicated analyses of antibody digestion. Consequently, peptide mapping was used to study protein sequences variant and PTM diffrences between the biosimilar and the reference proteins.Citation32
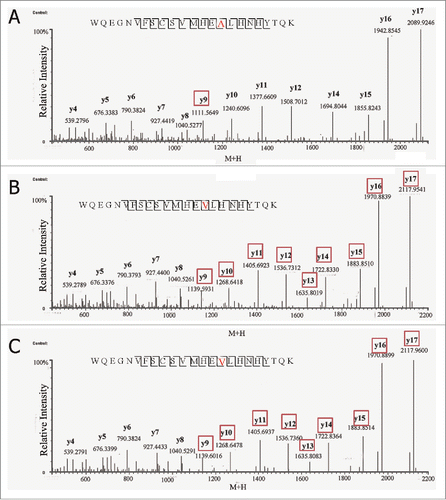
The tryptic peptide mapping chromatographic profiles () of the 2 clones were nearly consistent except for a new peak eluting at 49.10 min for clone A (). The extracted mass spectrum showed that the m/z of the peak was 708.81. The mass of the new tryptic peptide is 2,831.21 Da, and it did not match the desired antibody sequence according to the data analyzed by Biopharmalynx 1.3.3.Citation33 This molecular weight was +28 Da heavier than the T40 tryptic peptide of the heavy chain (WQEGNVFSCSVMHEALHNHYTQK, m/z 701.56). The MS/MS data analysis by collision-induced dissociation (CID) of the 2 peptides is shown in . Compared with the MS/MS spectrum of T40 tryptic peptide of clone B, clone A showed no mass shift in y5 to y8 ions, and a mass shift of +28 Da showed in y9 ion (). The proposal of the replacement of Ala by Val was offered as a result of the increased mass of +28 Da. This result should be further confirmed.
Figure 2. The tryptic peptide maps of Clone A and Clone B. The new peak (see arrow) at 49.10 min in Clone A is absent from Clone B.
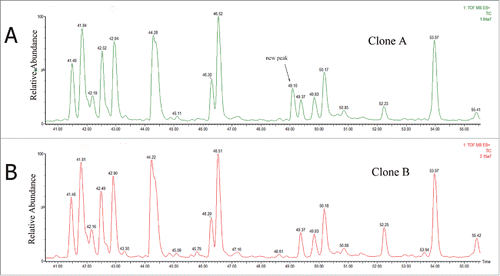
Figure 3. MS/MS spectra of precursor ions at (A) m/z of 701.56 for native peptide WQEGNVFSCSVMHEALHNHYTQK; and (B) m/z of 708.81 for variant peptide; and (C) m/z of 708.81 for synthetic peptide WQEGNVFSCSVMHEVLHNHYTQK.
Confirmation of Ala to Val substitution with synthetic peptides
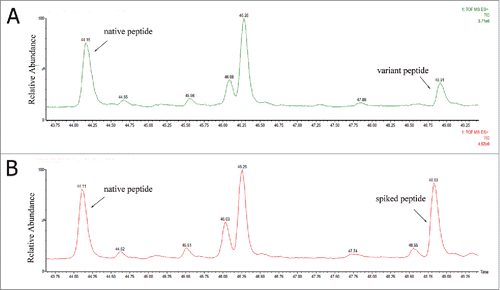
The synthetic peptide WQEGNVFSCSVMHEVLHNHYTQK, which was used for the identification of a Ala to Val sequence variant, was spiked into the clone A tryptic digests after alkylation. The chromatographs of both the synthetic peptide and the variant peptide in the spiked sample digest are shown in . The peak area increased dramatically after the sample was spiked with the synthetic peptide, and the retention time was almost the same. However, the retention time of the native peptide was earlier than the variant peptide, which was consistent with increased hydrophobicity when Ala was replaced by a Val residue. In , the variant peptide and corresponding synthetic peptide showed the same CID MS/MS spectra. These results again demonstrated that a variant peptide was present in clone A, and the assignment of the Ala to Val sequence variant was correct.
Figure 4. Total ion chromatogram of native peptide, WQEGNVFSCSVMHEALHNHYTQK and variant peptide, WQEGNVFSCSVMHEVLHNHYTQK in (A) non-spiked antibody tryptic digest and (B) spiked in WQEGNVFSCSVMHEVLHNHYTQK synthetic peptide.
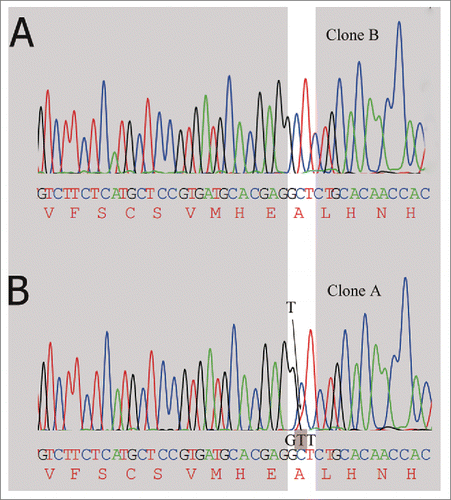
DNA sequencing confirmed the single base-pair mutation in GCT (Ala) to GTT (Val)
cDNA generated from clone A and the control clone B were subjected to DNA sequencing from both ends of the heavy chain. Sequencing of the PCR products generated 1290 base pairs of sequence data. Compared to the expected sequence, no differences were observed in the heavy chain sequence generated from clone B (), but the nucleobase thymine (T) signal curve of clone A showed a dissymmetry and the DNA sequencing electropherogram () for clone A showed mixed bases (C and T) at the opening reading frame, which suggested a fraction of the mRNA molecules corresponding to the cDNA were mutated from a C to a T. The presence of a mixed base results in a mutation of wild-type alanine (GCT) to valine (GTT). Thus, a small part of the codon coding for Ala were turned into a codon coding for Val. The DNA sequencing results were confirmed with repeated reactions, and all the sequencing data indicated a mutation in the clone A sequences, but not in the control clone B. This finding was also in agreement with the results from mass spectra data, where a +28 Da sequence variant was obtained from clone A.
Figure 5. Detection of alanine codon (GCT) change to valine codon (GTT) in mRNA encoding the high chain by DNA sequencing. (A) The clone A has no mutation at the highlighted position, only the expected nucleotide C. (B) Small amount of mutated T (shaded in the trace) is detected along with the wild type nucleotide C.
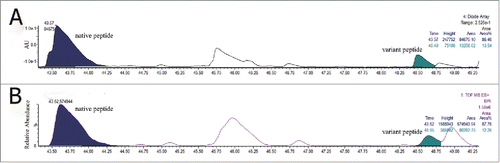
Relative quantitation of Ala to Val variants by LC–MS, UV
The amount of Ala to Val variant (WQEGNVFSCSVMHEVLHNHYTQK) in clone A was calculated by the peak area of their BPI and 214 nm UV chromatography (). The percentage of the peak area for the A424V sequence variant in clone A calculated from the 2 methods was 12.25% and 13.54%, respectively. The results demonstrated that the level of Ala to Val sequence variant was similar between the 2 quantitative methods.
Figure 6. Expanded UV (A) and BPI (B) of the variant peptide and native peptide of Clone A trypsin digested peptide map.
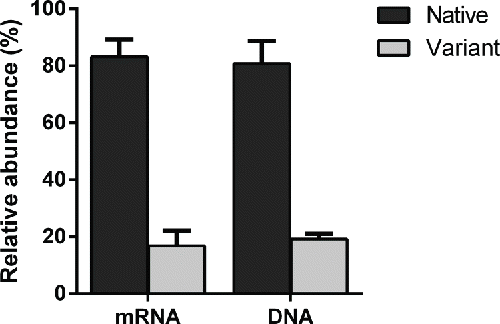
Relative quantitation of Ala to Val variants by RT-PCR and DNA sequencing
The genomic DNA and cDNA generated in both clone A and B were submitted to real-time PCR for the relative quantitation of Ala to Val variants, and the endogenous reference gene GAPDH was used for normalizing input DNA. In clone A, the relative amounts of native peptide mRNA were 83.2% compared with 16.8% of variants, and, at the genomic DNA level, the native were 80.8% while the variants were 19.2%. In clone B, no variant signal was detected in either the mRNA or genomic DNA levels (). The RT-PCR results were confirmed by repeated reactions.
Figure 7. The relative abundance of native and variant peptide at both gene level and transcription level in clone A.
Discussion
Nivolumab is a fully human IgG4 PD-1 antibody that binds to human and cynomolgus PD-1 with high affinity and blocks the interaction of PD-1 with both PD-L1 and PD-L2 ligands. It has been approved for use in melanoma and non-small-cell lung cancer in Japan, and it is also approved and marketed in the United States and Europe. Following patent expirations for brand products, biosimilars may in turn be approved and marketed. Biosimilar manufacturers should be vigilant regarding problems that may occur during the process of development, such as the sequence variance in the therapeutic mAb. In the early stage of the biosimilar development, we have used various methods to characterize the product, such as size-exclusion chromatography, cation ion exchange chromatography, hydrophobic chromatography and reversed phase chromatography. However, little difference between clone A and clone B was observed using these techniques (shown in Fig. S1). Therefore, great attention should be paid in monitoring and detecting potential sequence variant during the early development of antibody biosimilar products. Establishing an effective and sensitive method of detection is very important. Orthogonal state-of-the-art techniques such as LC-UV-MS should be implemented in the early stage of biosimilar development. Although it is difficult to assess the possible adverse reaction of the sequence variant in humans due to minor amounts of those variants, the potential immunogenicity and safety risks caused by the presence of sequence variants may be of concern.
The effects caused by sequence variant is likely to be variable because of the great diversity of possible mutation positions. For example, if the mutation position of the sequence variant is located in the complementarity-determining region of the light chain, it may affect the binding affinity.Citation6 A sequence variation located in the Fc region of the heavy chain may increase the immune response as a result of allo-immunization rather than affect the binding affinity.Citation34,35 For example, Harris et al. found a Y376Q variant in the heavy chain of an anti-human epidermal growth factor receptor-2 antibody expressed in CHO cells.Citation3 Zhang et al reported the mutation of TAA (stop codon) to GAA, which caused light chain extension.Citation36 Dorai et al. reported that a Phe to Leu sequence variant was observed in the recombinant peptide-antibody fusion protein, expressed in CHO cells.Citation11 In this study, due to the A424V mutation located at the C-terminal, the sequence variant may affect the function of the Fc region and cause unanticipated side effects.
Removing sequence variants found during biosimilar antibody development is a way to reduce the possibility of adverse reactions. Due to the little amounts of sequence variant and the similar properties with the normal therapeutic mAb, it is extremely difficult to remove the sequence variants during purification. The best way to prevent their occurrence is by monitoring the sequence variants in the initial phase of development. Monitoring and analyzing unintended amino acid substitutions plays an important role in ensuring the safety, efficacy and quality of biosimilar products. Therefore, the integrated analytical method should be implemented in order to detect the sequence variants in a timely fashion, and minimize such potential errors in the early-stage development of biosimilar product.
Since it is difficult to detect and quantify random sequence variations using conventional analytical characterization methods when the level of the sequence variant is low, one or more effective and sensitive analytical methods should be developed to solve the problems. With the wide application of accurate and sensitive mass spectrometry, it is likely to detect the trace sequence variants, which we have known before as low as the 0.01% level. However, peptide mapping with UV detection is still a simple yet powerful method to detect sequence variants in therapeutic mAbs produced from different clones when the mutation rate is higher than 1%.
The A424V variant levels obtained from the 2 detection methods are in close agreement with each other. There is only a little difference in the level of sequence variance between BPI and UV chromatography. The peak resolution in the BPI trace was slightly poorer, perhaps because the UV and LC-MS detectors were connected in series, in that order, and there was a small amount of diffusional band-broadening that occurred in the tubing connecting the 2 detectors. This result may also be caused by the differences in mass spectrometer ionization efficiency between native and mutation peptide.
RT-PCR can rapidly identify SNPs and detect DNA sequence variation. The mutant rate of clone A at the level of DNA and RNA can be quantified by RT-PCR. Through comparing the mutant rate at the level of DNA, RNA and protein, the difference between them was found to be marginal. This variety may result from effects of transcription and translation of the sequence variant. Our study provided a better understanding of the sequence variation mechanisms: the A424V was generated due to DNA mutation (SNP), not at the mRNA transcription step or due to an amino acid misincorporation. Using the integrate analysis methods to detect sequence variant, the Ala to Val sequence variant was observed in the Fc region of the heavy chain of the nivolumab biosimilar candidate by our lab through LC-MS peptide mapping with tandem mass spectrometry, but nivolumab (innovator product) does not have the same sequence variant and no other sequence variants were observed using this approach.
Materials and methods
Materials
We purchased the innovator's drug from Bristol-Myers Squibb and found the sequence information in the WHO Drug Information website (http://www.who.int/medicines/publications/druginformation/en/). Then, we confirmed the sequence through LC-MS/MS. The light chain and heavy chain genes obtained by total gene synthesis were cloned into eukaryotic vectors. Then, the light chain and heavy chain expression vectors were co-transfected into CHO cells. The nivolumab biosimilar candidate was manufactured in our lab. It was expressed in CHO cell lines and purified by affinity chromatography on Protein A-Sepharose (GE Healthcare), then desalted into PBS at 4°C. Peptide WQEGNVFSCSVMHEVLHNHYTQK was synthesized at Chinese Peptide Co., Ltd. Acetonitrile (LC/MS grade) was purchased from Fisher Scientific (USA); sodium iodide (NaI, purity ≥ 99.5%) and fibrinopeptide (GFP, HPLC grade, purity ≥ 99.5%) were all purchased from Sigma–Aldrich (St. Louis, MO). Dithiothreitol (DTT), iodoacetamide (MIA), and formic acid (FA, MS grade) were obtained from Fluka (GER). High-purity nitrogen (purity > 99.9 %) was purchased from Shanghai YiZhi Cold Engineering System Co., LTD; high purity argon (purity > 99.9 %) was purchased from Shanghai MAO Love Purifying Gas Co., LTD. Trypsin and IdeS were made in our laboratory.
Instrumentation
Separations were performed on ACQUITY UPLC®I-Class System equipped with a binary gradient pump, autosampler, temperature-controlled column compartment and a TUV detector (Waters, Milford, MA). All MS measurements were implemented on a Xevo G2-S system equipped with an ESI source (Waters, Milford, MA). The system was controlled by MassLynx 4.1 (Waters, Milford, MA). The analytic software is the Biopharmalynx 1.3 (Waters) with MaxEnt. The real-time PCR was performed on StepOnePlus System (Applied Biosystems, CA, USA).
Mass spectral data acquisition for reduced mAb
Antibody samples were incubated in a buffer of 50 mM NH4HCO3 solution (pH 8.2) containing 50 mM DTT at 37°C for 10 min. The final reduced samples were centrifuged at 17,000 g for 10 min, and the supernatant was transferred to a clean EP tube for LC–MS/MS analysis by reverse-phase LC-MS, the mobile phase A was water with 0.1% formic acid, while mobile phase B was acetonitrile with 0.1% formic acid and performed on a Waters C4 Column (2.1 × 50 mm, 1.7 µm) (Waters, Milford, MA) using a linear gradient (3–9 min, 25–35% B), and equilibrated with 20% mobile phase B with the column temperature of 60°C. The flow rate was maintained at 0.40 mL/min. Mass spectrometric analysis was performed on a Waters Xevo G2-S Q-TOF MS system with a positive ion mode. The desolvation gas and source temperature were set to 350°C and 120°C. The capillary and cone voltages were set at 3,000 and 40 V. The m/z scan range was set to 500–3,000. The system was controlled by MassLynx 4.1. The deconvolution of ESI mass spectra of reduced antibody samples were analyzed by Biopharmalynx 1.3.3 using MaxEnt 1 algorithm.
IdeS digesting analysis
The antibody samples were digested with IdeS (1:50 w/w) at 37°C for 30 min, and then IdeS digests were incubated with 50 mM DTT at 37°C for 10 min. The final IdeS digests were centrifuged at 17,000 g for 10 min, and the supernatant was transferred to a clean EP tube for LC–MS/MS analysis. The additional conditions were the same as above reduced samples mass analysis.
Peptide mapping analysis
The antibody samples were denatured with 8 M guanidine hydrochloride and further reduced with 15 mM DTT at 37°C for 1 h, after cooling the samples at room temperature, the antibody samples were alkylated with 50 mM IAM in the dark for 45 mins at room temperature. The reaction was stopped by adding 1 M DTT to the final concentration of 35 mM. Then samples were buffer exchanged to 50 mM NH4HCO3 solution (pH 8.2) prior to trypsin digestion. Antibody samples were digested with trypsin (1:50 w/w) at 37°C for 18 hours, and the tryptic digests were terminated at 0.5% FA. The final tryptic digests were centrifuged at 17,000 g for 10 mins, and the supernatant was transferred to a clean EP tube for LC–MS/MS analysis. The peptide mixture was separated on a BEH300 C18 column (2.1 × 100 mm, 1.7 µm) (Waters, Milford, MA) with a column temperature of 45°C using a 80-min linear gradient (1–37% B), and the total run time was 100 min. The column was equilibrated with 1% B in water, The LC mobile phases were the same as in above reduced samples mass analysis and the flow rate was maintained at 0.20 mL/min. Data were obtained with positive ionization. The eluted peptides were detected by MS with a changing low collision energy (6 V) and high collision energy (ramping from 25 to 45 V) ESI acquisition mode to obtain the precursor ions (MS) and their fragmentation data (MSE), respectively. The ion source setup was presented as follows: capillary voltage, 3,000 V; the desolvation temperature, 350°C; source temperature, 120°C; cone voltage, 40 V; cone gas flow, 50 L/h; and the desolvation gas flow, 800 L h−1; the m/z scan range was set to 50–3,000, respectively.
The system was controlled by MassLynx 4.1 (Waters, Milford, MA), and the LC-MS/MS data analysis was performed by BiopharmaLynx 1.3.3 software, using fully tryptic cleavage rules for the sequences of the mAb. The mass accuracy was set to no more than 30 ppm for both precursors and fragments. The identified peptides were confirmed by MSE spectra with at least 3 fragment ions.
DNA sequencing and Real-time PCR
Genomic DNA and total RNA were isolated from CHO cells using the QIAamp DNA Mini Kit and RNeasy Mini Kit (Qiagen, Hilden, Germany), respectively, according to the manufacturer's protocol. First-strand cDNA synthesis was done using SuperScript RT Kit (Life Technology, Carlsbad, USA). Real-time PCR was performed using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, USA), and reactions were run on an Applied Biosystems StepOnePlus Real-Time PCR System. GAPDH, being an endogenous reference gene, was used for normalizing input DNA. The primers specific for native peptide were synthesized as follows, sense primer 5′- CAGCGACATCGCCGTGGAGTG-3′ and antisense primer 5′- TGTGTAGTGGTTGTGCAGAA-3′. The primers specific for variant peptide were synthesized as follows, sense primer 5′- CAGCGACATCGCCGTGGAGTG-3′ and antisense primer 5′- TGTGTAGTGGTTGTGCAGAG-3′. The primers specific for GAPDH were synthesized as follows, sense primer 5′- TGTTACCAACTGGGACGACA-3′ and antisense primer 5′- CTGGGTCATCTTTTCACGGT-3′. Relative expression of genes was calculated and expressed by the 2−ΔΔCT method. The cDNA was used as the template in sequencing, which was done by BGI Shanghai.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Data.docx
Download MS Word (4.8 MB)Funding
This work was supported by grants from the Natural Science Foundation of China (81330061), Ministry of Science and Technology of China (973 projects 2010CB833605 and 863 projects 2011AA020114, 2014AA021004), State Key Project for New Drug Development (2013ZX09101021; 2013ZX09401303), Shanghai Key Laboratory of Cell Engineering (14DZ2272300), Rising-star program, Shanghai Leading Academic Discipline Project (B905, Shanghai Key technologies R&D Program of Biological medicine (15431906100) and Shanghai Excellent technical leader (13XD1424000).
References
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413-5; PMID:22531442; http://dx.doi.org/10.4161/mabs.19931
- Yu XC, Borisov OV, Alvarez M, Michels DA, Wang YJ, Ling V. Identification of codon-specific serine to asparagine mistranslation in recombinant monoclonal antibodies by high-resolution mass spectrometry. Anal Chem 2009; 81:9282-90; PMID:19852494; http://dx.doi.org/10.1021/ac901541h
- Harris RJ, Murnane AA, Utter SL, Wagner KL, Cox ET, Polastri GD, Helder JC, Sliwkowski MB. Assessing genetic heterogeneity in production cell lines: detection by peptide mapping of a low level Tyr to Gln sequence variant in a recombinant antibody. Biotechnology (N Y) 1993; 11:1293-7; PMID:7764191; http://dx.doi.org/10.1038/nbt1193-1293
- Wan M, Shiau FY, Gordon W, Wang GY. Variant antibody identification by peptide mapping. Biotechnol bioeng 1999; 62:485-8; PMID:9921157; http://dx.doi.org/10.1002/(SICI)1097-0290(19990220)62:4%3c485::AID-BIT12%3e3.0.CO;2-E
- Guo D, Gao A, Michels DA, Feeney L, Eng M, Chan B, Laird MW, Zhang B, Yu XC, Joly J, et al. Mechanisms of unintended amino acid sequence changes in recombinant monoclonal antibodies expressed in Chinese Hamster Ovary (CHO) cells. Biotechnol Bioeng 2010; 107:163-71; PMID:20506532; http://dx.doi.org/10.1002/bit.22780
- Wen D, Vecchi MM, Gu S, Su L, Dolnikova J, Huang YM, Foley SF, Garber E, Pederson N, Meier W. Discovery and investigation of misincorporation of serine at asparagine positions in recombinant proteins expressed in Chinese hamster ovary cells. J Biol Chem 2009; 284:32686-94; PMID:19783658; http://dx.doi.org/10.1074/jbc.M109.059360
- Yang Y, Strahan A, Li C, Shen A, Liu H, Ouyang J, Katta V, Francissen K, Zhang B. Detecting low level sequence variants in recombinant monoclonal antibodies. MAbs 2010; 2:285-98; PMID:20400866; http://dx.doi.org/10.4161/mabs.2.3.11718
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 15:7-12; PMID:10612815; http://dx.doi.org/10.1002/(SICI)1098-1004(200001)15:1%3c7::AID-HUMU4%3e3.0.CO;2-N
- Que AH, Zhang B, Yang Y, Zhang J, Derfus G, Amanullah A. Sequence variant analysis using peptide mapping by LC-MS/MS. BioProcess Int 2010; 8:52-60.
- Chapman JR. Protein and peptide analysis by mass spectrometry. Humana Press Totowa, NJ:, 1996.
- Dorai H, Sauerwald T, Campbell A, Kyung Y, Goldstein J, Magill A, et al. Investigation of product microheterogeneity: a case study in rapid detection of mutation in mammalian production cell lines. Bioprocess International 2007; 5:66.
- Langridge J. Mutation spectra and the neutrality of mutations. Aust J Biol Sci 1974; 27:309-19; PMID:4606829; http://dx.doi.org/10.1071/BI9740309
- Nene V, Glass RE. Genetic studies on the beta subunit of Escherichia coli RNA polymerase. VI. A redundant region in the beta polypeptide. Mol Gen Genet : MGG 1984; 196:64-7; PMID:6384732; http://dx.doi.org/10.1007/BF00334093
- Knowles JR. Tinkering with enzymes: what are we learning? Science 1987; 236:1252-8; PMID:3296192; http://dx.doi.org/10.1126/science.3296192
- Cupples CG, Miller JH. Effects of amino acid substitutions at the active site in Escherichia coli beta-galactosidase. Genetics 1988; 120:637-44; PMID:2906303
- Fu J, Bongers J, Tao L, Huang D, Ludwig R, Huang Y, Qian Y, Basch J, Goldstein J, Krishnan R, et al. Characterization and identification of alanine to serine sequence variants in an IgG4 monoclonal antibody produced in mammalian cell lines. J Chromatogr B Analyt Technol Biomed Life Sci 2012; 908:1-8; PMID:23122394; http://dx.doi.org/10.1016/j.jchromb.2012.09.023
- Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014; 2:846-56; PMID:24872026; http://dx.doi.org/10.1158/2326-6066.CIR-14-0040
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat RevCancer 2012; 12:252-64; http://dx.doi.org/10.1038/nrc3239
- Deeks ED. Nivolumab: a review of its use in patients with malignant melanoma. Drugs 2014; 74:1233-9; PMID:25022950; http://dx.doi.org/10.1007/s40265-014-0234-4
- Weber J, Grob J-J, Margolin KA, Ascierto PA, Sznol M, Ott PA, et al. A Phase III study (CheckMate 238) of adjuvant immunotherapy with nivolumab (NIVO) versus ipilimumab (IPI) after complete resection of stage IIIb/c or stage IV melanoma (MEL) in patients (pts) at high risk for recurrence. Journal Immunother cancer 2015; 3:P166; PMID:4645291 ; http://dx.doi.org/10.1186/2051-1426-3-S2-P166
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015; 16:375-84; PMID:25795410; http://dx.doi.org/10.1016/S1470-2045(15)70076-8
- Yu L, Remmele RL, He B. Identification of N-terminal modification for recombinant monoclonal antibody light chain using partial reduction and quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2006; 20:3674-80; PMID:17117408; http://dx.doi.org/10.1002/rcm.2790
- von Pawel-Rammingen U, Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 2002; 21:1607-15; PMID:11927545; http://dx.doi.org/10.1093/emboj/21.7.1607
- Vincents B, von Pawel-Rammingen U, Bjorck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry 2004; 43:15540-9; PMID:15581366; http://dx.doi.org/10.1021/bi048284d
- Wenig K, Chatwell L, von Pawel-Rammingen U, Bjorck L, Huber R, Sondermann P. Structure of the streptococcal endopeptidase IdeS, a cysteine proteinase with strict specificity for IgG. Proc Natl Acad Sci U S A 2004; 101:17371-6; PMID:15574492; http://dx.doi.org/10.1073/pnas.0407965101
- Chevreux G, Tilly N, Bihoreau N. Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal Biochem 2011; 415:212-4; PMID:21596014; http://dx.doi.org/10.1016/j.ab.2011.04.030
- Wagner-Rousset E, Janin-Bussat MC, Colas O, Excoffier M, Ayoub D, Haeuw JF, Rilatt I, Perez M, Corvaïa N, Beck A. Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. MAbs 2014; 6:173-84; PMID:25006625; http://dx.doi.org/10.4161/mabs.26773
- Strand J, Huang CT, Xu J. Characterization of Fc-fusion protein aggregates derived from extracellular domain disulfide bond rearrangements. J Pharm Sci 2013; 102:441-53; PMID:23242781; http://dx.doi.org/10.1002/jps.23421
- Dick LW, Mahon D, Qiu D, Cheng K-C. Peptide mapping of therapeutic monoclonal antibodies: Improvements for increased speed and fewer artifacts. J Chromatogr B 2009; 877:230-6; PMID:NOT_FOUND; http://dx.doi.org/10.1016/j.jchromb.2008.12.009
- Castro-Perez JM, Kamphorst J, DeGroot J, Lafeber F, Goshawk J, Yu K, Shockcor JP, Vreeken RJ, Hankemeier T. Comprehensive LC− MSE lipidomic analysis using a shotgun approach and its application to biomarker detection and identification in osteoarthritis patients. J Proteome Res 2010; 9:2377-89; PMID:20355720; http://dx.doi.org/10.1021/pr901094j
- Chakraborty AB, Berger SJ, Gebler JC. Use of an integrated MS–multiplexed MS/MS data acquisition strategy for high-coverage peptide mapping studies. Rapid Commun Mass Spectrom 2007; 21:730-44; PMID:17279597; http://dx.doi.org/10.1002/rcm.2888
- Xie H, Gilar M, Gebler JC. Characterization of protein impurities and site-specific modifications using peptide mapping with liquid chromatography and data independent acquisition mass spectrometry. Anal Chem 2009; 81:5699-708; PMID:19518054; http://dx.doi.org/10.1021/ac900468j
- Zhu L, Guo Q, Guo H, Liu T, Zheng Y, Gu P, et al. Versatile characterization of glycosylation modification in CTLA4-Ig fusion proteins by liquid chromatography-mass spectrometry. MAbs 2014; 6:1474-85; PMID:25484062; http://dx.doi.org/10.4161/mabs.36313
- Jefferis R, Lefranc M-P. Human immunoglobulin allotypes: possible implications for immunogenicity. MAbs 2009; 1:332-8; PMID:20073133; http://dx.doi.org/10.4161/mabs.1.4.9122
- Carter P, Presta L, Gorman CM, Ridgway J, Henner D, Wong W, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 1992; 89:4285-9; PMID:1350088; http://dx.doi.org/10.1073/pnas.89.10.4285
- Zhang T, Huang Y, Chamberlain S, Romeo T, Zhu-Shimoni J, Hewitt D, Zhu M, Katta V, Mauger B, Kao YH. Identification of a single base-pair mutation of TAA (Stop codon) –>GAA (Glu) that causes light chain extension in a CHO cell derived IgG1. MAbs 2012; 4:694-700; PMID:23018810; http://dx.doi.org/10.4161/mabs.22232