
ABSTRACT
Although recent advances have substantially improved the management of multiple myeloma, it remains an incurable malignancy. We now demonstrate that anti-CD138 molecules genetically fused to type I interferons (IFN) synergize with the approved therapeutic bortezomib in arresting the proliferation of human multiple myeloma cell lines both in vitro and in vivo. The anti-CD138-IFNα14 fusion protein was active in inducing increased expression of signal transducer and activator of transcription 1 (STAT1) and its phosphorylation while the cell death pathway induced by bortezomib included generation of reactive oxygen species. Interferon regulatory factor 4 (IRF4), an important survival factor for myeloma cells, was down regulated following combination treatment. Induction of cell death appeared to be caspase-independent because treatment with inhibitors of caspase activation did not decrease the level of cell death. The observed caspase-independent synergistic cell death involved mitochondrial membrane depolarization, and poly(ADP-ribose) polymerase-1 (PARP-1) cleavage, and resulted in enhanced induction of apoptosis. Importantly, using 2 different in vivo xenograft models, we found that combination therapy of anti-CD138-IFNα14 and bortezomib was able to cure animals with established tumors (7 of 8 using OCI-My5 or 8 of 8 using NCI-H929). Thus, the combination of anti-CD138-IFNα with bortezomib shows great promise as a novel therapeutic approach for the treatment of multiple myeloma, a malignancy for which there are currently no cures.
Introduction
Multiple myeloma (MM) is a cancer formed by a clonal proliferation of malignant plasma cells. MM is the second most frequent hematological malignancy after non-Hodgkin's lymphoma. Although novel drugs have improved survival in those with MM, it remains an incurable malignancy. Bortezomib (also known as Velcade® or PS-341) is a proteasome inhibitor approved by the Food and Drug Administration (FDA) for the treatment of MM. Although bortezomib has been shown to inhibit proliferation and induce apoptosis, the mechanisms are not completely understood. While originally recognized for its effects on NF-κB, recent studies have shown that the effects of bortezomib are complex,Citation1 and involve the generation of reactive oxygen species (ROS) and mitochondrial membrane depolarization.Citation2,3
Interferons (IFN) have been shown to inhibit tumor cell growth and induce apoptosis. Type I IFNs (IFNα and IFNβ) share a common receptor composed to 2 subunits, IFNAR1 and IFNAR2. Signaling through IFNAR involves Tyk2, Jak/STAT proteins,Citation1,4 as well as p38 MAPK and PI3 kinase pathways.Citation5,6 IFNα has been used for the treatment of MM, albeit with limited success. Higher doses of IFNα have been shown to be the most effective;Citation7 however, systemic toxicity makes it difficult to deliver therapeutically effective doses. To address this concern, we have used antibody-based fusion proteins in which IFN is genetically joined to an antibody that recognizes a tumor-associated antigen. This approach has been used effectively to target IFN to various tumors.Citation8-11 To target IFN to MM, we fused it to the B-B4 antibody that specifically binds CD138.Citation8,12 CD138, also known as syndecan-1, is highly expressed on mature plasma cells, making it a suitable target for MM.
Bortezomib has shown synergistic effects with many of the drugs used to treat MM, including alkylating agents, immunomodulatory drugs, corticosteroids and anthracyclines.Citation13 Synergy between IFNα and bortezomib has been seen in several different cancers, including melanomaCitation14 and bladder cancer cells, in which IFNα and bortezomib work through different mechanisms (apoptosis versus angiogenesis inhibition, respectively).Citation15 In the study reported here, we made the novel observation that antibody-targeted IFNα can synergize with bortezomib. Using anti-CD138-IFNα2, anti-CD138-IFNα14 and anti-CD138-IFNα2YNS for the treatment of MM, we found that all 3 fusion proteins effectively synergize with bortezomib in inhibiting MM cell proliferation in vitro. Treatment with anti-CD138-IFNα14 and bortezomib resulted in synergistic decreases in expression of the MM survival factor IRF4. Interestingly, MM cell death induced by bortezomib and anti-CD138-IFNα14 was independent of caspase activation. Instead, pathways that lead to mitochondrial dysfunction, such as ROS generation, changes to mitochondrial membrane potential (ΔΨm), and PARP-1 cleavage, appeared to be important for the synergistic induction of apoptosis and cell death in MM. Using two different xenograft models, we showed that anti-CD138-IFNα14 synergized with bortezomib to provide enhanced protection against tumor growth in vivo, actually curing mice with established tumors. Thus, the novel combination of the approved therapeutic, bortezomib, and anti-CD138-IFNα14 antibody fusion protein showed great promise against MM, a currently incurable malignancy, suggesting that this combination therapy should be examined in the clinic.
Results
Anti-CD138-IFNα synergizes with bortezomib in inhibiting cell growth
There are 12 human IFNαs with differing activities. Our first fusion proteins contained IFNα2Citation9,11 since it is the IFN most frequently used in the clinic. However, other IFNs with greater activity have the potential to be better therapeutics. Therefore, we also tested fusion proteins containing IFNα14, which was reported to have the highest anti-proliferative activity among the 12 naturally-occurring IFNα against ovarian cancer cells,Citation16 and IFNα2YNS, which carries 3 introduced mutations (H57Y, E58N, Q61S) conferring greater anti-tumor activity than wild-type IFNα2.Citation8,17 In addition, we tested whether the fusion proteins could synergize with bortezomib against MM.
Four human myeloma cell lines (U266, NCI-H929, OCI-My5 and ANBL-6) were used in this study. To confirm expression of CD138, a protein that is characteristic of MM, human myeloma cell lines (HMCLs) were incubated with anti-CD138, then stained using anti-human κ-PE, and analyzed by flow cytometry. As expected, all 4 HMCLs express CD138 (Fig. S1).
HMCLs were incubated with bortezomib alone, fusion protein alone, or fusion protein + bortezomib for 3 days, and the percentage of metabolically active cells was then determined using the MTS assay. For all 4 cell lines, the combination of fusion protein + bortezomib was much more effective in inhibiting cell growth than either alone. The results with anti-CD138-IFNα14 are shown in . Anti-CD138-IFNα2 and anti-CD138-IFNα2YNS showed similar results (Fig. S2). To confirm these results, HMCLs were incubated with a fixed concentration of fusion protein with or without bortezomib for 3 days. For all 4 HMCLs and all 3 fusion proteins, the combination of fusion protein + bortezomib was significantly more effective in inhibiting cell proliferation than either treatment alone (). In order to determine if the effect of the combination of anti-CD138-IFNα with bortezomib was synergistic, we calculated the combination index (CI)-isobologram equation, which allows quantitative determination of drug interactions. CI < 1, CI = 1 and CI > 1 indicate synergism, additive effect, and antagonism, respectively.Citation18 The CI was <1 for all treatments, indicating that the fusion proteins synergize with bortezomib in inhibiting HMCL proliferation (). When cell proliferation was assessed using [3H]-thymidine incorporation, similar results were observed (data not shown). Since all three 3 fusion proteins exhibited similar activities, we chose to use anti-CD138-IFNα14 for the rest of our studies.
Figure 1. Bortezomib and IFNα fusion proteins act synergistically in inhibiting proliferation in HMCLs. HMCLs were treated for 3 days with fusion protein or bortezomib. Percent proliferation was calculated relative to cells grown in the absence of treatment by MTS. Each cell line was examined 3 to 5 times. Representative experiments are shown with data presented as the mean of triplicate experiments with the error bars indicating the SEM. (A) OCI-My5, NCI-H929, and ANBL-6 cells were treated with 1.25 – 10 nM bortezomib or 1.25 – 10 pM anti-CD138-IFNα14. U266 cells were treated with 0.4 – 50 nM bortezomib or 0.4 – 50 pM anti-CD138-IFNα14. (B) Cells were treated with anti-CD138-IFNα2, anti-CD138-IFNα2YNS, or anti-CD138-IFNα14 in the presence or absence of bortezomib. The concentrations used were 3 pM fusion protein and 1 nM bortezomib for OCI-My5; 5 pM fusion protein and 5 nM bortezomib for NCI-H929 and ANBL-6; 1.5 pM anti-CD138-IFNα2, 0.5 pM anti-CD138-IFNα2YNS, 1.5 pM anti-CD138-IFNα14, and 1.5 nM bortezomib for U266. Panels A and B are from independent experiments.
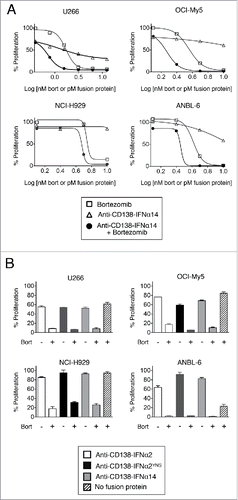
Table 1. Quantitative determination of drug interactions between bortezomib and IFNα fusion proteins.
Anti-CD138-IFNα14 can activate the STAT1 pathway
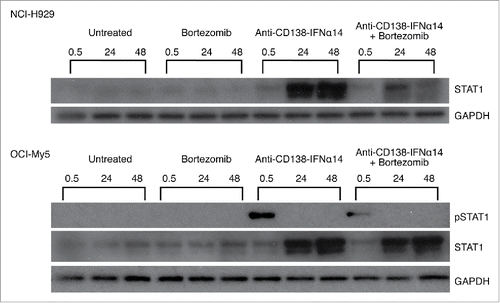
STAT proteins are part of a major IFNα signaling pathway. IFNα is known to both increase the expression of STAT1 and promote its activation through tyrosine phosphorylation (for a review see ref.Citation19). Activation of STAT1 is required for IFNα-mediated cell death.Citation20 Therefore, we wanted to determine if the fusion protein could be contributing to cell death through the activation of this pathway, and if there was any synergistic activation of STAT1 following combination treatment. Cells were treated for 0.5, 24 or 48 h with bortezomib or anti-CD138-IFNα14, and cell lysates were examined by Western blotting for levels of total STAT1 protein and phosphorylated STAT1 (pSTAT1). For both NCI-H929 and OCI-My5 cells, there was an increase in STAT1 protein at 24 and 48 h after treatment with anti-CD138-IFNα14, but not with bortezomib (). Similar increases in the level of STAT1 protein were seen with U266 and ANBL-6 (Fig. S3). pSTAT1 was seen following 0.5 h treatment in OCI-My5 and U266, but not NCI-H929 or ANBL-6. Thus anti-CD138-IFNα14 activates the STAT1 pathway as shown by increased expression of STAT1, but no synergistic activation of the STAT1 pathway was observed in cells treated with bortezomib + anti-CD138-IFNα14.
Figure 2. Activation of STAT1 in cells treated with bortezomib or anti-CD138-IFNα14. Cells were treated with anti-CD138-IFNα14, bortezomib or both for 0.5, 24 and 48 h. NCI-H929 was treated with 2.5 pM fusion protein or 5 nM bortezomib. OCI-My5 were treated with 3 pM anti-CD138-IFNα14 or 1.5 nM bortezomib. Total STAT1 protein and pSTAT1 levels in cell lysates were determined by Western blotting. Protein loading was monitored by probing the same membranes with anti-GAPDH.
IRF4 is downregulated by anti-CD138-IFNα14 and bortezomib
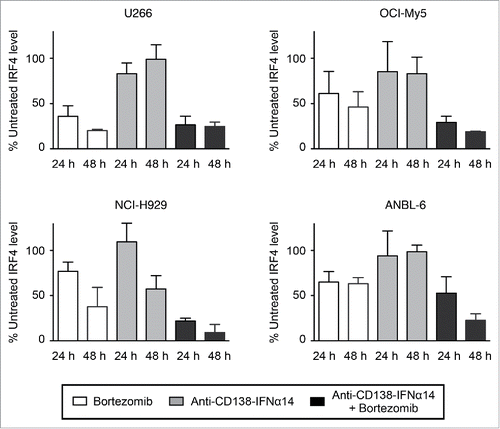
IRF4, a transcription factor whose expression is critical for MM survival, has been shown to be a master regulator targeting many genes involved in the survival and proliferation of MM cells (for a review see ref.Citation21). MM's dependence on this survival factor has been called IRF4 addiction.Citation22 To determine if changes in IRF4 expression were involved in synergistic cell death, cells were treated for 24 h and 48 h with bortezomib, anti-CD138-IFNα14, or both, and the expression of IRF4 detected by Western blotting. IRF4 levels were quantitated and expressed as a percentage of the untreated level (). Representative gels of the Western blots are shown in Treatment with either agent alone did lead to some decreases in IRF4 level; this was seen especially following bortezomib treatment. However, combination treatment with bortezomib and anti-CD138-IFNα14 led to synergistic decreases in IRF4 expression in NCI-H929, OCI-My5 and ANBL-6 cells, which was evident especially after the 48 h treatment. U266 cells appeared to be highly sensitive to bortezomib, and showed a large change in IRF4 levels following treatment with this single agent. These data suggest that the down regulation of the IRF4 survival factor seen following treatment with bortezomib and anti-CD138-IFNα14 may play an important role in the synergistic inhibition of MM cell growth.
Figure 3. IRF4 expression is down-regulated by bortezomib + anti-CD138-IFNα14 treatment. Cells were treated with anti-CD138-IFNα14, bortezomib or both for 24 and 48 h. U266 and OCI-My5 were treated with 3 pM anti-CD138-IFNα14 or 1.5 nM bortezomib. ANBL-6 was treated with 6 pM fusion protein or 2.3 nM bortezomib. NCI-H929 was treated with 2.5 pM fusion protein or 5 nM bortezomib. The levels of IRF4 in cell lysates were determined by Western blotting. IRF4 levels were quantitated using NIH ImageJ and expressed as a percentage of the untreated level as described in Materials and Methods.
The cytotoxic effects observed are caspase-independent
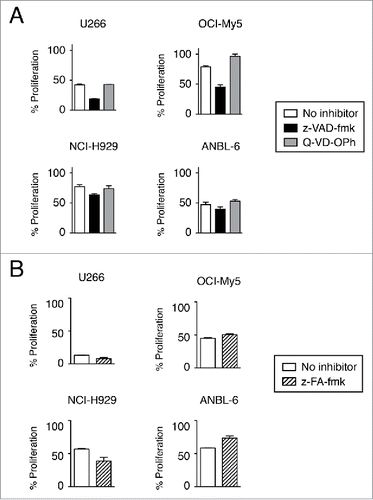
Although caspase activation is often considered to play an essential role in the induction of apoptosis, caspase-independent cell death has been described.Citation23 z-VAD-fmk is often used as a pan-caspase inhibitor, and has been shown to inhibit bortezomib-induced apoptosis.Citation14,24 However, when cells were treated with bortezomib and anti-CD138-IFNα14 in the presence of z-VAD-fmk, inhibition of cytotoxicity was not observed. Surprisingly, the opposite effect was seen in which z-VAD-fmk actually enhanced cytotoxicity (). Using a different pan-caspase inhibitor, Q-VD-OPh, we also found that inhibition of caspase activation had little to no effect on cytotoxicity except for in OCI-My5 in which there was a small increase in cell proliferation (). Unlike z-VAD-fmk, Q-VD-OPh treatment did not enhance cytotoxicity. Treatment of cells with z-FA-fmk, a cysteine protease inhibitor that selectively inhibits effector caspases, cathepsins, as well as cruzain and papain, also showed little or no effect on cell viability after bortezomib + anti-CD138-IFNα14 treatment. Although some increase in ANBL-6 proliferation was observed, it did not reach the level seen in the absence of treatment with bortezomib + anti-CD138-IFNα14 (). Analysis by Western blot after bortezomib or anti-CD138-IFNα14 treatment failed to detect the activated cleavage product of caspase-3 (data not shown), which is consistent with cell death being caspase-independent at the concentrations of bortezomib and IFNα14 tested.
Figure 4. Induction of cell death is not prevented by caspase inhibition. Cells were treated with anti-CD138-IFNα14 and bortezomib in the presence or absence of pan-caspase inhibitors for 3 days. U266 and OCI-My5 were treated with 3 pM anti-CD138-IFNα14 and 1.5 nM bortezomib. NCI-H929 and ANBL-6 were treated with 5 pM anti-CD138-IFNα14 and 4 nM bortezomib. Cells were assessed for changes in metabolic activity by MTS. Assays were done in triplicate with the error bars indicating the SEM. In some cases, the error bars cannot be seen because they are obscured by the plot. Percent proliferation was calculated relative to cells grown in the absence of treatment. (A) 50 μM z-VAD-fmk or 20 μM Q-VD-OPh. (B) 20 μM z-FA-fmk.
The mitochondrial cell death pathway is induced by bortezomib and IFNα fusion protein
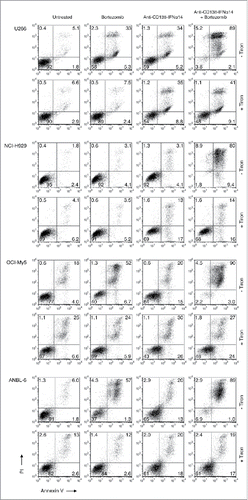
Caspase-independent cell death pathways involving signals that converge in the mitochondria have been reported.Citation3 High levels of mitochondrial reactive oxygen species (ROS) can activate apoptotic pathways, leading to cell death. Although ROS production has been shown to play an important role in the initiation of the bortezomib-induced apoptotic cascade in cancer cells,Citation2,3 the effects of IFNα on ROS production have not been determined. To inhibit ROS, we used tiron, an oxygen scavenger that has been shown to block bortezomib-induced cell death.Citation2 After treatment with anti-CD138-IFNα14, bortezomib or both for 3 days in the presence or absence of 1 mM tiron, cells were stained with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry to assess the level of apoptosis (). For all 4 cell lines, there were fewer Annexin V+ apoptotic cells following treatment with anti-CD138-IFNα14 + bortezomib in the presence of tiron than in its absence. Thus, ROS plays a role in the induction of apoptosis following treatment with anti-CD138-IFNα14 + bortezomib.
Figure 5. ROS generation plays a role in the induction of cell death by bortezomib + anti-CD138-IFNα14. Cells were treated for 3 days with anti-CD138-IFNα14 or bortezomib in the presence or absence of 1 mM tiron to inhibit ROS. U266 and OCI-My5 were treated with 3 pM anti-CD138-IFNα14 or 1 nM bortezomib. NCI-H929 and ANBL-6 were treated with 5 pM anti-CD138-IFNα14 or 4 nM bortezomib. Cells were stained with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry to assess the level of apoptosis.
Changes in the mitochondrial membrane potential (ΔΨm) are responsible for releasing apoptotic factors from the intermembrane space of the mitochondrion into the cytosol. Therefore, to investigate possible contributions of changes in ΔΨm to the observed cytotoxicity, HMCLs were treated with bortezomib, anti-CD138-IFNα14 or the combination for 48 h and analyzed by flow cytometry following staining with DiOC6(3). The cell lines had variable decreases to ΔΨm when treated with either bortezomib or anti-CD138-IFNα14 alone, with the most change seen in U266. However, combination treatment resulted in an increase in the percentage of cells with ΔΨm changes for all cell lines (). These data suggest that the effect on ΔΨm contributes to the observed synergistic inhibition of cell proliferation seen following treatment with bortezomib + anti-CD138-IFNα14.
Figure 6. The effect of bortezomib or anti-CD138-IFNα14 treatment on mitochondrial dysfunction. (A) Cells were treated with anti-CD138-IFNα14 or bortezomib for 2 days. The concentrations of anti-CD138-IFNα14 and bortezomib were: U266 (3 pM, 1 nM), NCI-H929 (5 pM, 3 nM), ANBL-6 (5 pM, 3.5 nM), and OCI-My5 (3 pM, 1.5 nM). After treatment, cells were incubated with DiOC6(3) and mitochondrial membrane potential ΔΨm measured by flow cytometry. (B) ANBL-6 and U266 cells were untreated (lane 1) or treated for 24 h with anti-CD138-IFNα14 (lane 2), bortezomib (lane 3), or both (lane 4). Western blot analysis of cell lysates was used to determine the levels of uncleaved and cleaved PARP. (C) OCI-My5 and NCI-H929 cells were treated for 0.5, 24 or 48 h with anti-CD138-IFNα14, bortezomib, or both and the levels of uncleaved and cleaved PARP were determined by Western blotting. For (B) and (C), protein loading was monitored by probing the same membranes with anti-GAPDH. PARP – full length. PARP* – cleaved.
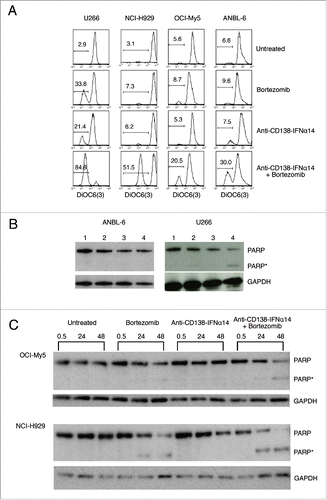
PARP plays a key role in triggering mitochondria to release their proapoptotic molecules by a mechanism that does not require caspase activation (for a review see ref.Citation25). A previous study showed that combined treatment of melanoma cells with bortezomib and IFNα resulted in enhanced cleavage of PARP compared to either treatment alone.Citation14 In initial experiments to determine if enhanced cleavage of PARP occurred in HMCLs, ANBL-6 and U266 cells were treated for 24 h with anti-CD138-IFNα14, bortezomib or anti-CD138-IFNα14 + bortezomib and cell extracts were examined by Western blot. Under these conditions, cleavage of PARP was not observed in ANBL-6 (), although, in a subsequent experiment, small amounts of cleavage were seen in cells treated with bortezomib or anti-CD138-IFNα14 + bortezomib (data not shown). However, for U266, treatment with both anti-CD138-IFNα14 + bortezomib resulted in cleavage of PARP (). In follow up studies, OCI-My5 and NCI-H929 cells were treated for 0.5, 24 and 48 h, and the levels of PARP cleavage assessed. In these cell lines, bortezomib, but not fusion protein treatment, resulted in a small amount of PARP cleavage, which was greatly enhanced following treatment with anti-CD138-IFNα14 + bortezomib (). These data suggest that, at least for some MMs, enhanced PARP cleavage contributes to the enhanced inhibition of cell growth following treatment with bortezomib + anti-CD138-IFNα14. Taken together, the involvement of ROS, decrease in mitochondrial membrane potential, and enhanced PARP cleavage, suggest that caspase-independent mitochondrial dysfunction plays a significant role in the synergistic inhibition of cell growth seen following treatment with anti-CD138-IFNα14 and bortezomib.
Anti-CD138-IFN fusion proteins synergize with bortezomib in eliciting apoptosis
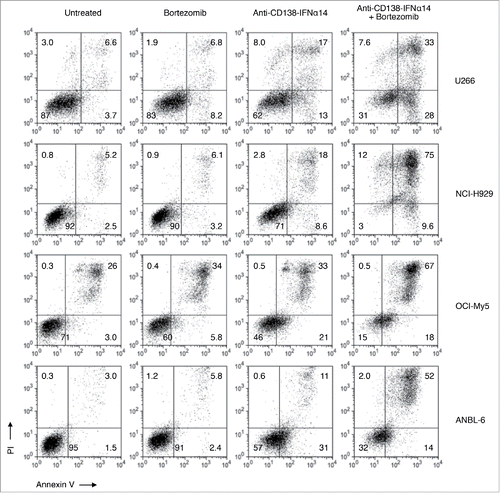
To determine if the synergistic inhibition of cell growth resulted at least partly from increased apoptosis, HMCLs incubated with anti-CD138-IFNα14, bortezomib, or anti-CD138-IFNα14 + bortezomib for 3 days were stained with Annexin V-FITC and PI and analyzed by flow cytometry. For all 4 HMCLs, higher levels of apoptosis were seen following combination treatment than with a single agent (). Similar results were obtained with anti-CD138-IFNα2 and anti-CD138-IFNα2YNS (data not shown). Treatment with anti-CD138 did not enhance apoptosis in the presence or absence of bortezomib (Fig. S5). Therefore, synergistic induction of apoptosis is seen following combination therapy with anti-CD138-IFN fusion proteins + bortezomib.
Figure 7. Bortezomib and anti-CD138-IFNα14 act synergistically to induce apoptosis in HMCLs. HMCLs were treated with anti-CD138-IFNα14 or bortezomib for 3 days. U266 and OCI-My5 were treated with 3 pM anti-CD138-IFNα14 or 1 nM bortezomib. NCI-H929 and ANBL-6 were treated with 5 pM anti-CD138-IFNα14 or 4 nM bortezomib. Cells were stained with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry to assess the level of apoptosis.
Combination of anti-CD138-IFNα14 and bortezomib provides enhanced protection in vivo
We showed that fusion protein + bortezomib treatment results in synergistic activity in vitro in inhibiting the proliferation of MM cells. The next step was to test their effectiveness in vivo. Our strategy of fusing IFN to antibody has several major advantages in vivo. While the half-life of IFNα is only 1 h,26 we have shown that fusing it to IgG extends the half-life to 8 h.Citation10,11 In addition, the anti-CD138 portion of the fusion protein provides a binding specificity to target MM and deliver therapeutically useful levels of IFN without systemic toxicity. Indeed, we have shown the effectiveness of this strategy in targeting not only MM,Citation8 but also lymphoma using anti-CD20-IFNα2.Citation9
Two different in vivo models were used. In one model, OCI-My5 cells were subcutaneously injected into SCID mice that were then treated on days 14, 16, 18, 21, 28, 35 and 42 (see Materials and Methods for details). Survival and tumor growth were monitored (). Phosphate-buffered saline (PBS)-treated mice all succumbed by day 44. Treatment with either bortezomib or fusion protein prolonged survival, but all mice succumbed by day 71 and day 76, respectively; anti-CD138-IFNα14 was somewhat more effective than bortezomib at the doses used (P = .03) in this model. Treatment with the combination of fusion protein + bortezomib was much more effective (P = < .0001 compared to single treatment) with 7 of 8 mice remaining tumor free. Only one mouse ultimately succumbed to a very slowly growing tumor. When this tumor was biopsied, extensive necrosis was observed, and the viable cells within the tumor continued to express CD138 (data not shown).
Figure 8. Synergistic protection against tumor growth is observed between anti-CD138-IFNα14 and bortezomib in vivo in murine models of MM. (A) OCI-My5 tumors were established in SCID mice by subcutaneous injection of cells. Mice were treated iv on days 14, 16, 18, and then ip on days 21, 28, 35 and 42 for a total of 7 treatments. (B) NCI-H929 tumors were established in NSG mice by subcutaneous injection of cells. Mice were treated iv on days 14, 16, 18 and then ip on days 21 and 28 for a total of 5 treatments. For both models, each group consisted of 8 mice. Treatment groups included PBS, 100 µg anti-CD138-IFNα14, 0.75 mg/kg bortezomib or 100 µg anti-CD138-IFNα14 + 0.75 mg/kg bortezomib. Tumor growth and survival were monitored.
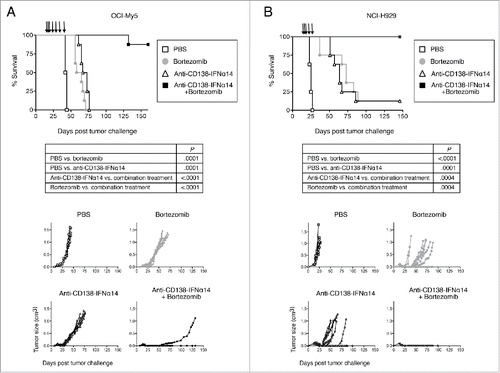
In the second model, NCI-H929 cells were subcutaneously injected into NOD-scid IL2rγnull (NSG) mice that were then treated on days 14, 16, 18, 21, and 28 (). PBS-treated mice all succumbed by day 27. Treatment with either bortezomib or fusion protein prolonged survival (P = .0001), but 7 of 8 mice succumbed by day 89 and day 86, respectively; there was no statistical difference between the efficacy of anti-CD138-IFNα14 and bortezomib (P = .58). The lone survivor in each group remained tumor free. Remarkably, treatment with the combination of anti-CD138-IFNα14 + bortezomib completely prevented tumor growth, with all 8 mice remaining tumor free. Therefore, combination treatment was able to cure animals of established tumors (P = .0004 compared to single treatment).
Discussion
IFNs were the first recombinant proteins to be used in the treatment of cancer (for a review see refs.Citation27,28). Compared to other cytokines, IFNs have an optimal combination of anti-tumor properties, including direct inhibition of tumor cell growth, enhancement of antibody-dependent cell-mediated cytotoxicity (ADCC) and antigen processing,Citation29 stimulation of T cell immunity,Citation30 and inhibition of angiogenesis.Citation27,28 Previous studies in our laboratoryCitation8-11 have shown that genetic fusion of IFNs to an antibody is an effective strategy to specifically target IFN to the tumor microenvironment, reduce systemic toxicity, and increase half-life.
Bortezomib is a proteasome inhibitor that is effective against MM, but its mechanisms of action remain unclear. Proteasome inhibition results in induction of the unfolded protein response (UPR). UPR reestablishes endoplasmic reticulum (ER) homeostasis; however, persistent ER stress can switch the cytoprotective functions of UPR into cell death-promoting mechanisms.Citation31 ROS production and oxidative stress are integral UPR components.Citation32
Both IFNα and bortezomib have separately been approved for MM therapy and shown to improve patient outcome; however, MM remains an incurable malignancy. In our study, we show that combination treatment of IFNα fusion protein + bortezomib leads to synergistic induction of cell death in HMCLs representing different molecular subtypes of myeloma. A significant reduction in cell proliferation was observed in vitro with combination treatment compared to either treatment alone. Importantly, significant synergistic inhibition of tumor growth was also observed in vivo in 2 different xenograft models using SCID (OCI-My5 model) and NSG (NCI-H929 model) mice, with mice actually being cured of established tumors (7 of 8 for OCI-My5 and 8 of 8 for NCI-H929). Thus, this combination therapy shows great potential for development as a novel therapeutic. It should be noted that these xenograft models use immunodeficient mice, and even greater efficacy may be seen in immunocompetent animals in which the immune-stimulatory activity of the IFN would be exerted. In the OCI-My5 model, ADCC mediated by the antibody portion of the fusion protein may contribute to increased survival since SCID mice possess NK cells.
To elucidate some of the mechanisms that contribute to synergy, we tested several different pathways known to be activated by bortezomib or IFNα. IFNα is known to activate STAT1 and promote cell death. STAT1 has been shown to play an essential role in the caspase-independent cell death of activated macrophages.Citation33 Anti-CD138-IFNα14 treatment did indeed increase STAT1 expression in all 4 HMCL with phosphorylated STAT1 observed in 2 of the 4 cell lines. However, treatment of cells with both anti-CD138-IFNα14 and bortezomib did not augment STAT1 activation. Similar results have also been reported for melanoma treated with bortezomib and IFNα2b.Citation34
Expression of IRF4, a member of the IRF family of transcription factors restricted to the immune system, has been found to be essential for the viability of MM cells,Citation21,22 with even small changes in IRF4 levels resulting in cell death. Treatment with anti-CD138-IFNα14 or bortezomib alone resulted in some decreases in IRF4 levels. However, treatment with anti-CD138-IFNα14 + bortezomib resulted in even greater decreases in IRF4 levels, especially at the 48 h treatment point for OCI-My5, NCI-H929 and ANBL-6 cell lines ( and Fig. S4). For the U266 cell line, combination treatment did not result in further decreases in IRF4, possibly due to the fact that, at the doses used, bortezomib alone drastically decreased IRF4 expression in U266. Thus, decreased IRF4 levels may be responsible in part for the efficacy of the combination treatment in preventing MM growth. Indeed, decreased IRF4 levels have been found to sensitize MM cells to bortezomib-induced apoptosis.Citation35
Caspase activation has been reported to play a role in both IFNα- and bortezomib-triggered apoptosis in MM cells.Citation36,37 In most cases, IFN- and bortezomib-induced cell death could be prevented by z-VAD-fmk.Citation36,38,39 Combined treatment with bortezomib + IFNα was also reported to induce synergistic apoptosis, which could be inhibited by z-VAD-fmk in melanoma and other tumor cell lines.Citation14 In contrast, in our HMCLs, we found that inhibition of caspase activation using a variety of inhibitors, including z-VAD-fmk, Q-VD-OPh, and z-FA-fmk, had little or no effect on the cytotoxic activity of anti-CD138-IFNα14, bortezomib or anti-CD138-IFNα14 + bortezomib (). In addition, we failed to see activation of caspase-3. Thus, we could not identify a role for caspase activation in IFNα- and bortezomib-induced apoptosis. These differences may reflect differences in the treatment conditions or differences in cell types.
Caspase-independent modes of cell death have been identified, such as those involving mitochondrial dysfunction. Although previous studies have shown that treatment with IFNα or bortezomib can lead to ROS generation and mitochondrial dysfunction,Citation3,40 no one has previously investigated the consequences of the combined treatment with bortezomib and targeted IFNα14. Our experiments with the ROS inhibitor, tiron, showed that ROS plays a role in cell death induced by anti-CD138-IFNα14 + bortezomib (). In addition, we found that combination treatment with bortezomib and anti-CD138-IFNα14 resulted in enhanced changes to ΔΨm compared to treatment with either agent alone (). A previous study has shown that bortezomib-induced disruption of mitochondrial membrane potential and increase in ROS was not affected by z-VAD-fmk,Citation3 consistent with our findings that caspases are not involved. Enhanced cleavage of PARP was seen in 3 of the 4 HMCLs following treatment with anti-CD138-IFNα14 + bortezomib (). Activated PARP-1 has been shown to promote apoptosis by triggering mitochondria to release their proapoptotic molecules by a mechanism that does not require caspase activation (for a review see ref.Citation25). Both IFNα2b and bortezomib have been reported to induce PARP-1 cleavage in anaplastic large cell lymphoma and KB epidermoid carcinoma cells.Citation41,42 Taken together, these data suggest that bortezomib and IFNα work, at least in part, through the mitochondrial pathway of apoptosis, leading to synergistic cell death. This may involve apoptosis inducing factor (AIF), which is known to be activated independently of caspases and is released upon disruption of ΔΨmCitation43 and activation of PARP-1.Citation25
We have shown previously that targeting IFNα and IFNβ fusion proteins is an effective strategy against lymphoma and MM.Citation8-11 Based on these studies, we initiated Phase 1 clinical testing of anti-CD20-IFNα to treat B-cell lymphomas (ClinicalTrials.gov Identifier NCT02519270), underscoring the therapeutic potential of this approach. One of the goals of the study described here was to determine the effect of combining treatment using IFN fusion proteins with the FDA-approved drug, bortezomib. We found that treatment of MM in vitro and in vivo with anti-CD138-IFNα14 + bortezomib resulted in synergistic cell death. Synergy between bortezomib and IFNα2b has also been reported for melanoma and combination treatment has gone through a successful Phase 1 trial against metastatic melanoma.Citation34 Our studies support moving forward with clinical testing of combination therapy with bortezomib and anti-CD138-IFN fusion proteins for the treatment of multiple myeloma.
Materials and methods
Cells and chemicals
HMCLs were obtained through the generous gift of Dr. W. Michael Kuehl and Dr. Diane Jelinek. Their CD138 expression was confirmed by flow cytometry. HMCLs were cultured as described previously.Citation8 Bortezomib (NDC 63020-049-01) was obtained from Millennium Pharmaceuticals. z-VAD-fmk (51-69361U) was purchased from BC Pharmingen. Q-VD-OPh (A1901) was purchased from Apexbio.
Construction of vectors and protein production
Construction of anti-CD138, anti-CD138-IFNα2 and anti-CD138-IFNα2YNS has been described.Citation8 The antibody portion of the fusion proteins is IgG1κ. Anti-CD138-IFNα14 was produced by replacing the IFNα2 sequence with that of IFNα14 (NCBI reference sequence: NP_002163.2). Fusion proteins were produced and purified from Chinese hamster ovary cells following transfection of H and L chain expression vectors as described previously.Citation8
Determination of cellular metabolic activity
HMCLs were treated with varying concentrations of fusion protein, bortezomib or both at 37°C for 3 days. Metabolic activity was determined using MTS solution (Promega, G3581) by measuring absorbance at 490 nm using a Synergy HT Multi-Detection Microplate Reader (BioTek Instruments). GraphPad Prism (GraphPad Software) was used to analyze data as described previously.Citation8 Data are expressed as percent proliferation with untreated cells being 100%. The starting concentration was 10 nM bortezomib and 10 pM anti-CD138-IFNα14, which was serially diluted 1:2 for OCI-My5, NCI-H929, and ANBL-6. For U266, the starting concentration was 50 nM bortezomib and 50 pM anti-CD138-IFNα14.
In a different experiment (), HMCLs were incubated with anti-CD138-IFNα14, anti-CD138-IFNα2 or anti-CD138-IFNα2YNS in the presence or absence of bortezomib at 37°C for 3 days. U266 cells were treated with 1.5 pM anti-CD138-IFNα2, 0.5 pM anti-CD138-IFNα2YNS, 1.5 pM anti-CD138-IFNα14, and 1.5 nM bortezomib. NCI-H929 and ANBL-6 cells were treated with 5 pM fusion protein and 5 nM bortezomib. OCI-My5 cells were treated with 3 pM fusion protein and 1 nM bortezomib. Metabolic activity was determined in triplicate using MTS as described above.
Inhibition of caspases
HMCLs were incubated with anti-CD138-IFNα14 and bortezomib in the presence or absence of caspase inhibitors, z-VAD-fmk (50 μM), Q-VD-OPh (20 μM), or z-FA-fmk (20 μM). U266 and OCI-My5 were treated with 3 pM anti-CD138-IFNα14 and 1.5 nM bortezomib. NCI-H929 and ANBL-6 were treated with 5 pM anti-CD138-IFNα14 and 4 nM bortezomib. Metabolic activity was determined by MTS assay in triplicate as described above. The percent proliferation was calculated relative to cells grown in the absence of treatment.
Western blots
Cells were treated for 0.5, 24 or 48 h with anti-CD138-IFNα14, bortezomib or both. U266 and OCI-My5 were treated with 3 pM anti-CD138-IFNα14 and 1.5 nM bortezomib. ANBL-6 was treated with 6 pM fusion protein and 2.3 nM bortezomib. NCI-H929 was treated with 2.5 pM fusion protein and 5 nM bortezomib. Cells were lysed using 50 mM Tris-HCl pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate containing a protease inhibitor cocktail (Roche Applied Science, 05892791001) and PhosSTOP (Roche Applied Science, 04906845001). The lysates were reduced with β-mercaptoethanol and separated by SDS-PAGE. Following transfer to 0.45 μM PVDF membrane (Millipore, IPVH09120) and blocking with 3% bovine serum albumin (BSA) in PBS + 0.1% Tween 20, membranes were incubated with rabbit anti-IRF4 (Epitomics, EP190), anti-STAT1 (Cell Signaling, 9172S), anti-pSTAT1 Y705 (Cell Signaling, 9171S), anti-PARP-1 (Cell Signaling, 9532S) or rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Sigma-Aldrich, G9545). Secondary anti-rabbit IgG-HRP (Cell Signaling, 7074S) was used, and the blots were developed using enhanced chemiluminescence (Thermo Fisher Scientific, 34078).
For IRF4 experiments, films were scanned and analyzed. Briefly, films were scanned using an EPSON Perfection 4490 scanner with data collected as a TIFF file. The TIFF file was opened in Adobe Photoshop and converted into a gray scale JPEG file that was then analyzed using NIH ImageJ (http://www.yorku.ca/yisheng/Internal/Protocols/ImageJ.pdf). For each gel, the region of interest (ROI) was defined, and the pixel density of each ROI and corresponding negative region determined. Similar analysis was done for the loading control for each gel. After appropriate backgrounds were subtracted, the relative signal was determined by normalizing to the loading controls. For each time point, the normalized value obtained for the untreated sample was set as 100%, and all values for the treated samples were expressed as percentage of the untreated sample. SEM was calculated from duplicate experiments.
Measurement of ΔΨm
Cells were treated with anti-CD138-IFNα14, bortezomib, or both for 2 days at 37°C. The concentrations of anti-CD138-IFNα14 and bortezomib were: U266 (3 pM, 1 nM), NCI-H929 (5 pM, 3 nM), ANBL-6 (5 pM, 3.5 nM), and OCI-My5 (3 pM, 1.5 nM). Cells were incubated with 10 nM 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3); Enxo, EN2-52303) for 20 min at 37°C. ΔΨm was measured using flow cytometry.
Apoptosis assay
Cells were treated with anti-CD138-IFNα14, anti-CD138, anti-CD138-IFNα2, or bortezomib for 3 days at 37°C. The concentration of anti-CD138-IFNα14 and bortezomib were: U266 (3 pM, 1 nM), NCI-H929 (5 pM, 4 nM), ANBL-6 (5 pM, 4 nM), and OCI-My5 (3 pM, 1 nM). The concentrations of anti-CD138, anti-CD138-IFNα2 and bortezomib were: U266 and OCI-My5 (3 pM, 1.5 nM), NCI-H929 (5 pM, 5 nM), and ANBL-6 (5 pM, 4nM). After treatment, cells were stained with Alexa Fluor 488-labeled Annexin V and propidium iodide (PI) using the Vybrant Apoptosis Kit #2 (Molecular Probes, V13241) and analyzed by flow cytometry.
To inhibit ROS, cells were treated and analyzed for apoptosis as described except that they were treated in the presence or absence of 1 mM tiron (Sigma-Aldrich, 72553-25G).
Murine tumor models
6–8 week old female NSG mice were used to establish NCI-H929 tumors and SCID mice were used to establish OCI-My5 tumors. Mice were inoculated subcutaneously with 1×107 OCI-My5 or 5×105 NCI-H929 on their backs. Mice were treated with PBS, 100 μg anti-CD138-IFNα14, 0.75 mg/kg bortezomib or 100 μg anti-CD138-IFNα14 + 0.75 mg/kg bortezomib. The treatments on days 14, 16, and 18 post tumor challenge were intravenous (iv). For NCI-H929, mice were also treated intraperitoneally (ip) on days 21 and 28 for a total of 5 treatments. For OCI-My5, mice were also treated ip on days 21, 28, 35 and 42 for a total of 7 treatments. Each group consisted of 8 mice. Bidirectional tumor growth was measured, and mice were sacrificed when tumors reached 1.4 cm as per institutional guidelines. All animal studies were performed in compliance with the US Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and were approved by the UCLA Animal Research Committee.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Authors' contributions
Contribution: A.V. and K.R.T designed and performed experiments. E.M.Y, and S.L.M designed research, analyzed data and wrote the manuscript. A.L and J.M.T analyzed data and provided discussions.
Supplementary_Figures_1_-_5.zip
Download Zip (5.5 MB)Acknowledgments
This work is dedicated to Peter Dalis, former Director of Athletics at UCLA, and Dr. Tetsuo Otsuki, former Professor of Chemistry at Occidental College, who died of multiple myeloma.
Funding
This work was supported by a Senior Research Award and the Dean Assink MMRF Senior Research Award from the Multiple Myeloma Research Foundation, and by the National Institutes of Health grant CA162964.
References
- Parmar S, Platanias LC. Interferons: mechanisms of action and clinical applications. Curr OpinS Oncol 2003; 15:431-9; PMID:14624225; http://dx.doi.org/10.1097/00001622-200311000-00005
- Llobet D, Eritja N, Encinas M, Sorolla A, Yeramian A, Schoenenberger JA, Llombart-Cussac A, Marti RM, Matias-Guiu X, Dolcet X. Antioxidants block proteasome inhibitor function in endometrial carcinoma cells. Anticancer Drugs 2008; 19:115-24; PMID:18176107; http://dx.doi.org/10.1097/CAD.0b013e3282f24031
- Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem 2003; 278:33714-23; PMID:12821677; http://dx.doi.org/10.1074/jbc.M302559200
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem 1998; 67:227-64; PMID:9759489; http://dx.doi.org/10.1146/annurev.biochem.67.1.227
- Uddin S, Platanias LC. Mechanisms of type-I interferon signal transduction. J Biochem Mol Biol 2004; 37:635-41; PMID:15607020; http://dx.doi.org/10.5483/BMBRep.2004.37.6.635
- Kaur S, Uddin S, Platanias LC. The PI3′ kinase pathway in interferon signaling. J Interferon Cytokine Res 2005; 25:780-7; PMID:16375606; http://dx.doi.org/10.1089/jir.2005.25.780
- Lauta VM. A review of the cytokine network in multiple myeloma: diagnostic, prognostic, and therapeutic implications. Cancer 2003; 97:2440-52; PMID:12733143; http://dx.doi.org/10.1002/cncr.11072
- Yoo EM, Trinh KR, Tran D, Vasuthasawat A, Zhang J, Hoang B, Lichtenstein A, Morrison SL. Anti-CD138-targeted interferon is a potent therapeutic against multiple myeloma. J Interferon Cytokine Res 2015; 35:281-91; PMID:25353626; http://dx.doi.org/10.1089/jir.2014.0125
- Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-α via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood 2010; 115:2864-71; PMID:20139095; http://dx.doi.org/10.1182/blood-2009-10-250555
- Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-β fusion protein therapy of murine B-cell lymphomas. J Immunother 2013; 36:305-18; PMID:23719241; http://dx.doi.org/10.1097/CJI.0b013e3182993eb9
- Huang TH, Chintalacharuvu KR, Morrison SL. Targeting IFN-α to B cell lymphoma by a tumor-specific antibody elicits potent antitumor activities. J Immunol 2007; 179:6881-8; PMID:17982079; http://dx.doi.org/10.4049/jimmunol.179.10.6881
- Wijdenes J, Vooijs WC, Clement C, Post J, Morard F, Vita N, Laurent P, Sun RX, Klein B, Dore JM. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br J Haematol 1996; 94:318-23; PMID:8759892; http://dx.doi.org/10.1046/j.1365-2141.1996.d01-1811.x
- Romano A, Conticello C, Di Raimondo F. Bortezomib for the treatment of previously untreated multiple myeloma. Immunotherapy 2013; 5:327-52; PMID:23557417; http://dx.doi.org/10.2217/imt.13.14
- Lesinski GB, Raig ET, Guenterberg K, Brown L, Go MR, Shah NN, Lewis A, Quimper M, Hade E, Young G, et al. IFN-α and bortezomib overcome Bcl-2 and Mcl-1 overexpression in melanoma cells by stimulating the extrinsic pathway of apoptosis. Cancer Res 2008; 68:8351-60; PMID:18922907; http://dx.doi.org/10.1158/0008-5472.CAN-08-0426
- Papageorgiou A, Kamat A, Benedict WF, Dinney C, McConkey DJ. Combination therapy with IFN-α plus bortezomib induces apoptosis and inhibits angiogenesis in human bladder cancer cells. Mol Cancer Ther 2006; 5:3032-41; PMID:17172406; http://dx.doi.org/10.1158/1535-7163.MCT-05-0474
- Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, Pestka S, Schreiber G. Binding and activity of all human α interferon subtypes. Cytokine 2011; 56:282-9; PMID:21856167; http://dx.doi.org/10.1016/j.cyto.2011.07.019
- Kalie E, Jaitin DA, Abramovich R, Schreiber G. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J Biol Chem 2007; 282:11602-11; PMID:17310065; http://dx.doi.org/10.1074/jbc.M610115200
- Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem 1981; 115:207-16; PMID:7227366; http://dx.doi.org/10.1111/j.1432-1033.1981.tb06218.x
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014; 14:36-49; PMID:24362405; http://dx.doi.org/10.1038/nri3581
- Arulampalam V, Kolosenko I, Hjortsberg L, Bjorklund AC, Grander D, Tamm KP. Activation of STAT1 is required for interferon-α-mediated cell death. Exp Cell Res 2011; 317:9-19; PMID:20937272; http://dx.doi.org/10.1016/j.yexcr.2010.10.002
- Shaffer AL, Emre NT, Romesser PB, Staudt LM. IRF4: Immunity. Malignancy! Therapy? Clin Cancer Res 2009; 15:2954-61; PMID:19383829; http://dx.doi.org/10.1158/1078-0432.CCR-08-1845
- Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. IRF4 addiction in multiple myeloma. Nature 2008; 454:226-31; PMID:18568025; http://dx.doi.org/10.1038/nature07064
- Chipuk JE, Green DR. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol 2005; 6:268-75; PMID:15714200; http://dx.doi.org/10.1038/nrm1573
- Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009; 114:1046-52; PMID:19436050; http://dx.doi.org/10.1182/blood-2009-01-199604
- Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004; 23:2785-96; PMID:15077142; http://dx.doi.org/10.1038/sj.onc.1207517
- Peleg-Shulman T, Tsubery H, Mironchik M, Fridkin M, Schreiber G, Shechter Y. Reversible PEGylation: a novel technology to release native interferon alpha2 over a prolonged time period. J Med Chem 2004; 47:4897-904; PMID:15369394; http://dx.doi.org/10.1021/jm0497693
- Borden EC. Review: Milstein Award lecture: interferons and cancer: where from here? J Interferon Cytokine Res 2005; 25:511-27; PMID:16181052; http://dx.doi.org/10.1089/jir.2005.25.511
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 2007; 6:975-90; PMID:18049472; http://dx.doi.org/10.1038/nrd2422
- Santini SM, Lapenta C, Logozzi M, Parlato S, Spada M, Di Pucchio T, Belardelli F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med 2000; 191:1777-88; PMID:10811870; http://dx.doi.org/10.1084/jem.191.10.1777
- Belardelli F, Gresser I. The neglected role of type I interferon in the T-cell response: implications for its clinical use. Immunol Today 1996; 17:369-72; PMID:8783497; http://dx.doi.org/10.1016/0167-5699(96)10027-X
- Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol 2010; 2010:930509; PMID:20145727; http://dx.doi.org/10.1155/2010/930509
- Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal 2009; 11:2409-27; PMID:19388824; http://dx.doi.org/10.1089/ars.2009.2625
- Kim HS, Lee MS. Essential role of STAT1 in caspase-independent cell death of activated macrophages through the p38 mitogen-activated protein kinase/STAT1/reactive oxygen species pathway. Mol Cell Biol 2005; 25:6821-33; PMID:16024814; http://dx.doi.org/10.1128/MCB.25.15.6821-6833.2005
- Markowitz J, Luedke EA, Grignol VP, Hade EM, Paul BK, Mundy-Bosse BL, Brooks TR, Dao TV, Kondalasula SV, Lesinski GB, et al. A phase I trial of bortezomib and interferon-α-2b in metastatic melanoma. J Immunother 2014; 37:55-62; PMID:24316557; http://dx.doi.org/10.1097/CJI.0000000000000009
- Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, Shaffer AL, Louie T, Chen I, Randolph S, et al. Prolonged early G1 arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood 2012; 120:1095-106; PMID:22718837; http://dx.doi.org/10.1182/blood-2012-03-415984
- Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, Einhorn S, Grander D. Mechanisms of interferon-α induced apoptosis in malignant cells. Oncogene 2002; 21:1251-62; PMID:11850845; http://dx.doi.org/10.1038/sj.onc.1205179
- Gu H, Chen X, Gao G, Dong H. Caspase-2 functions upstream of mitochondria in endoplasmic reticulum stress-induced apoptosis by bortezomib in human myeloma cells. Mol Cancer Ther 2008; 7:2298-307; PMID:18723477; http://dx.doi.org/10.1158/1535-7163.MCT-08-0186
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003; 8:237-49; PMID:12766484; http://dx.doi.org/10.1023/A:1023668705040
- Dong H, Chen L, Chen X, Gu H, Gao G, Gao Y, Dong B. Dysregulation of unfolded protein response partially underlies proapoptotic activity of bortezomib in multiple myeloma cells. Leuk Lymphoma 2009; 50:974-84; PMID:19391038; http://dx.doi.org/10.1080/10428190902895780
- Yanase N, Ohshima K, Ikegami H, Mizuguchi J. Cytochrome c release, mitochondrial membrane depolarization, caspase-3 activation, and Bax-α cleavage during IFN-α-induced apoptosis in Daudi B lymphoma cells. J Interferon Cytokine Res 2000; 20:1121-9; PMID:11152579; http://dx.doi.org/10.1089/107999000750053799
- Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD30+ anaplastic large cell lymphoma. Leukemia 2007; 21:838-42; PMID:17268529; http://dx.doi.org/10.1038/sj.leu.2404528
- Quesada P, Malanga M, Di Meglio S, De Lorenzo S, Fabbrocini A, Garbi C, Bianco AR, Pepe S. Recombinant IFN-alpha2b treatment activates poly (ADPR) polymerase-1 (PARP-1) in KB cancer cells. Eur J Cancer 2003; 39:2103-9; PMID:12957467; http://dx.doi.org/10.1016/S0959-8049(03)00433-7
- Ly JD, Grubb D, Lawen A. The mitochondrial membrane potential (delta psi m) in apoptosis; an update. Apoptosis 2003; 8:115-28; PMID:12766472; http://dx.doi.org/10.1023/A:1022945107762