
ABSTRACT
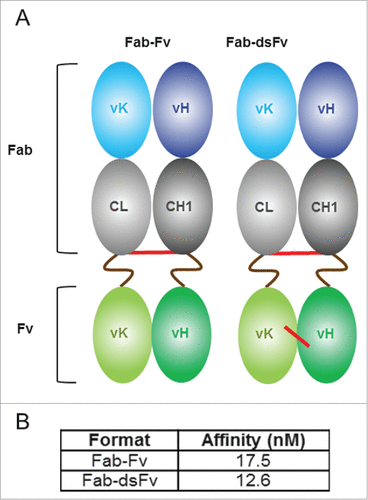
An antibody format, termed Fab-dsFv, has been designed for clinical indications that require monovalent target binding in the absence of direct Fc receptor (FcR) binding while retaining substantial serum presence. The variable fragment (Fv) domain of a humanized albumin-binding antibody was fused to the C-termini of Fab constant domains, such that the VL and VH domains were individually connected to the Cκ and CH1 domains by peptide linkers, respectively. The anti-albumin Fv was selected for properties thought to be desirable to ensure a durable serum half-life mediated via FcRn. The Fv domain was further stabilized by an inter-domain disulfide bond. The bispecific format was shown to be thermodynamically and biophysically stable, and retained good affinity and efficacy to both antigens simultaneously. In in vivo studies, the serum half-life of Fab-dsFv, 2.6 d in mice and 7.9 d in cynomolgus monkeys, was equivalent to Fab'-PEG.
Abbreviations
| Fab | = | Antigen-binding fragment |
| Fv | = | fragment variable |
| G4S | = | (glycine)4-serine |
| ds | = | disulfide |
| VL | = | variable light |
| VH | = | variable heavy |
| Cκ | = | constant light |
| CH1 | = | constant heavy |
| FcRn | = | neonatal Fc receptor |
| IgG(s) | = | immunoglobulin(s) |
| dAb | = | domain antibody |
| VHH | = | variable domain of a heavy only antibody |
| scFv | = | single chain Fv |
| HSA | = | human serum albumin |
| Fab' | = | Fab prime |
| F(ab')2 | = | bivalent Fab |
| MSA | = | mouse serum albumin |
| CSA | = | cynomolgus serum albumin |
| Fab'-PEG | = | PEGylated Fab' |
Introduction
Monoclonal antibodies are important agents for treatment of human disorders and diseases. Although immunoglobulins (IgGs) are the primary format utilized, the modular nature of IgGs has enabled drug developers to reformat antibodies to accommodate the need for therapeutics with customized pharmacological properties. Thus, antibodies with tailored characteristics such as affinity and avidity, valency, serum half-life, biodistribution, tissue penetration, effector functions, and presence of bi- or tri-target binding specificities, have been engineered by modular design or genetic mutation.Citation1-3
Antigen-binding fragments (Fabs) of antibodies have been approved for therapeutic as well as diagnostic uses, e.g., Reopro®, Lucentis®, Digibind®, Thromboview.Citation4-6 The distinguishing properties of Fabs include small size, monovalent antigen binding, lack of FcR binding, general lack of complex glycosylation and robust biophysical properties. These can confer a clinical advantage over IgG in certain indications. Compared to a ∼150 kDa full length antibody, the small size of a Fab fragment, at ∼50 kDa, results in rapid, large volume biodistribution and high levels of tissue penetration.Citation7-9 Monovalent target binding or lack of FcγR binding minimizes direct and indirect immune activation, respectively, and hence toxicological risk.Citation10-15 In addition, the lack of glycosylation enables the use of diverse production hosts.
Parentally administered Fab has a short serum half-life, typically 7 to 20 hours in humans.Citation7 While this may be irrelevant or advantageous in some applications, it invokes impractical dosing regimens in the treatment of chronic disorders. Consequently, approaches to extend the serum half-life of small recombinant therapeutic molecules, including Fabs, to levels similar to that of an IgG have become increasingly important.Citation16-17 One established half-life extension strategy is to increase the hydrodynamic volume of the molecule by coupling to inert polymers such as polyethylene glycol, as reviewed by Chapman,Citation18 or other mimetic hydrophilic polymers. Indeed, fusion or conjugation to large disordered peptides has been well validated experimentally and in the clinic.Citation18-24 Another approach is to prolong serum half-life by exploiting neonatal Fc receptor (FcRn)-mediated recycling, as used by endogenous substrates such as IgGs and serum albumin. The therapeutic molecule is fused or coupled to ligands, such as Fc domains or to serum albumin, that bind FcRn directly.Citation25-26
Alternatively, FcRn recycling can be engaged indirectly by fusion or coupling to a moiety that binds non-covalently to Fc or albumin. Thus, moieties such as IgG-binding domains or albumin-binding moieties, such as organic molecules (AlbuTag), fatty acids (myristic acid), peptides, binding domains from natural sources (Streptococcal protein G) and antibody modular domains (AlbudAbs, nanobodies) have been documented.Citation25-36
The use of serum albumin as a tool to prolong serum half-life of therapeutic proteins has gained popularity because of the status of serum albumin as a natural delivery vehicle with desirable properties. Albumin is a stable protein, and has been deduced by differential scanning calorimetry to have a Tm of 63.1°C at pH 7.4.Citation37-38 It is an abundant protein (34-54 g/L) that accounts for 65% of total plasma proteins;Citation39 in an average 70 kg male, ∼55-65% of albumin is found in the interstitial fluid (14-16 L) with the residual located in the plasma (3 L).Citation40-41
Albumin has a plasma half-life of 19 d in humans primarily because of FcRn-mediated recycling.Citation42 Genetic fusion to albumin or albumin domains is effective in conferring long half-life,Citation43-47 and various therapeutics as albumin fusions have been approved for marketing.Citation47 However, expression and purification, and the identification of relevant preclinical models for such fusions, are not straightforward, and this can limit their generic adoption.Citation43 The half-life of albumin fusions can be fine-tuned by means of inclusion of mutations that influence FcRn binding.Citation48 Non-covalent binding to albumin was shown to confer a long half-life to proteins of interest.Citation36,43 A range of albumin-binding modalities have been demonstrated, including peptides, single antibody domains (dAb, VHH), single chain Fv (scFv), Fab and albumin-binding domains (ABD, 3 helix bundles). The binding approach may be favorable in a number of respects. It can aid facile expression and purification depending upon the specific fusion partner. Effective species cross-reactive albumin binding enables the study of a clinical candidate in animal models without the need for human albumin transgenic animals or production of rodent albumin parallel reagents. With conventional blocking or neutralizing antibody therapy, a fast association rate with a slow dissociation rate is desirable to facilitate prolonged immediate and durable antibody-antigen interaction.Citation49 However, in this case, where a therapeutic drug binds to a carrier protein such as albumin, a faster dissociation rate resulting in a more dynamic antibody-albumin interaction may be more appropriate, depending on some targets, and may offer subtly different tissue penetration properties.
Previously, half-life extension of an antibody fragment has been achieved in a bivalent Fab' (F(ab)2) format where a Fab prime (Fab') directed against rat serum albumin (RSA) was cross linked by in vitro conjugation to an anti-tumor necrosis factor (TNF) Fab'.Citation43 This format proved to be effective in rats, showing a 5-fold longer half-life than an equivalent anti-TNF F(ab)2. However, a major obstacle in conjugation methodology is that the resulting products of random chemical conjugation are heterogeneous and require in-depth process development and purification, which have manufacturing and financial implications. By comparison, Dennis and colleaguesCitation22 pioneered genetic fusion of albumin binding peptides to Fabs. This approach results in a homogenous product but low affinity albumin binding, and non-uniform cross-species reactivity were technical complications.
Here, we describe an alternative bispecific antibody fragment called a Fab-dsFv that was engineered to exploit the modular nature of an IgG with the view to extending the serum half-life of a Fab. The Fab-dsFv consists of a Fab fused to an Fv derived from an anti- human serum albumin (HSA) binding antibody.Citation50 The anti-HSA antibody was selected because of its properties, which are essential to facilitate half-life extension via potential albumin recycling by FcRn. The Fv was additionally engineered to contain an inter-domain disulfide (ds) bond in order to optimize biophysical properties. Hence, as a genetic fusion and high affinity, cross reactive albumin-binding molecule, it offers both simplified production and pre-clinical translation properties.
In the Fab-dsFv format, the variable light (VL) and variable heavy (VH) domains of anti-HSA Fv are individually linked to the respective constant light (Cκ) and constant heavy (CH1) domains of the Fab region via peptide linkers (). The use of 2 linkers was intentionally adopted to reduce the theoretical risk of loss of extended half-life due to linker peptide proteolysis in serum, as documented with some scFvs or fusions.Citation51-56
Figure 1. Fab-Fv and Fab-dsFv formats and SPR analysis. Panel (A) an illustrative representation of the initial Fab-Fv and final Fab-dsFv format. The Fab-Fv format is composed of a Fab with an inter-chain disulfide bond linked to an anti-HSA Fv domain via 16 amino-acid S(G4S)3 linkers (brown wavy line). Disulfide bonds are illustrated (red line). The Fab-dsFv format retains the inter-chain disulfide bond between the constant domains and additionally contains a disulfide bond incorporated within the Fv domain at VH44 and VL100 residues. Panel (B) SPR analysis to measure the affinity of Fab-Fv and Fab-dsFv formats to the normal form of HSA (pH 7.4). The bispecific proteins were captured on the chip using a human F(ab')2-specific goat Fab and HSA (ChromPure) normal form (at pH 7.4) was titrated over the captured antibody from 50 nM. Each assay cycle consisted of 1 min injection to capture the antibody prior to an association phase consisting of a 3 min injection of the albumin; subsequent dissociation was monitored for 10 min. Kinetic parameters were determined by simultaneous global-fitting of the resulting sensorgrams (best fit) to a standard 1:1 binding model using Biacore T200 evaluation software v1.
The Fab-dsFv format was validated using a variety of in vitro and in vivo tests, including effective neutralization of the target antigen in a disease model and long serum half-life in rodents and a primate. The data presented here provide a viable alternative half-life extension technology executed by selection and engineering of the modular domains of an antibody.
Results
Construction and characterization of bispecific antibodies
The bispecific format was designed such that the constant regions of a Fab were linked to the anti-albumin Fv domain (645 Fv) derived from the humanized graft of antibody CA645.Citation23 This anti-albumin graft was selected for its desirable properties, including binding to albumin across rodent and primate species with similar affinities, the ability to retain binding at a pH ranging from 5 to 7.5 to engage FcRn recycling, and the ability to bind albumin without occluding the binding of known drugs, compounds or FcRn.Citation50
The light and heavy constant chains of an IgG1 isotype Fab were connected to the respective VH and VL domains of the Fv via 16 amino-acid long serine-glycine rich (S(G4S)3) peptide linkers, as shown in . G4S-based linkers were selected based on preliminary studies indicating flexible linkers were advantageous in terms of expression (data not shown), and although linker length was observed not to affect affinity of the 645 Fv to HSA, a minimum length of 3 x G4S is required for increased monomeric propensity (data not shown). The interchain disulfide bridge between the light and heavy constant domains was retained in the initial Fab-Fv format (). The protein was expressed in a mammalian transient expression system and purified by protein G affinity chromatography. The protein was buffer exchanged and concentrated to either a low (∼1 mg/mL) or high (∼5 mg/mL) concentration in phosphate-buffered saline (PBS; pH 7.4) and equilibrated at 4°C for at least 24 h. Size-exclusion HPLC revealed that Fab-Fv existed as monomers at low protein concentration or multimers at high concentration. These multimeric forms appear to be stable while maintained at a high concentration, and only appear to resolve to lower order or monomeric forms following dilution to lower concentrations and a period of equilibration. This suggests that the anti-albumin Fv region is in dynamic equilibrium, and this is concentration-dependent. A variant known as Fab-dsFv was subsequently constructed where a disulfide bridge was introduced between VH44 and VL100Citation57 (Kabat definition) of the respective VH and VL domains of the anti-HSA Fv (). Although Fab-dsFv expression still resulted in a combination of monomers, dimers and trimers (data not shown), this strategy solved the issue of concentration-dependent multimerization. The affinity of the 645 Fv for HSA of the monomeric fraction of Fab-dsFv was not affected by the presence of the additional disulfide (). Interestingly, isolated fractions of dimeric and trimeric proteins showed avidity of binding to HSA with respect to order of multimerization (data not shown).
Biophysical analysis of bispecific Fab-dsFv
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
The presence or lack of particular disulfide bonds resulted in a shift in the apparent molecular weight (MW) when non-reduced and reduced Fab-Fv variants were analyzed by sodium dodecyl sulfate-PAGE (SDS-PAGE). Non-reduced Fab-Fv and its variants were predicted to have a MW of ∼73-74 kDa, with free light and heavy chains at ∼36 and ∼37 kDa, respectively. In all cases however, non-reduced proteins appeared to consistently migrate with dissimilar mobilities. Fab-Fv, which lacks the Fv disulfide (), was observed to migrate to ∼97 kDa () whereas FabΔinter-dsFv (), which lacks the interchain disulfide bond between the constant domains but retains the Fv disulfide bond, demonstrated a further shift to ∼66 kDa (). Fab-dsFv (), which contains both disulfide bonds migrated close to its predicted MW at ∼79 kDa (). Reduced light and heavy chains of all variants migrated to their predicted MWs at ∼36-37 kDa ().
Figure 2. Analysis of disulfide Fab-Fv variants. (A) an illustrative representation of disulfide Fab-Fv variants where disulfide bonds are shown in red and S(G4S)3 linkers in brown lines: (1) the initial format Fab-Fv, contains the inter-chain disulfide bond between the constant domains (2) FabΔinter-dsFv where the inter-chain disulfide bond between the constant domains was removed but a disulfide bond has been introduced within the anti-HSA Fv domain at VH44 and VL100 residues. (3) The final format Fab-dsFv contains both the inter-chain disulfide bond and the anti-HSA Fv disulfide bond at VH44 and VL100 residues. (B) SDS-PAGE of purified proteins separated under non-reducing conditions: (1) Fab-Fv (∼3 µg) (2) FabΔinter-dsFv (∼3 µg) (3) Fab-dsFv (∼5 µg) and purified proteins separated under reducing conditions: (1R) Fab-Fv (∼3 µg) (2R) FabΔinter-dsFv (∼3 µg) and (3R) Fab-dsFv (∼5 µg). Novex Mark12 wide-range protein standards were used as markers (lane M). Full length FL; heavy chain H; light chain L; heavy + light chain H+ L.
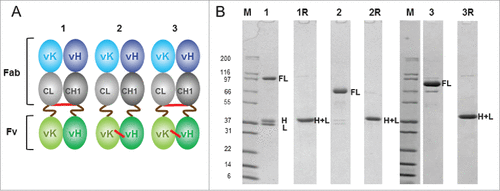
The shift in mobility of non-reduced proteins appears to be dependent on the presence, and position of the disulfide bond(s). Indeed, the presence of intact disulfide bonds and more specifically, the compact nature of some S-S bridged conformations have been shown to affect SDS-PAGE migration rates.Citation58-60 Thus, the Fab-Fv molecule bearing only the interchain disulfide bond appears to be less compact, FabΔinter-dsFv is more compact and Fab-dsFv approaches the predicted MW on SDS-PAGE.
A doublet at ∼36-38 kDa (), consistent with incomplete disulfide bond formation between the light and heavy chains, was also observed on non-reduced gels of 2 of the variants, more significantly for Fab-Fv than FabΔinter-dsFv. Incomplete disulfide bonding appears to be generated during expression (data not shown), and has previously been reported for an IgG-scFv bispecific format, where 18-amino acid long peptide linkers connected to the terminal cysteine residues of the Cκ domains were sufficient to cause incomplete disulfide bond formation between light and heavy chains of an IgG. The addition of an alternative disulfide bond between the VL and VH domains of the IgG appeared to improve the interchain covalent linkage.Citation61 Poor disulfide formation was confirmed when 11 amino acid- long S(G4S)2 peptide linkers were linked to the C-terminal residue of both constant domains of a Fab (data not shown). This suggests that the efficiency of the interchain disulfide bond formation can be disrupted when a polypeptide is extended from either or both C-terminal cysteine residues involved in the interchain disulfide bond process.
Size-exclusion high performance liquid chromatography
Size-exclusion high performance liquid chromatography (SE HPLC) of minimally purified (protein G affinity chromatography only) Fab-Fv, FabΔinter-dsFv and Fab-dsFv disulfide variants displayed a combination of monomers and higher order species (). The monomeric and each of the higher order species appeared to elute similarly during HPLC (indicating similar hydrodynamic volumes) despite showing migration disparities during SDS-PAGE, which we interpret as ‘gel-shift’ events. The presence of an interchain or Fv disulfide bond appears to affect the degree of multimerization, with Fab-dsFv, which contains both disulfide bonds, being the most monomeric (). The total expressed pool of Fab-dsFv consisted of 47.7 ± 14.2% monomer, 19.3 ± 3.1% dimer and 33 ± 13.4% trimer, with the apparent MWs of 79.4 ± 7.5 kDa, 202.6 ± 6.8 kDa and 346.3 ± 21.4 kDa, respectively, based on MW standards and at least 3 replicates. This indicates that both interchain and Fv disulfide bonds are essential for improved properties displayed by the finalized format, the Fab-dsFv molecule.
Figure 3. Size exclusion HPLC analysis of disulfide variants and Fab-dsFv. Disulfide variants and Fab-dsFv were purified by Protein G chromatography and prior to analysis. (A) purified Fab-Fv (![]()
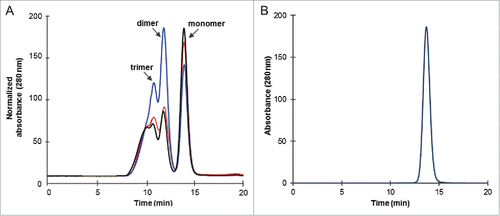
The monomeric fraction of Fab-dsFv was isolated by ion exchange chromatography followed by gel filtration and equilibrated in PBS (pH 7.4) prior to analysis by SE HPLC on Superdex 200. The purified material was 100% monomeric with a single peak eluting at 13.7 min (). A MW of 86.9 ± 0.8 kDa (from 3 replicates), based on MW standards (not shown) was calculated. No protein loss or proteolytic clipping was detected and this was further confirmed by SDS-PAGE (). Monomeric Fab-dsFv was used for all further studies.
Mass spectrometry
The identity of Fab-dsFv was confirmed by nanoscale liquid chromatography coupled to mass spectrometry (nLC-MS; ). An observed mass of 73616.0 Da was detected for the whole molecule, 3.8 Da (58 ppm) lower than the expected mass (73619.8 Da) as deduced from the amino acid sequence and the expected disulfide bonding pattern (). This value is within the accuracy specifications of the mass spectrometer. The observed mass is therefore consistent with the expected mass of Fab-dsFv.
Figure 4. Intact mass of purified Fab-dsFv by nLC-MS. Purified Fab-dsFv was analyzed by nLC-MS to obtain the intact mass. Main panel: combined mass spectrum showing the m/z envelope of Fab-dsFv ions. Inset panel: De-convoluted mass spectrum produced by MaxEnt algorithm (Waters, USA).
Isoelectric focusing
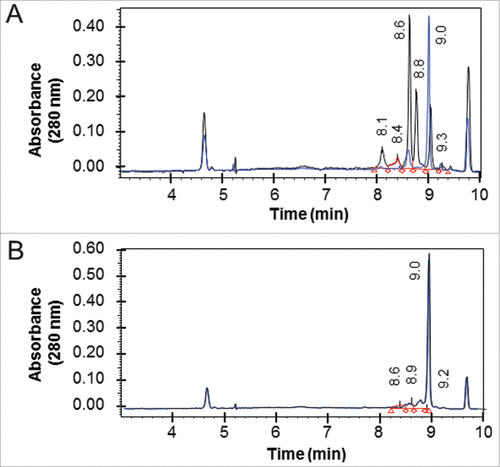
A high experimental pI of 9.0 was observed for the Fab-dsFv molecule. This can be advantageous from a manufacturing perspective because it enables efficient DNA, host cell protein and endotoxin removal. The Fab-dsFv molecule was found to be less heterogeneous with respect to acidic and basic species compared with the equivalent Fab' fragment ( vs B), with the majority of the sample resolving at pI of 9.0. Mild conditions were used to reduce and alkylate surface accessible thiol adducts, and unlike the Fab' molecule, under these conditions Fab-dsFv showed no major changes in the overall cIEF profile, indicating the absence of reducible thiol adducts.
Figure 5. Isoelectric focusing of Fab-dsFv. The pI of non-reduced (black line) and reduced and alkylated (blue line) of purified (A) Fab' fragment and (B) Fab-dsFv was determined by iCE280 capillary isoelectric focusing. Samples were mixed with methylcellulose, pharmalytes (pH3-10) and synthetic pI markers prior to separation. For mild reduced and alkylated samples, iodoacetamide was used as the alkylating agent after reduction with Tris(3-hydroxypropyl)phosphine (THPP). Profiles were continuously detected at an absorbance of 280 nm. The pI values of the non-reduced sample are indicated on each graph. Calibrated electropherograms were analyzed using Empower 2 (Waters).
Molecular stability of Fab-dsFv
Thermostability following exposure to pH
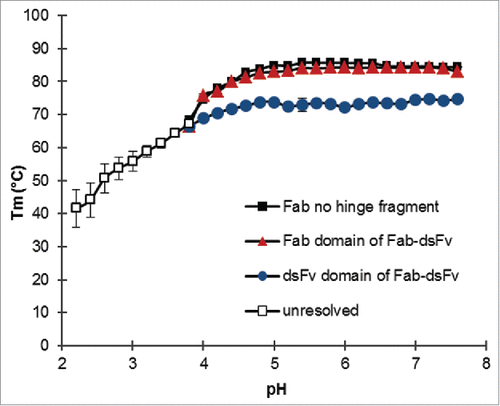
The FcRn recycling mechanism is known to exhibit optimal activity at acidic pH, and it is thus desirable that the bispecific format exhibits stability at low pH as well as neutral pH. Also, it is essential that the molecule is stable from aggregation at low pH steps that occur during manufacture (affinity purification and viral inactivation). The molecular stability of bispecific proteins in the presence of a pH gradient was measured by determination of the midpoint melting temperature (Tm). The Thermofluor assay was used to demonstrate whether unfolding transitions of the individual Fab and dsFv domains of the format are affected by pH, and subsequently whether the format is pH stable. The phosphate / citrate buffer system was selected to maintain a pH range between pH 2.6 and pH 7.4. A correction for pH changes due to increasing temperature was not made because this effect is small in the chosen buffer system. Sypro-orange concentrations remained the same throughout the experiment, and therefore Tm data is valid and comparable. The fluorescence intensity across the pH titration did not change in the absence of Fab-dsFv, indicating that Sypro orange was not sensitive to pH (data not shown). A Fab no-hinge variant of Fab-dsFv was used as the reference for the Fab domain. Fab-dsFv showed a typical pH titration profile where 2 transition events, one tracking the unfolding of the Fab no-hinge fragment and a second assumed to be the anti-HSA dsFv, were tracked against pH (). The Fab and dsFv domains of the bispecific format exhibited good thermal stability above pH 5; the Tm of the Fab domain was measured as 84 ± 0.4°C and 82.9 ± 0.4°C at pH 7.4 and pH 5, respectively, comparable to the Tm of the Fab no-hinge fragment (84.3 ± 0°C at pH 7.4, 84.9 ±0 .1°C at pH 5). The Tm of the dsFv was 74.3 ± 0.4°C and 73.8 ± 0.5°C at pH 7.4 and pH 5, respectively, ∼10°C lower than the Fab domain. By comparison, the dsFv thermal stability value is ∼4 - 5°C above the CH2 domain of an IgG1 antibody (pH 7.4), which is generally considered the most stable isotype based on the Tm of its CH2 and CH3 domains.Citation62 The thermal stability of both entities could not be resolved below pH 4. Overall, the separate entities of Fab-dsFv appear to be pH stable above pH 5. Indeed, surface plasmon resonance (SPR) analysis indicated that affinity to albumin in the sub-picomolar range was maintained at the pH range (pH 5 to 7) tested.Citation50
Figure 6. Thermostability of Fab-dsFv following exposure to pH. A thermofluor assay was used to determine the midpoint melting temperature (Tm) transition states of the individual Fab (![]()
Resistance to aggregation
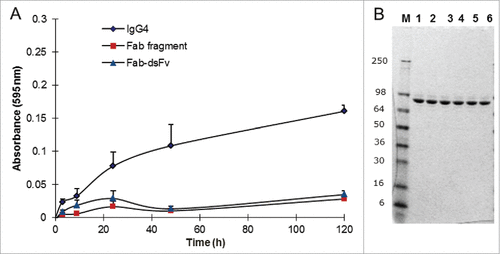
As a novel format, it is important that the antibody is stable in terms of minimal degradation or formation of high MW species while under mechanical stress, e.g., ultrafiltration steps during manufacture.Citation63 Purified Fab-dsFv in PBS (pH 7.4) was vortexed at 1,400 rpm at 25°C for up to 120 hours. A corresponding Fab no hinge fragment and an IgG4 antibody were used as benchmark controls. At each time point, the samples were centrifuged briefly, mixed to resuspend any pellet and the absorbance at 595 nm was obtained. At the same time, a 10 µL aliquot was removed from each replicate for storage at −20°C for subsequent SDS-PAGE analysis. Fab-dsFv demonstrated a similar aggregation profile to the Fab, where minimal aggregation (<0.1 A595) was observed at the 120 h time point (). In comparison to the full-length IgG4, which typically exhibited a greater propensity to aggregate under these conditions, aggregation of Fab-dsFv was negligible. Prior to SDS-PAGE analysis, samples were thawed, centrifuged and re-suspended in Tris-Glycine SDS-PAGE loading gel buffer for gel analysis. A volume equivalent to ∼2 µg of protein was loaded per track. Fab-dsFv samples showed that there was no degradation or appearance of protein fragments at any time point compared to the untreated control (). Thus, the Fab-dsFv antibody was found to be resistant to aggregation and degradation when stressed by agitation over 5 d.
Figure 7. Fab-dsFv resistance to aggregation. Analysis of Fab-dsFv Fab (no hinge) fragment and IgG4 in a vortexing aggregation assay. (A) Purified protein at 1 mg/mL was vortexed at 1400 rpm for over 120 h in PBS (pH 7.4) at 25°C. Samples were taken at 0, 1.5, 3, 24, 48 and 120 h and the absorbance at 595 nm was measured and plotted against time: Fab-dsFv (![]()
Long term stability of Fab-dsFv
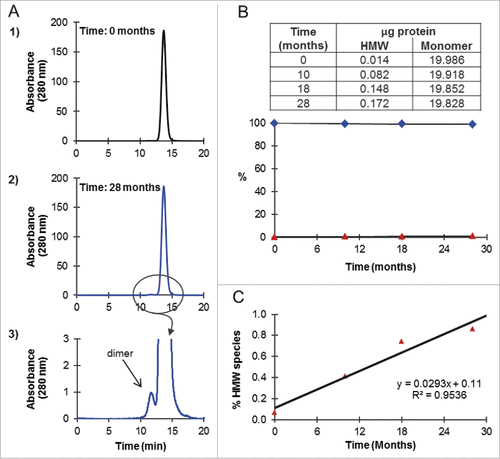
Long-term stability of highly purified Fab-dsFv was assessed by incubating the antibody at 10 mg/mL in PBS (pH 7.4) at 4°C for 28 months. At intervals of 0, 10, 18 and 28 months, samples (∼20 µg) were analyzed by SE HPLC on Superdex 200. shows the SE chromatograms at the start (time zero) and at the end (28 months) of the experiment. At time zero (), a single peak eluting at 13.7 min and corresponding to monomer with a MW of 86.9 ± 0.8 kDa, based on MW standards (not shown), was observed. Over the course of 28 months, an aberration to the baseline was detected, cumulating in the resolution of a minor peak eluting at 11.7 min at the end of the time course (). Based on MW standards, this peak had a MW of 206.3 ± 1.0 kDa, corresponding to a dimer, and was practically indiscernible on the chromatogram (), with < 0.2 µg of the total 20 µg load or < 1%, resolving as a dimer after 28 months at 4°C (). Aggregates, higher order species or clipped species were not detected. Thus, the Fab-dsFv molecule was shown to be > 99% monomeric over this storage period. The % rate of dimer formation of 0.029%/month was calculated from the slope of a plot of % HMW species against time (), confirming that the Fab-dsFv molecule is stable for long-term storage in this formulation.
Figure 8. Long-term stability of Fab-dsFv. Purified Fab-dsFv (n = 1) at 10 mg/mL in PBS (pH 7.4) was incubated at 4°C for 27 months. At ∼9 monthly intervals the sample was mixed and an aliquot (∼20 µg) was analyzed by SE HPLC on Superdex 200. (A) elution of Fab-dsFv at 1) the start of the experiment and 2) at the end of the experiment at 18 months, where 3) shows the zoomed profile of the peak corresponding to dimer. (B) the amount of HMW species (![]()
Independent and simultaneous binding of Fab-dsFv to the Fab antigen and serum albumin
SPR binding assays were utilized to monitor the independent or simultaneous binding of Fab-dsFv to the Fab antigen or serum albumin. No difference in affinity or on- or off- rates was observed between the Fab (45.9 pM) or Fab-dsFv (40.8 pM) binding to the target antigen (). Similarly, binding of Fab-dsFv to serum albumin across species was similar with HSA at 2.93 ± 0.52 nM, mouse serum albumin (MSA) at 4.16 ± 0.13 nM and cynomolgus serum albumin (CSA) at 2.66 ± 0.18 nM (). Equally, on- and off rates of binding to albumin were within an acceptable range between species.
Table 2. Independent and simultaneous binding SPR kinetics and affinity of Fab-dsFv for the target antigen and serum albumin. The binding affinities and kinetic parameters for the interactions of antibodies were determined by SPR with HBS-EB buffer (pH 7.4) as the running buffer at 25°C. The antibody samples were captured to the sensor chip surface via a human F(ab')2-specific goat Fab. A) Binding kinetics and affinity (KD) of captured Fab or Fab-dsFv to the target antigen. Values are the arithmetic mean and standard deviation (s.d) was determined from four independent titrations. B) Binding kinetics and affinity (KD) of captured Fab-dsFv to HSA (normal form at pH 7.4) MSA or CSA. Values are the arithmetic mean and s.d was determined from three independent titrations. For both A) and B) the association rate (kon) was determined by a 3 min injection of the target antigen or albumin over captured antibody after which dissociation rate (koff) was monitored for 30 min for the target antigen and 10 min for albumin. C) Simultaneous binding of captured Fab-dsFv to the target antigen HSA or a mixed solution of target antigen and HSA. The kon rate was determined by injecting HSA the target ligand or a mixed solution of both HSA and the target antigen over the captured antibody for 3 min. The koff rate was monitored for 30 min. Kinetic parameters were determined by simultaneous global-fitting of the resulting sensorgrams to a standard 1:1 binding model using Biacore T200 evaluation software v1. The affinity (KD) was calculated from the koff and kon rates.
Table 1. Protein Identification of Fab-dsFv by mass spectrometry. The Observed mass [M] of the whole protein was detected by nLC-MS.Footnote1 The theoretical mass of the light and heavy chains, and the resulting mass of the intact molecule was calculated from the amino acid sequence of Fab-dsFv using the ExPASy Compute pI/Mw tool http://web.expasy.org/compute_pi/. Footnote2 The expected mass was calculated with respect to the number of H+ derived from the disulfide bonding pattern: each chain of Fab-dsFv contains 3 intra-domain disulfide bonds, in addition to the inter-chain disulfide between light and heavy chain, and the disulfide bond between the VL and VH domains of the anti HSA-Fv. All masses are presented as Da units.
The simultaneous binding of the target antigen and HSA to Fab-dsFv was assessed. The bispecific protein was first captured to a BIAcore sensor chip by immobilized anti-human F(ab').Citation2 In preliminary experiments, the order in which ligands were titrated (HSA followed by the target antigen, or vice versa) was found to be not relevant, and use of a mixed solution of both ligands was comparable and valid. Subsequently, the target antigen (25 nM) alone, HSA (50 nM) alone or a combined solution of the target antigen and HSA, were titrated over the captured Fab-dsFv antibody. The binding responses for all 3 experiments were measured; the results are shown in . The binding response for the combined target antigen/HSA solution was shown to be equivalent to the sum of the responses of the independent injections. This confirms that Fab-dsFv is capable of simultaneous binding of both antigens.
Mouse pharmacokinetics
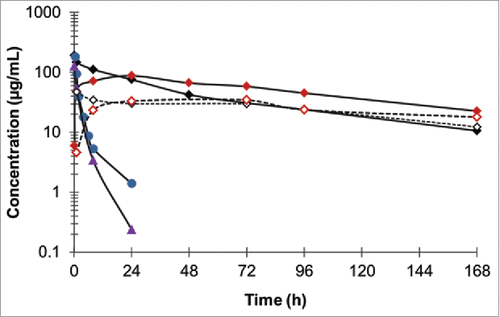
The serum half-life of Fab-dsFv was first established in BALB/c mice dosed by a single subcutaneous (s.c.) or intravenous (i.v.) injection at 10 mg/kg and measured in blood sera taken at various time points over 168 h. A non-half-life extended F(ab')2, a Fab-dsFvB, where dsFvB is an unrelated, non-albumin-binding dsFv, and a PEGylated Fab' against the target ligand (Fab'-PEG) were used as controls. Fab-dsFv showed more durable serum exposure compared to the F(ab')2 or Fab-dsFvB, when i.v. administered (). A 20-fold lower clearance rate (CL) at 1.3 ± 0.1 mL/h/kg was observed for Fab-dsFv compared to 29.5 ± 2.7 mL/h/kg and 22.9 ± 0.1 mL/h/kg for F(ab')2 and Fab-dsFv, respectively. Accordingly, Fab-dsFv showed a 16-fold and 34-fold longer serum half-life of 62 ± 4 h compared to 4 ± 0.3 h for F(ab')2 and 1.8 ± 0.1 h for Fab-dsFvB. Interestingly, Fab-dsFv showed similar CL at 1.3 ± mL/h/kg compared to 2.6 ± 0.1 mL/h/kg for Fab'-PEG, with half-lives at 62 ± 4 h and 65 ± 5 h, respectively. Comparable observations also were made when these molecules were administered via the s.c. route, indicating that Fab-dsFv has similar overall pharmacokinetics (PK) properties to Fab'-PEG.
Figure 9. Pharmacokinetics of Fab-dsFv in BALB/C mice. Fab-dsFv at a single dose of 10 mg/kg was administered via the i.v (![]()
Cynomolgus monkey pharmacokinetics
To better understand the PK of Fab-dsFv, healthy cynomolgus monkeys (2 males and 2 females per group) received i.v. (bolus, 2 mL/kg) doses of 30, 3 and 0.3 mg/kg of Fab-dsFv. The effect of Fab-dsFv on serum albumin levels was assessed at multiple time points during the first week, and then every other week to determine that the binding of the Fv domain to albumin did not adversely affect albumin levels in vivo.
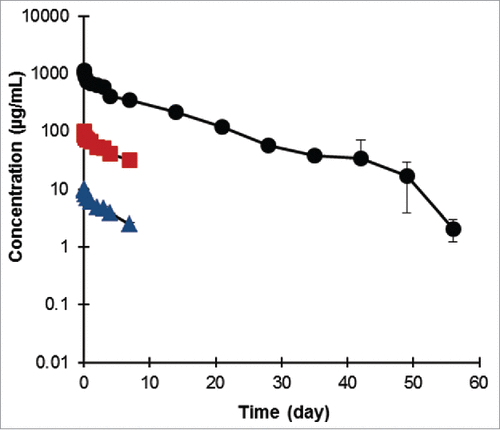
The bispecific antibody was well tolerated, with no clinical signs, no effects on body weight, hematology or clinical pathology parameters. shows the mean PK profile of the Fab-dsFv antibody in the plasma of animals up to 56 d as measured using a Meso-Scale Discovery detection assay. No change in serum albumin levels was observed due to administration of Fab-dsFv as a single i.v. dose at 30, 3 or 0.3 mg/kg (data not shown). At both lower doses, inter-animal variation was observed within the sample population. Because the sample size was small and statistical variation large, data from the 2 groups where LLOQ <0.03 or n < 3 were not reported. Further tests to establish the cause of the variation were not performed, although it should be noted that inter-animal variation associated with low dose regimens of biologics in cynomolgus monkeys is frequently reported.Citation64-68 The 30 mg/kg dose group displayed relatively small variation and a mean clearance rate of 3.49 ± 0.21 mL/day/kg was observed. The mean half-life of this group was calculated to be 7.87 d.
Figure 10. Pharmacokinetics of Fab-dsFv in cynomolgus monkey. The presence of Fab-dsFv in diluted plasma samples was measured at multiple time points and detected in a MSD immunoassay. Six male and 6 female cynomolgus monkeys were allocated to 3 treatment groups (2 per sex per group) and received 1 i.v. dose of Fab-dsFv at 0.3 (![]()
Allometric scaling
The PK data from the cynomolgus monkey was analyzed using a mixed effects approach. Data displaying LLOQ <0.03 or n < 3 were excluded from the calculation. The PK parameters, clearance in mL/day (CL), intercompartmental clearance (Q) and volume of the distribution of the central (V1) and peripheral compartment (V2) were allometrically scaled. A body weight of 3.3 kg for monkey and 70 kg for human was used. The formula used for scaling is based on the simple allometric equation: CL = a*BW^b, where CL = clearance in mL/day, a = normalization constant, BW = body weight in kg, and b = the scaling exponent. This is an accepted method for scaling the PK of protein therapeutics.Citation69 CL and Q were scaled using an exponent of 0.75. This value is consistent with the allometric scaling of endogenous metabolic and flow rates,Citation70 and is also consistent with the allometric scaling used for monoclonal IgGs.Citation68 Distribution volumes were scaled with an exponent of 1, as protein therapeutics typically distribute to common endogenous fluid volumes (plasma, extracellular fluid). The calculation of the half-life was derived from the scaled values of CL, Q, V1, and V2 using a 2-compartment model. This predicts a half-life in man for Fab-dsFv in the range of 14 – 17 d.
Mouse model of human T cell proliferation (Hu-NSG model)
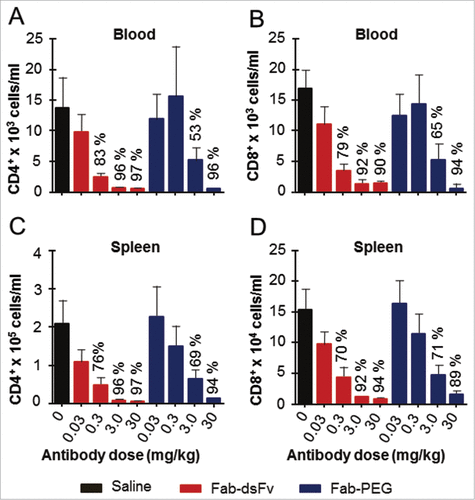
An in vivo model of human T cell proliferation was used to determine the in vivo efficacy and potency of the Fab-dsFv antibody to bind its target antigen and elicit a biological activity. The target antigen is exemplified by a T-cell receptor. A single dose of Fab-dsFv or Fab'-PEG at 0.03, 0.3, 3 or 30 mg/kg was administered subcutaneously 1 day prior to the transfer of human peripheral blood mononuclear cells (PBMCs) to NOD SCID gamma (NSG) mice. After a predetermined period of 14 days, blood, spleen and peritoneal cavity samples were taken for fluorescence-activated cell sorting (FACS) analysis of the relative number of human CD4+ and CD8+T cells in these compartments. Proliferation of both CD4+ and CD8+ T cell numbers was inhibited in a dose-dependent manner with both therapeutic entities compared to the robust proliferation noted in mice receiving vehicle alone (). When administered at day −1, 3 mg/kg of Fab-dsFv resulted in 96% and 92% inhibition of human CD4+ and CD8+ proliferation found in the spleen, respectively. Similar levels of inhibition were also found in blood. Administration of 3 mg/kg of Fab'-PEG resulted in 69% and 71% inhibition of CD4+ and CD8+ T cells in the spleen, respectively, and 63% and 65% inhibition of CD4+ and CD8+ T cells in the blood, respectively. These data indicate that, when administered on the day prior to cell transfer, a dose of 3 mg/kg of Fab-dsFv results in complete inhibition of T cell proliferation in the Hu-NSG model, whereas a dose of 30 mg/kg of the Fab'-PEG was required to confer similar efficacy. Although Fab-dsFv was shown to have similar affinities to the Fab for the target antigen (), and comparable serum half-lives with Fab'-PEG (), differences in penetration in terms of the hydrodynamic volumeCitation18 or albumin bindingCitation71 could be contributing factors. Significant inhibition of T-cell proliferation was also noted when Fab-dsFv was dosed at 0.3 mg/kg (76% and 70% inhibition of CD4+ and CD8+ T cells in the spleen, respectively, and 83% and 79% inhibition of CD4+ and CD8+ T cells in the blood, respectively). Similar results were obtained for the peritoneal cavity (data not shown). Data indicates that Fab-dsFv is broadly as efficacious as Fab'-PEG.
Figure 11. Fab-dsFv administered prior to cell transfer dose dependently inhibits CD4+ and CD8+ T cell proliferation in the Hu-NSG model. Mice were dosed subcutaneously with 0.03, 0.3, 3 or 30mg/kg Fab-dsFv or Fab-PEG one day prior to transfer of 10 million human PBMCs into the peritoneal cavity. A saline only dose was used as a control. After 14 days, mice were bled by cardiac puncture under terminal anesthesia and then killed by cervical dislocation. The number of human CD4+ and CD8+ cells in the blood (panels (a) and (b) respectively) and the spleen (panels (c) and (d), respectively) was then determined by FACS analysis. Data (n = 10) is expressed as means ± SEM and statistical analysis is by one way ANOVA with Bonferroni post test.
Discussion
In order to confer sufficient serum half-life to a Fab that it could be considered for therapeutic uses, we designed a small bispecific antibody format known as a Fab-dsFv. We used a high-affinity, cross-species reactive humanized anti-HSA antibody, CA645 that displayed desirable properties, including the ability to support FcRn-mediated recycling via albumin binding, and the ability to bind to albumin without occluding the binding of known drugs.Citation23 This antibody's binding site was structurally identified as HSA domain II,Citation50 and deduced by modeling to bind distinctly from the binding sites of known major drugs and compounds such as warfarin, salicylate and bilirubinCitation72-74 on domain II, heminCitation75 and metal ionsCitation76 on or near domain I, and FcRn, which binds primarily to domain IIICitation39,77 and domain I.Citation78
The Fab-dsFv format consists of the anti-HSA Fv region from CA645 fused to the C-termini of a Fab through 2 flexible linkers. Two linkers rather than one, as commonly observed with fusions with albumin or albumin-binding domains, were used to mitigate the risk of losing the extended serum half-life characteristic should the linker be susceptible to serum proteolysis, i.e., extended serum half-life should be retained even with the loss of one peptide linker. Loss of scFv activity upon proteolytic clipping at the G4S linker interconnecting V-regions in vivo has been previously reported.Citation51-56 Extensive studies to engineer protease-resistant peptide linkers, where immunogenicity issues are a concern, were thus avoided through use of this alternative route to ensure serum stability. Furthermore, a G4S-rich linker has been used in numerous therapeutic fusions to date, and found not to be immunogenic.Citation79-82 The anti-HSA Fv was further stabilized though a disulfide bond that afforded stability essential to minimize V-region dynamic exchange and the formation of concentration-dependent multimeric forms. It is crucial that the format remains monomeric to prevent crosslinking when blocking cell surface targets in vivo.Citation10-15 Disulfide stabilization also contributed to thermal stability. The Fv disulfide bond critically served as a safeguard, allowing the Fv to remain intact and attached to the Fab if one linker was vulnerable to proteolytic attack in vivo. Surprisingly, inclusion of a second disulfide bond within the anti-HSA Fv provided another positive aspect in terms of increased monomer of specifically, covalently linked Fab-dsFv.
The target antigen affinity and Fab stability were both unaffected by the presence of the C-terminal Fv domain. Fab-dsFv has been shown to retain the desirable binding properties attributed to the parent antibody CA645. It bound HSA with high affinity and demonstrated broad reactivity to albumin from a range of species with similar affinities. This has the advantage of evading the need to generate rodent preclinical models transgenic for HSA and human FcRn, which have both been shown to bind albumin in a species-dependent manner.Citation33 Importantly, Fab-dsFv bound albumin at pH 5, a property critical for utilization of the FcRn recycling mechanism.Citation83-84 The format also exhibited behaviors and properties important for biophysical stability, including negligible aggregation upon prolonged storage, making it unlike scFvs and Fab-scFvs, which have the propensity to aggregate.Citation85-86
In addition, Fab-dsFv was shown to simultaneously bind the target antigen and HSA without occlusion of binding of the other ligand. In the SPR experimental design, the binding responses to the individual ligands and the response to binding where both ligands are injected as a mixed solution was measured. The latter case is an in vitro representation of the environment, where the drug can encounter both soluble ligands simultaneously, and covers all binding scenarios where either both ligands bind simultaneously, or where an affinity-dependent target-mediated response is engaged.
In terms of ligand engagement, Fab-dsFv was shown to bind to both the target antigen and serum albumin simultaneously in vitro and presumed to do so in vivo, as demonstrated by enhanced PK and efficacy in preclinical models, similar to Fab'-PEG. The molecule shows substantial serum half-life in rodents (2.6 d and 3 d via the respective i.v. and s.c. routes) and non-human primate (7.9 d) comparable to Fab'-PEGCitation87 and therapeutic fusions with albumin-binding peptides, albumin or albumin domain fusions.Citation88-89 Allometric scaling using the PK of cynomolgus monkey and body weight has predicted this molecule to have a plasma half-life of 14 to 17 d in humans. Thus, the extended half-life of Fab-dsFv is likely primarily due to the nM affinity for albumin and the pH insensitivity of albumin binding and intrinsic biophysical stability of the molecule at neutral and acidic pH.
Fab-dsFv expressed well in Chinese hamster ovary (CHO) cells and was purified by standard means, thereby overcoming some of the disadvantages with other albumin fusion and albumin-binding technologies, such as the bispecific F(ab)2 by Smith et al.Citation43 We have accumulated considerable experience with the Fab-dsFv format, having made numerous variants with different therapeutic Fabs that could be expressed in concentrations in the range of hundreds of mgs/L in mammalian transient expression platforms. All show similar extended serum half-lives profiles as the Fab-dsFv reported here (data not shown). Generation of stable cell lines resulted in > 2 g/L yields from fed-batch fermentations, and the protein has been purified in a form suitable for human clinical studies (data not shown). The Fab-dsFv format is thus a viable option for clinical use where target and disease biology dictate a need for a monovalent antigen-binding therapeutic with enhanced serum half-life, but the absence of direct FcR engagement.
Materials and methods
Materials
All materials and reagents were sourced from Life Technologies unless otherwise stated. Purified target antigen, Fab (no hinge) fragments, F(ab')2, IgGs and Fab'-PEG (conjugated to 40 kDa PEG) against the target antigen or other antigens were prepared in-house.
Antibody discovery and humanization
For the Fab of the humanized bispecific Fab-Fv antibody format, antibodies were raised against an immune target in female Sprague Dawley rats using a modified selected lymphocyte antibody method.Citation90-92 The immunization, screening process and humanization of the lead anti-HSA antibody has been described elsewhere.Citation23
Bispecific antibody engineering
Both heavy and light chain genes of the bispecific molecule were designed in silico and chemically synthesized (DNA 2.0 Inc.). Briefly, the DNA sequence of the C-terminus of CH1 domain of the Fab was fused to the VH variable domain of the Fv via a 16 amino-acid long polyglycine/serine linker consisting of S(G4S)3. Similarly the DNA sequence of the Fab light chain, encoding the C-terminus of the Cκ domain was fused to the DNA sequence of the VL domain of the Fv via a S(G4S)3linker. A disulfide bond was introduced between the VH and VL domains of the Fv. Cysteines at VH44-VL100 positions,Citation57 according to Kabat numbering, were introduced into the respective VH and VL domains using Quikchange Lightning site-directed PCR mutagenesis (Agilent).
Antibody expression
Chemically synthesized antibody genes were sub-cloned into UCB's proprietary mammalian expression vectors as single genes for transient expression. All antibody formats were transiently expressed in HEK-293 or CHO-S XE cellsCitation93 using standard lipid transfection or electroporation methods, respectively. Culture supernatants were harvested by centrifugation and 0.2 μm filter sterilized. Expression titers were measured by Protein G HPLC using a HiTrap Protein G column (GE Healthcare) according to manufacturer's recommendations, against a standard curve of a purified Fab.
Protein purification
Protein G affinity chromatography was used to purify antibody proteins from culture supernatants. Briefly, supernatants were loaded on a HiTrap Protein G (GE Healthcare) and then washed with PBS pH 7.4. The bound material was eluted with 0.1 M glycine pH 2.7, and neutralized with 2 m Tris-HCl (pH 8.5) prior to buffer exchange into PBS pH 7.4. Samples were fractionated by ion exchange chromatography with salt gradient elution. The fractions collected were neutralized and buffer exchanged into PBS pH 7.4. The eluted protein was quantified by absorbance at 280 nm and stored at 4 °C for further analysis. Monomeric fractions were isolated by size exclusion chromatography using a HiLoad 16/60, Superdex 200 column (GE Healthcare) equilibrated with PBS, pH 7.4. Fractions containing monomeric protein were pooled, quantified, concentrated and stored at 4°C.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Samples were prepared by the addition of 4 x Novex NuPAGE LDS sample buffer and either 10X NuPAGE sample reducing agent or 100 mM N-ethylmaleimide (Sigma-Aldrich), and were heated to 100°C for 3 min. Prepared samples containing 3 µg (for 15- well gels) or 5 µg of purified protein (for 10-well gels) were loaded onto a 15 well Novex 4-20% Tris-glycine SDS-polyacrylamide gel and separated at a constant voltage of 125 V for 110 min in Tris-glycine SDS running buffer. Novex Mark12 wide-range protein standards were used as standards. The gel was stained with Coomassie brilliant blue (Sigma-Aldrich) in 10% methanol, 7.5% acetic acid for 1 h and destained with several changes of distilled water.
Size exclusion HPLC
Purified proteins were analyzed by Superdex 200 or TSK G3000 depending on resolution required. For Superdex 200 SE HPLC, samples (∼20 µg) were loaded on to a Superdex 200 10/300 GL Tricorn column (GE Healthcare) and eluted with an isocratic gradient of PBS pH 7.1 at 1 mL/min. Continuous detection was by absorbance at 280 nm. For G3000 SE HPLC, purified protein samples (∼20 µg) were loaded on to a TSKgel G3000SW, 10 µm, 7.5 mm ID x 300 mm column (Tosoh) and developed with an isocratic gradient of 0.2 M phosphate pH 7 at 1 mL/min. Continuous detection was by absorbance at 280 nm. Gel filtration standards (Biorad) were also loaded for MW estimation.
Mass spectrometry
Highly purified monomeric Fab-dsFv (10.5 mg/ml) in PBS (pH 7.4) was diluted to 0.7 mg/mL in H2O then sub-diluted to ∼50 µg/mL in solvent A (2% methanol, 0.3% formic acid in H2O). The diluted sample (4 µL) was loaded onto a 75 µm (diameter) x 50 mm (length) column of PLRP-S 5µ reverse-phase matrix (Polymer Labs) equilibrated with 96% solvent A / 4% solvent B (25% acetonitrile, 75% propan-1-ol, 0.1% formic acid) operated at 400 nL/min. Protein was eluted from the column with a linear gradient of 4-80% solvent B via a nanospray interface into a Waters Qtof Type I mass spectrometer (Waters, USA). TofMS spectra were collected in positive-ion mode over the range 500-2600 m/z during elution. Data was analyzed using MassLynx software and spectra were de-convoluted using the MaxEnt algorithm (Waters, USA).
Surface plasmon resonance/dual affinity assay
The binding affinities and kinetic parameters for the interactions of antibodies were determined by SPR conducted on a Biacore T100 or a Biacore 3000 using CM5 sensor chips (GE Healthcare Bio-Sciences AB) and HBS-EP (10 mM HEPES (pH7.4), 150 mM NaCl, 3 mM EDTA, 0.05% v/v surfactant P20) running buffer. All experiments were performed at 25°C. The antibody samples were captured to the sensor chip surface using a human F(ab')2-specific goat Fab (Jackson ImmunoResearch). Covalent immobilization of the capture antibody was achieved by standard amine coupling chemistry to a level of 6000-7000 response units (RU).
Human albumin, ChromPure (Jackson ImmunoResearch), cynomolgus albumin (Abcam), and murine albumin (Sigma-Aldrich), were titrated over the captured antibody from 50 nM. Each assay cycle consisted of firstly capturing the antibody sample using a 1 min injection, before an association phase consisting of a 3 min injection of the target antigen or albumin, after which dissociation was monitored for 10 min for albumin and 30 min for target antigen. After each cycle, the capture surface was regenerated with 2 × 1 min injections of 40 mM HCl followed by 30 s of 5 mM NaOH. The flow rates used were 10 μl/min for capture, 30 μl/min for association and dissociation phases, and 10 μl/min for regeneration.
The potential for the bispecific antibody to bind simultaneously to both HSA and the target antigen was assessed by capturing the antibody to the sensor chip surface, before performing either separate 3 min injections of 5 µM HSA: 50 nM target antigen or a mixed solution of both 5 µM HSA and 50 nM target antigen.
For kinetic assays, a blank flow-cell was used for reference subtraction and buffer-blank injections were included to subtract instrument noise and drift.
Kinetic parameters were determined by simultaneous global-fitting of the resulting sensorgrams to a standard 1:1 binding model using Biacore T200 evaluation software v1.
Antibody biophysical characterization
(i) Thermal Stability
A thermofluor assay was used to monitor the thermal stability of purified proteins. The reaction mix contained 5 µl of 30x SYPRO® Orange dye diluted with water from 5000 x stock solution and 45 µl of the purified protein sample at 0.11 mg/mL in PBS pH 7.4. Aliquots (10 µl) of the mixture were dispensed in quadruplicate into a 384 PCR optical well plate and were run on a 7900HT Fast Real-Time PCR System (Agilent). The peltier-based thermal cycling system was set at 20°C to 99°C with a ramp rate of 1.1°C/min. A charge-coupled device (CCD) monitored the fluorescence changes in the wells. The intensity increases in fluorescence were plotted and the inflection point of the slope(s) was used to generate the thermostability transition midpoint (Tm). pH stability was studied using citrate/phosphate buffers ranging from pH 2.6 to pH 7.6 in 0.2 pH unit increments (ionic strength was equalized by the addition of NaCl). A volume of 1 µl of a 5 mg/mL protein solution was added to 44 µl of buffer, following a 1 hour incubation period SYPRO® Orange dye was added to the mixture as described above.
(ii) Isoelectric point
The pI of both non-reduced, and reduced and capped purified antibody was determined using capillary isoelectric focusing (iCE280). Purified antibody at 2 mg/mL was mixed with 0.35% methylcellulose, 4% pH 3 to 10 pharmalytes (GE Healthcare) and synthetic pI markers (pI 4.65 and 9.77, Protein Simple) and separated by iCE280 isoelectric focusing (pre-focusing at 1,500 V for 1 min followed by focusing at 3,000 V for 6 min and measured at an absorbance of 280 nm). Reduced and alkylated samples were prepared by adding a final concentration of 2 mM Tris(3-hydroxypropyl)phosphine (THPP; Sigma Aldrich) to 2 mg/mL antibody and incubated at room temperature for 30 minutes. A final concentration of 20 mM iodoacetamide (Sigma-Aldrich) was added and samples were reincubated at room temperature in the dark for 90 minutes. The samples were then mixed with 0.35% methylcellulose, 4% pH 3-10 Pharmalytes (GE Healthcare) and synthetic pI markers (4.65 and 9.77) and separated as described above. Calibrated electropherograms were exported and analyzed using Empower 2 (Waters).
(iv) Shaking aggregation assay
Purified antibodies were subjected to vortex stress to provide information on aggregation stability at an air-liquid interface. This served to mimic shear that the molecule would potentially be subjected to during manufacture. Aggregation stability was monitored by measuring the turbidity of the sample at 595 nm with time. A 250 µl aliquot of the antibody at 1 mg/mL (in PBS, pH 7.4) concentration was transferred to 1.5 mL conical capped plastic tubes in triplicate, and vortexed at 1400 rpm, at 25°C in an Eppendorf Thermomixer Comfort. The extent of aggregation (turbidity) was measured at 0, 1.5, 3, 24, 48 and 120 h by reading the absorbance at 595 nm using a Varian Cary 50 Bio-spectrophotometer. Mean and standard deviation of the absorbance was obtained for each sample and plotted against time (h). SDS-PAGE analysis (as described previously) on a volume equivalent to a ∼2 µg load of protein was also performed at various time points.
Mouse pharmacokinetics
Six male BALB/c mice weighing 25-30 g were injected subcutaneously or intravenously with a single dose of the antibody at 10 mg/kg bodyweight. Serial blood samples (35 µl) were collected from the tail vein at 0.25, 1, 8, 24, 48, 72, 96 and 167 h post-dose. To obtain sera, blood samples were centrifuged for 5 min at 10,000 rpm at room temperature and analyzed for the antibody concentration by ELISA. An antibody against the target antigen and an anti-human kappa-horseradish peroxidase conjugate (Stratech) were used as the capture and secondary antibody, respectively. A purified sample of the target antigen was used as the standard. Plates were developed using TMB peroxidase solution (Sigma-Aldrich) and read at 450 nm (reference at 630 nm). PK parameters were calculated from the final dataset using Phoenix WinNonlin 6.2 (Pharsight).
Cynomolgus monkey pharmacokinetics model
This study was carried out by Charles River Laboratories, Edinburgh. Six male and 6 female cynomolgus monkeys were allocated to 3 treatment groups (2 per sex per group) to receive 1 i.v. dose of Fab-dsFv at 0.3, 3 or 30 mg/kg. Blood samples were taken from all monkeys at the following time points: pre-dose, 5 min, 1 h, 6 h, 12 h, 24 h, 48 h, 72 h, 96 h, 7 days, 14 days, 21 days, 28 days, 35 days, 42 days, 49 d and 56 d post dosing. Serum samples were analyzed for Fab-dsFv concentration by an immunoassay using a MSD SULFO-TAG™ (Meso Scale Discovery, USA) developed in-house. PK parameters were calculated from the final data set using Phoenix WinNonlin 6.2 (Pharsight). Data displaying LLOQ <0.03 or where n < 3 was not used in PK calculations.
Analysis of serum concentrations of in vivo administered antibody by Meso-Scale Discovery immunoassay
An immunoassay using a MSD SULFO-TAG™ (Meso Scale Discovery, USA) was specifically developed in-house to detect minute quantities of antibody drug present in serum samples. Serum samples were incubated with a biotin-labeled conjugate of the target antigen for 1 h prior to capture of the resulting complex on a streptavidin-coated MSD gold 96-well plate (Meso Scale Discovery, USA). The plate was washed with 3 cycles of PBS/0.1% tween 20 to remove unbound conjugate. An anti-human kappa antibody (Stratech) conjugated to MSD SULFO-TAG™ (Meso Scale Discovery, USA) was used to detect bound antibody drug in a further 0.5 h incubation. Following a 3 cycle wash with PBS/0.1% tween 20, MSD read buffer was added to the plate prior to its reading on an MSD sector imager 6000 (Meso Scale Discovery, USA). Fab-dsFv concentrations were calculated using the electrochemiluminescent signal as a proportional value to the amount of bound antibody.
Allometric scaling
The calculation of the predicted half-life in humans was derived from PK data from the cynomolgus monkey where the scaled values of CL in mL/day, intercompartmental clearance (Q), volume of the distribution of the central (V1) and peripheral compartment (V2) were calculated using a 2 compartment model. Data displaying LLOQ <0.03 or where n < 3 was not used in the calculation. The formula used for scaling was CL = a*BW^b, a = normalization constant, BW = body weight in kg, and b = the scaling exponent. CL and intercompartmental clearance (Q) were scaled using an exponent of 0.75. Distribution volumes were scaled with an exponent of 1. The PK parameters (CL, Q V1 and V2) were calculated by a non-linear mixed effects approach (NONMEM v7, ICON).
Mouse model of human T cell proliferation (Hu-NSG model)
NSG mice were purchased from Charles River Laboratories (UK) and housed in pathogen-free conditions under a 12h light, 12h dark cycle and provided with food and water ad libitum. Animal experiments were performed in accordance with the UK Animals (Scientific Procedures) Act 1986. Human peripheral blood was obtained by venipuncture from consenting healthy blood donors. PBMCs were isolated by the Ficoll-Paque PLUS (GE Healthcare Bio-Sciences AB) density centrifugation gradient method. Mice were dosed s.c with 0.03, 0.3, 3 or 30 mg/kg of Fab-dsFv one day prior to transfer of 10 million human PBMCs into the peritoneal cavity. After 14 d mice were bled by cardiac puncture under terminal anesthesia and then killed by cervical dislocation. The number of human CD4+ and CD8+ cells in the spleen and blood was then determined by FACS analysis. Data (n = 10) is expressed as means + SEM and statistical analysis is by one way ANOVA with Bonferrroni post-test.
Disclosure of potential confllicts of interest
The following hold shares in UCB: Adams R, Carrington B, Wild G, Marshall D, Lawson AD, Heywood S and Humphreys DP.
Acknowledgments
We would like to express our thanks to Jay Tibbitts for helpful discussions on the interpretation of the cynomolgus monkey PK data.
References
- Colcher D, Pavlinkova G, Beresford G, Booth BJ, Choudhury A, Batra SK. Pharmacokinetics and biodistribution of genetically-engineered antibodies. Q J Nucl Med 1998; 42:225-241; PMID:9973838
- McDonagh C F, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, Zhang B, Luus L, Overland R, Nguyen S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther 2012; 11:582-593; PMID:22248472; http://dx.doi.org/10.1158/1535-7163.MCT-11-0820
- Kontermann RE. Dual targeting strategies with bispecific antibodies. MAbs 2012; 4:182-197; PMID:22453100; http://dx.doi.org/10.4161/mabs.4.2.19000
- Asadi H, Yan B, Dowling R, Wong S, Mitchell P. Advances in medical revascularisation treatments in acute ischemic stroke. Thrombosis 2014; 2014:714218; PMID:25610642; http://dx.doi.org/10.1155/2014/714218
- Dedania VS, Bakri SJ. Current perspectives on ranibizumab. Clin Ophthalmol 2015; 9:533-542; PMID:25848203; http://dx.doi.org/10.2147/OPTH.S80049
- Houshmand S, Salavati A, Hess S, Ravina M, Alavi A. The role of molecular imaging in diagnosis of deep vein thrombosis. Am J Nucl Med Mol Imaging 2014; 4(5):406-425; PMID:25143860
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes limitations and hopes for the future. Br J Pharmacol 2009; 157:220-33; PMID:19459844; http://dx.doi.org/10.1111/j.1476-5381.2009.00190.x
- Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res 1990; 50 (3 Suppl):814s-819s; PMID:2404582
- Nelson A. Antibody fragments: hope and hype. MAbs 2010; 2(1):77-83; PMID:20093855; http://dx.doi.org/10.4161/mabs.2.1.10786
- Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med 2000; 6(2):114; PMID:10655072; http://dx.doi.org/10.1038/72162
- Langer F, Ingersoll SB, Amirkhosravi A, Meyer T, Siddiqui FA, Ahmad S, Walker JM, Amaya M, Desai H, Francis JL. The role of CD40 in CD40L- and antibody-mediated platelet activation. Thromb Haemost 2005; 93(6):1137-46; PMID:15968400; http://dx.doi.org/10.1160/TH04-12-0774
- Robles-Carrillo L, Meyer T, Hatfield M, Desai H, Dávila M, Langer F, Amaya M, Garber E, Francis JL, Hsu YM, et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol 2010; 185(3):1577-83; PMID:20585032; http://dx.doi.org/10.4049/jimmunol.0903888
- Wakefield ID, Harari O, Hutto D, Burkly L, Ferrant J, Taylor F, et al. An assessment of the thromboembolic potential of CDP7657 a monovalent Fab' PEG anti-CD40L antibody in rhesus macaques. Arthritis Rheum 2010; 62:1243; http://dx.doi.org/10.1002/art.29009
- Hsu YM, Su L. Biogen Idec MA Inc. CD40L-binding agent based platelet aggregation assays. US patent 9297810B2, Mar 29 2016.
- Shock A, Burkly L, Wakefield I, Peters C, Garber E, Ferrant J, Taylor FR, Su L, Hsu YM, Hutto D, et al. CDP7657 an anti-CD40L antibody lacking an Fc domain inhibits CD40L-dependent immune responses without thrombotic complications: an in vivo study. Arthritis Res Ther 2015; 17:234; PMID:26335795; http://dx.doi.org/10.1186/s13075-015-0757-4
- Kontermann RE. Strategies to extend plasma half-lives of recombinant antibodies. Biodrugs 2009; 23:93-109; PMID:19489651; http://dx.doi.org/10.2165/00063030-200923020-00003
- Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol 2011; 22:868-876; PMID:21862310; http://dx.doi.org/10.1016/j.copbio.2011.06.012
- Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review.Adv Drug Deliv Rev 2002; 54:531-545; PMID:12052713; http://dx.doi.org/10.1016/S0169-409X(02)00026-1
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Biodrugs 2014; 28:15-23; PMID:24687235; http://dx.doi.org/10.1007/s40259-013-0064-z
- Jevsevar S, Kunstelj M, Porekar VG. PEGylation of therapeutic proteins. Biotechnol J 2010; 5:113-128; PMID:20069580; http://dx.doi.org/10.1002/biot.200900218
- Schlapschy M, Binder U, Börger C, Theobald I, Wachinger K, Kisling S, Haller D, Skerra A. PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng Des Sel 2013; 26:489-501; PMID:23754528; http://dx.doi.org/10.1093/protein/gzt023
- Constantinou A, Chen C, Deonarain MP, Kontermann RE. Polysialic acid and polysialylation to modulate pharmacokinetics in Therapeutic proteins: strategies to modulate their plasma half-life. Kontermann R, Wiley, 2012.
- Adams R, Griffin L, Compson JE, Jairaj M, Baker T, Ceska T, West S, Zaccheo O, Dave E, Lawson AD, et al. Extending the half-life of a Fab fragment through generation of a humanized anti-human serum albumin Fv domain: an investigation into the correlation between affinity and serum half-life. MAbs 2016:in press.
- Hassouneh W, MacEwan SR, Chilkoti A. Fusions of elastin-like polypeptides to pharmaceutical proteins. Methods Enzymol 2012; 502:215-237; PMID:22208987; http://dx.doi.org/10.1016/B978-0-12-416039-2.00024-0
- MacEwan SR, Chilkoti A. Elastin-like polypeptides: biomedical applications of tuneable biopolymers. Biopolymers 2010; 94:60-77; PMID:20091871; http://dx.doi.org/10.1002/bip.21327
- Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med 2012; 4:1015-1028; PMID:22837174; http://dx.doi.org/10.1002/emmm.201201379
- Evans L, Hughes M, Waters J, Cameron J, Dodsworth N, Tooth D, Greenfield A, Sleep D. The production characterisation and enhanced pharmacokinetics of scFv-albumin fusions expressed in Saccharomyces cerevisiae. Protein Expr Purif 2010; 73:113-124; PMID:20546898; http://dx.doi.org/10.1016/j.pep.2010.05.009
- Hutt M, Färber-Schwarz A, Unverdorben F, Richter F, Kontermann RE. Plasma half-life extension of small recombinant antibodies by fusion to immunoglobulin-binding domains. J Biol Chem 2011; 287:4462-4469; PMID:22147690; http://dx.doi.org/10.1074/jbc.M111.311522
- Trüssel S, Dumelin C, Frey K, Villa A, Buller F, Neri D. New strategy for the extension of the serum half-life of antibody fragments. Bioconjug Chem 2009; 20: 2286-2292; PMID:19916518; http://dx.doi.org/10.1021/bc9002772
- Home P, Kurtzhals P. Insulin detemir: from concept to clinical experience. Expert Opin, Pharmacother 2006; 7:325-43; PMID: 16448327; http://dx.doi.org/10.1517/14656566.7.3.325
- Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem 2002; 277:35035-35043; PMID:12119302; http://dx.doi.org/10.1074/jbc.M205854200
- Nguyen A, Reyes AE, Zhang M, McDonald P, Wong WL, Damico LA, Dennis MS. The pharmacokinetics of an albumin-binding Fab (AB,Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel 2006; 9:291-297; PMID:16621915; http://dx.doi.org/10.1093/protein/gzl011
- Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsén L, Ekblad C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J Biol Chem 2011; 286:5234-5241; PMID:21138843; http://dx.doi.org/10.1074/jbc.M110.164848
- Andersen JT, Cameron J, Plumridge A, Evans L, Sleep D, Sandlie I. Single-chain variable fragment albumin fusions bind the neonatal Fc receptor (FcRn) in a species-dependent manner: implications for in vivo half-life evaluation of albumin fusion therapeutics. J Biol Chem 2013; 288:24277-24285; PMID:23818524; http://dx.doi.org/10.1074/jbc.M113.463000
- Hopp J, Hornig N, Zettlitz KA, Schwarz A, Fuss N, Müller D, Kontermann RE. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng Des Sel 2010; 23:827-834; PMID:20817756; http://dx.doi.org/10.1093/protein/gzq058
- Holt LJ, Basran A, Jones K, Chorlton J, Jespers LS, Brewis ND, Tomlinson IM. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng Des Sel 2008; 21:283-288; PMID:18387938; http://dx.doi.org/10.1093/protein/gzm067
- Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR, van Dongen GA. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther 2008; 8:2288-2297; PMID: 18723476; http://dx.doi.org/10.1158/1535-7163.MCT-07-2384
- Picó G. Thermodynamic aspects of the thermal stability of human serum albumin. Biochem Mol Biol Int 1995; 36:1017-1023; PMID:7580997
- Picó GA. Thermodynamic features of the thermal unfolding of human serum albumin. Int J Biol Macromol 1997; 20:63-73; PMID:Can't; http://dx.doi.org/10.1016/S0141-8130(96)01153-1
- Peters T Jr. All About Albumin: Biochemistry Genetics and Medical Applications. San Diego CA: Academic Press, 1996; http://dx.doi.org/10.1002/food.19970410631
- Jusko WJ, Gretch M. Plasma and tissue protein binding of drugs in pharmacokinetics. Drug Metab Rev 1976; 5(1):43-140; PMID:829788; http://dx.doi.org/10.3109/03602537608995839
- Paulev PE (Ed) Body fluids and their regulation in New human physiology, Copenhagen: Copenhagen Medical Publishers; 2000.
- Müller D, Karle A, Meissburger B, Höfig I, Stork R, Kontermann RE. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem 2007; 282:12650-12660; PMID:17347147; http://dx.doi.org/10.1074/jbc.M700820200
- Smith BJ, Popplewell A, Athwal D, Chapman AP, Heywood S, West SM, Carrington B, Nesbitt A, Lawson AD, Antoniw P, et al. Prolonged in vivo residence times of antibody fragments associated with albumin. Bioconjug Chem 2001; 12: 750-756; PMID:11562193; http://dx.doi.org/10.1021/bc010003g
- Kenanova VE, Olafsen T, Salazar FB, Williams LE, Knowles S, Wu AM. Tuning the serum persistence of human serum albumin domain III:diabody fusion proteins. Protein Eng Des Sel 2010; 23:789-798; PMID:20802234; http://dx.doi.org/10.1093/protein/gzq054
- Sleep D, Cameron J, Evans LR. Albumin as a versatile platform for drug half-life extension. Biochim Biophys Acta 2013; 1830:5526-5534; PMID:23639804; http://dx.doi.org/10.1016/j.bbagen.2013.04.023
- Zhao S, Zhang Y, Tian H, Chen X, Cai D, Yao W, Gao X. Extending the serum half-life of G-CSF via fusion with the domain III of human serum albumin. Biomed Res Int 2013; 2013:107238; PMID:24151579; http://dx.doi.org/10.1155/2013/107238
- Elsadek B, Kratz F. Impact of albumin on drug delivery–new applications on the horizon. J Control Release 2012; 157:4-28; PMID:21959118; http://dx.doi.org/10.1016/j.jconrel.2011.09.069
- Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A, Bunting K, Antunes F, Williamson R, Athwal S, et al. Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. J Biol Chem 2014; 289: 13492-13502; PMID:24652290; http://dx.doi.org/10.1074/jbc.M114.549832
- Ritter MA, Ladyman HM (Ed) Monoclonal Antibodies. Cambridge University Press, 1995; http://dx.doi.org/10.1385/1-59259-076-4:267
- Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, Rollence ML, Wood JF, Schott ME, Milenic DE, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng 1993; 6: 989-95 ; PMID:8309948; http://dx.doi.org/10.1093/protein/6.8.989
- Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001; 305:989-1010; PMID:11162109; http://dx.doi.org/10.1006/jmbi.2000.4265
- Krauss J, Arndt MA, Zhu Z, Newton DL, Vu BK, Choudhry V, Darbha R, Ji X, Courtenay-Luck NS, Deonarain MP, et al. Impact of antibody framework residue VH-71 on the stability of a humanised anti-MUC1 scFv and derived immunoenzyme. Br J Cancer 2004; 90:1863-70; PMID:15150594
- Kim SH, Lee YS, Hwang SY, Bae GW, Nho K, Kang SW, Kwak YG, Moon CS, Han YS, Kim TY, Kho WG. Effects of PEGylated scFv antibodies against Plasmodium vivax duffy binding protein on the biological activity and stability in vitro. J Microbiol Biotechnol 2007; 17:1670-1674; PMID:18156783; http://dx.doi.org/10.1016/j.pnsc.2008.12.007
- Zhang J, Yun J, Shang Z, Zhang X, Pan B. Design and optimization of a linker for fusion protein construction. Progress in Natural Science 2009; 19:1197-1200; http://dx.doi.org/10.1073/pnas.90.16.7538
- Brinkmann U, Reiter Y, Jung S-H, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Nat Acad Sci 1993; 90:7538-7542; PMID:8356052; http://dx.doi.org/10.1073/pnas.90.16.7538
- Wozniak-Knopp G, Stadlmann J, Rüker F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS One 2012; 7:e30083; PMID:22272277; http://dx.doi.org/10.1371/journal.pone.0030083
- Pitt-Rivers R, Impiombato FS. The binding of sodium dodecyl sulphate to various proteins. Biochem J 1968; 109:825-30; PMID:4177067; http://dx.doi.org/10.1042/bj1090825
- Dunker AK, Kenyon AJ. Mobility of sodium dodecyl sulphate - protein complexes. Biochem J 1976; 153(2):191-7; PMID:1275884; http://dx.doi.org/10.1042/bj1530191
- Therien AG, Grant FE, Deber CM. Interhelical hydrogen bonds in the CFTR membrane domain. Nat Struct Biol 2001; 8(7):597-601; PMID:11427889; http://dx.doi.org/10.1038/89631
- Orcutt KD, Ackerman ME, Cieslewicz M, Quiroz E, Slusarczyk AL, Frangioni JV, Wittrup KD. A modular IgG-scFv bispecific antibody topology. Protein Eng Des Sel. 2010 Apr;23(4):221-8; PMID:17321501; http://dx.doi.org/10.1093/protein/gzp077
- Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun 2007; 355(3):751-7; PMID:17321501; http://dx.doi.org/10.1016/j.bbrc.2007.02.042
- Eppler A, Weigandt M, Hanefeld A, Bunjes H. Relevant shaking stress conditions for antibody preformulation development. Eur J Pharm Biopharm 2010; 74(2): 139-47; PMID:19922795; http://dx.doi.org/10.1016/j.ejpb.2009.11.005
- Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol 2006;72(1):1-10; PMID:16469301; http://dx.doi.org/10.1016/j.bcp.2005.12.041
- Cao Y, Jusko WJ. Incorporating target-mediated drug disposition in a minimal physiologically-based pharmacokinetic model for monoclonal antibodies. J Pharmacokinet Pharmacodyn 2014; 41(4):375-87; PMID:25077917; http://dx.doi.org/10.1007/s10928-014-9372-2
- Ryan PC, Sleeman MA, Rebelatto M, Wang B, Lu H, Chen X, Wu CY, Hinrichs MJ, Roskos L, Towers H, et al. Nonclinical safety of mavrilimumab an anti-GMCSF receptor alpha monoclonal antibody in cynomolgus monkeys: relevance for human safety. Toxicol Appl Pharmacol 2014: 279(2):230-9; PMID:24937321; http://dx.doi.org/10.1016/j.taap.2014.06.002
- Ponce R, Abad L, Amaravadi L, Gelzleichter T, Gore E, Green J, Gupta S, Herzyk D, Hurst C, Ivens IA, et al. Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regul Toxicol Pharmacol 2009; 54(2):164-82; PMID:19345250; http://dx.doi.org/10.1016/j.yrtph.2009.03.012
- Zorbas M, Hurst S, Shelton D, Evans M, Finco D, Butt M. A multiple-dose toxicity study of tanezumab in cynomolgus monkeys. Regul Toxicol Pharmacol 2011; 59(2):334-42; PMID:21130822; http://dx.doi.org/10.1016/j.yrtph.2010.11.005
- Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs 2011; 3(1):61-6; PMID:20962582; http://dx.doi.org/10.4161/mabs.3.1.13799
- West GB, Brown JH. The origin of allometric scaling laws in biology from genomes to ecosystems: towards a quantitative unifying theory of biological structure and organization. J Exp Biol 2005; 208(Pt 9):1575-92; PMID:15855389; http://dx.doi.org/10.1242/jeb.01589
- Dennis MS, Jin H, Dugger D, Yang R, McFarland L, Ogasawara A, Williams S, Cole MJ, Ross S, Schwall R. Imaging tumors with an albumin-binding Fab, a novel tumor-targeting agent. Cancer Res. 2007; 67(1):254-61; PMID:17210705; http://dx.doi.org/10.1158/0008-5472.CAN-06-2531
- Sudlow G, Birkett DJ, Wade DN. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975; 11:824-832; PMID:1207674; http://dx.doi.org/10.1371/journal.pcbi.1000231
- Sudlow G, Birkett DJ, Wade DN. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976; 12:1052-1061; PMID:1004490
- Kragh-Hansen U, Chuang VT, Otagiri M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol Pharm Bull. 2002; 25(6):695-704; PMID:12081132; http://dx.doi.org/10.1248/bpb.25.695
- Dockal M, Carter DC, Rüker F. The three recombinant domains of human serum albumin. Structural characterization and ligand binding properties. J Biol Chem. 1999; 274(41):29303-10; PMID:10506189; http://dx.doi.org/10.1074/jbc.274.41.29303
- Bal W, Sokołowska M, Kurowska E, Faller P. Binding of transition metal ions to albumin: sites, affinities and rates. Biochim Biophys Acta. 2013; 1830(12):5444-55; PMID:23811338; http://dx.doi.org/10.1016/j.bbagen.2013.06.018
- Chaudhury C, Brooks CL, Carter DC, Robinson JM, Anderson CL. Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry 2006; 45:4983-4990; PMID:16605266; http://dx.doi.org/10.1021/bi052628y
- Sand KM, Bern M, Nilsen J, Dalhus B, Gunnarsen KS, Cameron J, Grevys A, Bunting K, Sandlie I, Andersen JT. Interaction with both domain I and III of albumin is required for optimal pH-dependent binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2014; 289(50):34583-94; PMID:25344603; http://dx.doi.org/10.1074/jbc.M114.587675
- Camacho CJ, Katsumata Y, Ascherman DP. Structural and Thermodynamic Approach to Peptide Immunogenicity. PLoS Comput Biol 2008; 4(11):e1000231; PMID:19023401; http://dx.doi.org/10.1371/journal.pcbi.1000231
- Wyeth-Ayerst, Gemtuzumab ozogamicin summary for presentation to the FDA's Oncologic Division Advisory Committee, 2000; PMID:10429243; http://dx.doi.org/10.1038/11717
- Carrasco-Triguero M, Yi JH, Dere R, Qiu ZJ, Lei C, Li Y, Mahood C, Wang B, Leipold D, Poon KA, Kaur S. Immunogenicity assays for antibody-drug conjugates: case study with ado-trastuzumab emtansine. Bioanalysis 2013; 5(9):1007-23; PMID:23641693; http://dx.doi.org/10.4155/bio.13.64
- Chamow SM, Ryll T, Lowman HB, Farson D. Eds. Therapeutic Fc-fusion proteins. Wiley-Blackwell; 2014, pp400.
- Sand KM, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT. Unravelling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front Immunol. 2014; 5:682; PMID:25674083
- Sleep D. Albumin and its application in drug delivery. Expert Opin Drug Deliv 2015; 12(5):793-812; PMID:25518870; http://dx.doi.org/10.1517/17425247.2015.993313
- Ramm K, Gehrig P, Plückthun A. Removal of the conserved disulfide bridges from the scFv fragment of an antibody: effects on folding kinetics and aggregation. J Mol Biol 1999; 290:535-46; PMID:10390351; http://dx.doi.org/10.1006/jmbi.1999.2854
- Honegger A. Engineering antibodies for stability and efficient folding in Chernajovsky Y & Nissim A (eds) Therapeutic Antibodies Part 2, Springer Berlin Heidelberg; 2008, pp 47-68; PMID: 23335490; http://dx.doi.org/10.1002/btpr.1693
- Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nature Biotechnol. 1999; 17:780-783; http://dx.doi.org/10.1038/11717
- O'Connor-Semmes RL, Lin J, Hodge RJ, Andrews S, Chism J, Choudhury A, Nunez DJ. GSK2374697 a novel albumin-binding domain antibody (AlbudAb) extends systemic exposure of exendin-4: first study in humans–PK/PD and safety. Clin Pharmacol Ther 2014; 96:704-712; PMID:25238251; http://dx.doi.org/10.1038/clpt.2014.187
- Herring B, Hamilton BJ, Jespers LS, Holt LJ, Malgorzata P. Glaxo group Ltd. Drug fusions and conjugates. US patent 8779103B2, Jul 15 2014.
- Lightwood DJ, Carrington B, Henry AJ, McKnight AJ, Crook K, Cromie K, Lawson AD. Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J Immunol Methods 2006; 316:133-143; PMID:17027850; http://dx.doi.org/10.1016/j.jim.2006.08.010
- Clargo AM, Hudson AR, Ndlovu W, Wootton RJ, Cremin LA, O'Dowd VL, Nowosad CR, Starkie DO, Shaw SP, Compson JE, et al. The rapid generation of recombinant functional monoclonal antibodies from individual antigen-specific bone marrow-derived plasma cells isolated using a novel fluorescence-based method. MAbs 2014; 6:143-159; PMID:24423622; http://dx.doi.org/10.4161/mabs.27044
- Tickle S, Howells L, O'Dowd V, Starkie D, Whale K, Saunders M, Lee D, Lightwood D. A fully automated primary screening system for the discovery of therapeutic antibodies directly from B cells. J Biomol Screen 2015; 20(4):492-497; PMID:25548140; http://dx.doi.org/10.1177/1087057114564760
- Cain K, Peters S, Hailu H, Sweeney B, Stephens P, Heads J, Sarkar K, Ventom A, Page C, Dickson A. A CHO cell line engineered to express XBP1 and ERO1-Lα has increased levels of transient protein expression. Biotechnol Prog 2013; 29:697-706; PMID:23335490; http://dx.doi.org/10.1002/btpr.1693