
ABSTRACT
Post-translational modifications (PTMs) strongly influence the structure and function of proteins. Lysine side chain acetylation is one of the most widespread PTMs, and it plays a major role in several physiological and pathological mechanisms. Protein acetylation may be detected by mass spectrometry (MS), but the use of monoclonal antibodies (mAbs) is a useful and cheaper option. Here, we explored the feasibility of generating mAbs against single or multiple acetylations within the context of a specific sequence. As a model, we used the unstructured N-terminal domain of APE1, which is acetylated on Lys27, Lys31, Lys32 and Lys35. As immunogen, we used a peptide mixture containing all combinations of single or multi-acetylated variants encompassing the 24–39 protein region. Targeted screening of the resulting clones yielded mAbs that bind with high affinity to only the acetylated APE1 peptides and the acetylated protein. No binding was seen with the non-acetylated variant or unrelated acetylated peptides and proteins, suggesting a high specificity for the APE1 acetylated molecules. MAbs could not finely discriminate between the differently acetylated variants; however, they specifically bound the acetylated protein in mammalian cell extracts and in intact cells and tissue slices from both breast cancers and from a patient affected by idiopathic dilated cardiomyopathy. The data suggest that our approach is a rapid and cost-effective method to generate mAbs against specific proteins modified by multiple acetylations or other PTMs.
Abbreviations
| BSA | = | Bovine serum albumin |
| KLH | = | Keyhole Limpet Hemocyanin |
| LC-MS | = | Liquid Chromatography- Mass Spectrometry |
| mAb | = | Monoclonal antibody |
| OPD | = | o-Phenylenediamine dihydrochloride |
| OVA | = | Ovalbumin |
| PVDF | = | Polyvinylidene fluoride |
| RP-HPLC | = | Reverse Phase High performance liquid chromatography |
| SDS-PAGE | = | Sodium Dodecyl Sulfate - PolyAcrylamide Gel Electrophoresis |
Introduction
The introduction of post-translational modifications (PTMs) in proteins is a major mechanism for regulating their function, localization, stability, immunogenicity and other relevant properties. Among these, acetylation of the εNH2 of selected lysines, a process regulated by a fine balance between acetylases and de-acetylases, is one of the most important and widespread, playing a relevant role in protein-DNA binding, secondary or higher order structure modification and sub-cellular localization.Citation1,2
The ability to detect acetylated lysines in specific proteins or sets of proteins is thus critical to the investigation of the interplay between the occurrence of PTMs and cell biology. Several robust tools for studying protein acetylation are used by biochemists and cell biologists, including MSCitation3 and batteries of antibodies that allow the detection of acetylated lysines independent of flanking sequences.Citation4-6 To date, no examples have been reported for the detection of specific acetylated sequences with single antibodies. The occurrence of acetylation(s) in a given protein may be monitored using non-specific anti-acetyl antibodies in combination with anti-protein antibodies. However, the exact array of acetylated/free lysines in a specific stretch of amino acids is a much more difficult task and implies several steps of protein isolation, digestion by specific proteases and fine mapping of modified residues by MS.Citation3,7 The detection of multiple acetylations (di-, tri- or even tetra-acetylations) within short lysine-rich protein stretches is an even more challenging task because of the occurrence of highly similar structures with identical molecular mass.
Monoclonal antibodies (mAbs), in contrast, are highly specific reagents capable of recognizing small and large macromolecules, and are able to distinguish among very subtle molecular differences. Many reports have described the use of antibodies to detect lysine acetylation,Citation4-6, 8-13 but no studies have yet reported on the generation of antibodies that recognize peptide/protein sequences acetylated on specific sites. Several recent proteomics/acetylome studies suggest a relevant role for the lysine acetylation/deacetylation status of proteins in many eukaryotic cellular processes such as chromatin structure and gene activity, cell growth, differentiation, apoptosis, drug resistance and tumor growth.Citation1
We therefore explored the feasibility of generating antibodies against a specific array of acetylated lysines using as immunogens a pool of synthetic peptides representing all combinations of acetylated/non-acetylated synthetic fragments. Immunization with this pool of peptides should potentially elicit a population of antibodies from which, through an appropriate and targeted screening assay, some that are very specific for a given subset of antigens can be isolated. For this purpose, we focused our attention on the multiply acetylated 24–39 region of apurinic/apyrimidinic endonuclease 1,Citation14-23 also known as redox effector factor-1 (APE1/Ref-1 or APE1). APE1 is a multifunctional and essential protein in mammals that acts as a key regulator of cellular responses to oxidative stress and contributes to the maintenance of genome stability.Citation14-15 APE1 undergoes extensive acetylation during cell response to genotoxic damage, which may control its functional activation and subcellular distribution.Citation16-22 It has been recently found that APE1 acetylation promotes its own extracellular secretionCitation23 and may trigger apoptosis of cancer cells via RAGE binding.Citation24 In particular, APE1 K6/K7 may undergo acetylation during cell response to genotoxic insults, and the acetylation status of these lysines, controlled by the sirtuin 1 (SIRT1) deacetylase activity, is important in modulating protein DNA-repair functions by regulating the kinetics of its interaction with other enzymes involved in base excision repair.Citation16
Other lysines, K27/K31/K32/35, which are located in the region spanning amino acids 24–35 of the unstructured N-terminal domain, may also undergo acetylation in vivo. They are believed to be crucial for APE1 interaction with rRNA, with nucleophosmin (NPM1), with other different protein partners and for the modulation of its catalytic activity.Citation16-22 Very recently it has been demonstrated that APE1 acetylated on K27/K31/K32 is essential for sustained cell proliferation and survival of tumor cells.Citation25 Because genotoxic stress may induce PTMs, shifting the equilibrium between these non-acetylated and acetylated APE1 forms toward the latter and more active form, the protein N-terminal domain represents a specific region of relevant biological significance.Citation20-22 In addition, acetylation at the mentioned lysine residues may significantly affect APE1 protein stability. Since acetylation competes with ubiquitination for the same residues, it may also represent the switch for controlling the protein turnover rateCitation26 and a mechanism to protect it from N-terminal proteolysis and to support tumor growth.Citation25
Nevertheless, the acetylation status of such lysines is still not well understood, and indeed multiple/single specific modifications may occur depending on the cell metabolic status and on the presence/absence of different stimuli. To obtain new antibody reagents to that could be valuable for future studies, we generated mAbs against a pool of acetylated peptides containing all possible combinations of mono-, di-, tri- and tetra-acetylated variants of the APE1 peptide 24–39. The peptide pool, which also included the non-acetylated peptide, has been conjugated to keyhole limpet hemocyanin (KLH) and used for immunization. The resulting antibodies were screened for selective binding to acetylated peptides, and we selected those with binding ability for specific variants. The new antibodies have been tested on cell extracts, intact cells and tumor slices to evaluate their potential in analytical and functional studies.
Results
Acetylated peptide antigens were prepared and characterized as a mixture and fractionated by RP-HPLC
A mixture of 16 distinct peptides corresponding to all combinations of 4 acetylated/non acetylated lysines along the sequence K24KSK*TAAK*K*NDK*EAAG39 of APE1 was prepared by the solid phase method incorporating equimolar mixtures of Fmoc-L-Lys(Boc)-OH/Fmoc-L-Lys(Ac)-OH at the requested positions. All sequences generated are reported in and in Table S1A (entries 1–16). Synthesis progression was tightly monitored during chain assembly. For this purpose, small resin aliquots were removed after incorporation of lysine mixtures. Following cleavage reactions to free the corresponding peptides, the resulting compounds were characterized by liquid chromatography (LC)-MS, obtaining the expected molecular weight (MW) and approximate equimolar distributions of molecules (Fig. S1A-L and data not shown).
Figure 1. (A) Schematic composition of the acetylated peptides library. (B) RP-HPLC fractionation of the 16 distinct peptides corresponding to all combinations of 4 acetylated/non-acetylated lysines over the sequence K24KSK*TAAK*K*NDK*EAAG39 of APE1. Fractions were collected every 0.5 minutes identifying the pools of acetylated/non-acetylated peptides as illustrated on the top of the chromatogram. Peaks have been labeled according to the molecules they contain.
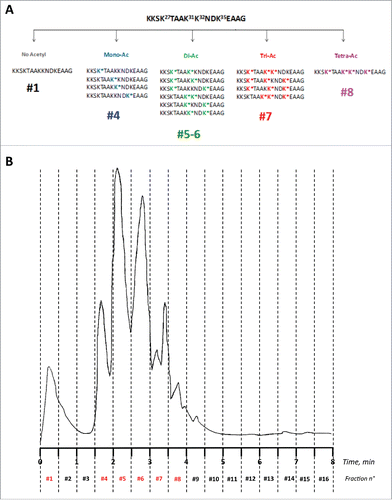
After RP-HPLC fractionation () and lyophilization, the 16 fractions were further characterized by LC-MS (Fig. S1B-K). Due to the high similarity in structure and hydrophobicity, peptides bearing the same number of acetylated lysines could not be separated and were recovered within the same fractions, except the di-acetylated peptides, which were retrieved under 2 well-separated fractions (See Table S1A for the expected and experimental MW of all peptides; see also ). In particular, the non-acetylated peptide was recovered within fraction #1 (flowthrough, MWcalcd/expm: 1672.96/1672.97 amu; Δmass = 0.01 amu); all the monoacetylated peptides (4 species) were recovered in fraction #4 (MWcalcd/expm: 1714.97/1714.98 amu; Δmass = 0.01 amu); the di-acetylated peptides (6 species) were recovered in fraction #5 and #6 (MWcalcd/expm: 1756.98/1757.05 amu; Δmass = 0.07 amu); the tri-acetylated peptides (4 species) were recovered in fraction #7 (MWcalcd/expm: 1798.99/1799.00 amu; Δmass = 0.01 amu); the single tetra-acetylated peptide was recovered in fraction #8 (MWcalcd/expm: 1841.00/1841.02 amu; Δmass= 0.02 amu). Sequences are shown in Table S1A and , the HPLC traces of isolated fractions are shown in Fig. S1A-F and the corresponding mass spectra are shown in Fig. S1G-K. The remaining fractions did not contain any material or contained unrelated side products. After lyophilization, we recovered 1.5 mg in fraction #1; 1.8 mg in fraction #4; 2.8 mg in fraction #5; 3.6 mg in fraction #6; 3.5 mg in fraction #7 and 1.7 mg in fraction #8. The mixture of c-Myc (138–161)-derived acetylated peptides was characterized by direct infusion MS (Table S1B; see also Fig. S2 for the deconvoluted mass spectrum). Single mono-acetylated APE1(24–39) peptides, peptide APE1(24–39)K4pleA, and the peptide N_1–13, prepared by solid phase chemical synthesis, were obtained in high yield and purified to homogeneity by semipreparative RP-HPLC. MWs and purities were confirmed by high-resolution LC-MS analyses (Table S1AB; Fig. S3A–J).
MAbs against the pool of acetylated peptides were generated and characterized
After the first immunization, blood samples were taken from the 2 mice and tested at T = 30d, 40 d and 60d. A sufficiently high antibody titer was detected in mice after 60 d from the first immunization as compared to the pre-immune serum. The immunoreactivity of both immune and pre-immune sera from a representative mouse toward coated Ac-APE1(24–39)-bovine serum albumin (BSA; reported as the OD at 415 nm from the ELISA assay) at increasing dilutions is shown in Fig. S4A. The same experiment, carried out with APE1(24–39)-BSA, is reported in Fig. S4B. As shown, we detected a high titer of antibodies against the pool of acetylated peptides, whereas only slight differences in immunoreactivity between pre- and post-immune sera were detected with APE1(24–39)-BSA, suggesting that no or few antibodies were generated against the non-acetylated peptide. This was also likely due to partial subtraction of the non-acetylated peptide during a preliminary purification step introduced to remove salts and other low MW contaminants. Under the conditions used (see Methods), the non-acetylated peptide was indeed eluted at the column flowthrough ( and Fig. S1B).
Mice were sacrificed and splenectomized. After hybridoma generation and clone stabilization, as described in the section of Methods, we isolated 12 clones whose supernatants were immunoreactive toward the BSA-conjugated antigen and not reactive toward the glutaraldehyde self-conjugated BSA (BSA2) control (not shown). Supernatants from 8 of these clones showed similar immunoreactivity toward both APE1(24–39)-BSA and Ac-APE1(24–39)-BSA (Fig. S4C), suggesting that they contained mAbs that recognized the peptide(s) on residues other than acetylated lysines. Supernatants from 4 other clones, 2E6, 2G10, 1D4, 1B6 (hereafter also anti-APE1 mAbs), contained antibodies that reacted only with the pool of acetylated peptides, and these were selected for further studies. Clones were adapted to grow in serum-free OPTIMEM medium and allowed to produce mAbs in 100 mL flasks for at least 4 weeks. Concentrated supernatants (about 50 mL) were collected and antibody concentration was determined by the Bradford method. Supernatants were also characterized by 10% SDS-PAGE under reducing and non-reducing conditions; the expected bands were observed in all the samples (Fig. S5). Supernatants from the 4 clones were purified by Protein G affinity chromatography and concentrated as described in the Methods section, yielding multi-mgs of the proteins.
Generated mAbs showed no selectivity for a specific acetylated antigen
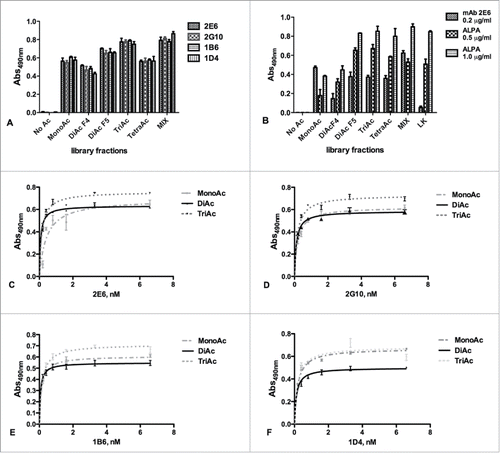
We next investigated whether the selected anti-APE1 mAbs recognized one or more specific peptide antigens used for their generation. Peptides from the different isolated library fractions, conjugated to BSA, were coated at 0.02 µg/mL and binding to anti-APE1 mAbs was tested at 0.2 µg/mL (about 1.3 nM) antibody concentration. As shown in , all mAbs bound the mono-acetylated, the di-acetylated and the tri-acetylated peptides with a similar strength, whereas they bound less well the tetra-acetylated peptide. As expected, these antibodies did not recognize the non-acetylated peptide. The same antigens, together with a mixture of unrelated c-Myc (138–161)-derived acetylated peptides (Table S1B), used as control, were tested for binding to the 2E6 mAb and to the commercially available anti-acetyl-lysine antibody (ALPA) at both 0.5 µg/mL and 1.0 µg/mL. As shown in , ALPA did bind all acetylated peptides, with the mono-acetylated ones recognized less strongly under these conditions. Remarkably, mAb 2E6 did not bind to the c-Myc (138–161) acetylated peptides, suggesting a strong selectivity of the new anti-APE1 mAbs against acetylated APE1 peptides. To further investigate the recognition ability of the 4 mAbs for the different antigens, dose-response binding assays were performed using the entire set of mAbs and the pool of mono-acetylated, di-acetylated and tri-acetylated peptides. All antibodies bound the immobilized peptides in a dose-dependent and saturable way (). Also, no relevant differences were observed among the various antibodies for the binding to the mixture of mono-acetylated, di-acetylated and tri-acetylated peptides, as assessed by fitting the binding curves and estimating the apparent KDs (Table S2).
Figure 2. (A) Screening of the HPLC fractions containing differently acetylated APE1 peptides using the 4 mAbs (2E6, 2G10, 1B6, 1D4) previously shown to discriminate between the acetylated and non-acetylated peptides (Fig. SM1). The antibodies appeared more specific for the mono-acetylated, di-acetylated (fraction 5) and tri-acetylated peptides. Also the non-acetylated peptide and the restored MIX of all molecules were used as control. Experiments were performed as quadruplicate and expressed as value ± standard deviation (sd). (B) Binding of 2E6 and ALPA antibodies to the APE1-derived acetylated peptides. As expected, no binding was detected for the non-acetylated molecule for both antibodies, whereas ALPA binding correlated with the extent of peptide acetylation. Experiments were performed as triplicate and expressed as values ± sd. Dose-dependent binding assay of 2E6 (C), 2G10 (D), 1B6 (E), 1D4 (F) antibodies to the fractions containing the APE1- derived mono-, di-, and tri-acetylated peptides. Concentrations ranged between 0.2 and 6.6 nM. The extrapolated apparent KDs are reported in Table S2.
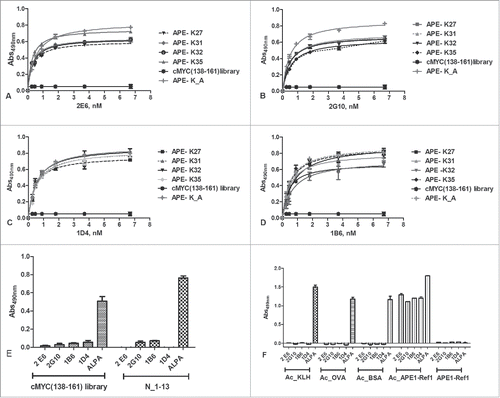
To verify whether the anti-APE1 mAbs recognized single antigens with some higher specificity, we next tested antibody binding to the purified mono-acetylated peptides. For this purpose, we coated every single molecule at 1.0 µg/mL (about 7 nM) and the binding to each antibody was probed at concentrations between 0.2 nM and 6.7 nM (). By fitting the binding curves, we estimated the KDs underlying such interactions and the values are reported in Table S3. The affinity constants ranged between around 280 pM (2G10 versus K27 mono-acetylated peptide) and 760 pM (1B6 vs. K27 mono-acetylated peptide), demonstrating that the interaction was very strong for all 4 mono-acetylated molecules.
Figure 3. (A-D) Dose-dependent binding assay of 2E6, 2G10, 1B6, 1D4 and ALPA antibodies to the isolated unconjugated mono-acetylated peptides. Antibody concentrations ranged between 0.2 and 6.6 nM. The c-Myc (138–162) acetylated peptides were used as negative controls. (E) Binding of anti-APE1 and ALPA antibodies to Myc (138–162) derived and another unrelated acetylated peptide (N_1–13), used as negative controls for anti-APE-1 antibodies. As expected, no binding was detected for anti-APE1 antibodies. ALPA, instead, bound in a dose-response manner all the acetylated peptides. Experiments were performed in triplicate and expressed as values ± sd. (F) Binding of 4 anti-APE1 and ALPA antibodies to acetylated proteins. As expected, no binding was detected for the anti-APE1 antibodies toward the unrelated proteins, whereas they bound the chemically acetylated APE-1. ALPA strongly bound all the acetylated proteins.
Anti-APE1 mAbs recognize acetylated K27, K31, K32, K35 lysines only in the context of the APE1 sequence
The specificity of the anti-APE1 mAbs was further investigated by evaluating their capability to bind a set of unrelated acetylated peptides. For this purpose, we used a pool of c-Myc (138–161)-derived acetylated peptides, containing 8 distinct molecules, and an unrelated acetylated cyclic peptide named N_1–13 used for other ongoing projects (see Table S1B). In addition, we investigated the recognition of chemically acetylated proteins, including the unrelated KLH, BSA, ovalbumin (OVA) and rh-APE1, residues 1–318. Proteins were chemically acetylated in vitro and characterized by SDS-PAGE (Fig. S6AB) as reported elsewhere.Citation5 The acetylation of rh-APE1 was also assessed by LC-MS (see Fig. S7, Figs. S8A–C and S9A–C). This analysis confirmed that the 29 lysines in rh-APE1 were all acetylated, since the shift of MW observed upon the chemical reaction was consistent with the incorporation of about 34 acetyl groups (Δmass ≈1426 amu, before and after acetylation). The binding of anti-APE1 antibodies and of ALPA was monitored by ELISA after coating the unrelated peptides at 10 μg/mL and the acetylated proteins at 1.0 µg/mL. Data clearly show the unrelated acetylated peptides and proteins were recognized only by ALPA and not by the anti-APE1 mAbs (). In contrast the recombinant, chemically acetylated APE1 protein was strongly bound by both anti-APE1 and ALPA antibodies. Remarkably, the unmodified APE1, which does not bear any acetylation (it is produced in E. coli), did not interact with the anti-APE1 antibodies. In the same assay, we also tested APE1(24–39)K4pleA, a peptide variant whereby K27, K31, K32, and K35 were replaced by alanine, a residue that could mimic an acetylated lysine side chain. As shown in , the antibodies did interact with APE1(24–39)K4pleA in a manner very similar to the isolated monoacetylated peptides, suggesting that the methyl group of the alanine side chain is able to adapt to some extent to the complementarity-determining regions of the antibodies.
The new anti-APE1 mAbs specifically detect the acetylated protein in Western blotting, immunofluorescence and immunohistochemistry analyses
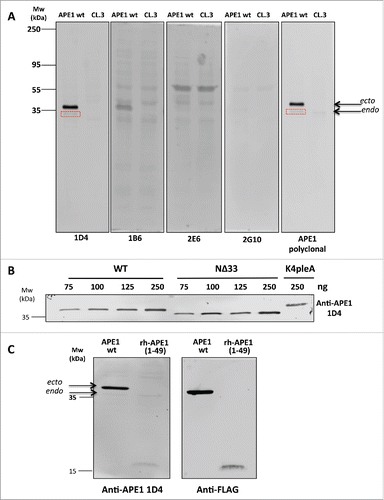
To evaluate the potential of the new anti-APE1 mAbs to detect acetylated APE1 in cells, we probed the same amounts of extracts of FLAG-tagged APE1-overexpressing HeLa cells or the same cells after APE1 knock-down with the 4 selected antibodies. In these cells, APE1 is acetylated by endogenous enzymes.Citation20,21 Data show that mAb 1D4 stained both ectopically overexpressed and endogenous APE1 with the same efficiency as the commercial polyclonal antibody, while no protein was detected after protein knock-down (). The endogenous protein was only faintly detected by both the commercial antibody and 1D4. The other mAbs did not effectively detect the protein and were not further considered in the subsequent experiments. To further confirm antibody specificity, we dose-dependently detected full-length FLAG-tagged APE1 or FLAG-tagged APE1 mutants immunopurified from cell extracts overexpressing them.Citation21 Recombinant proteins were probed with anti-APE1 mAb 1D4. We found that it detected in a dose-dependent fashion the full-length protein (), as well as the variant lacking the N-terminal 33 residues, still containing K35. The antibody also detected the APE1 mutant K4pleA, in which charged lysines K27, K31, K32, and K35 were replaced by alanines, which mimic constitutive acetylation. Also, this protein was clearly detected by 1D4 at the highest concentration (), a result in line with the binding of anti-APE1 to the synthetic APE1(24–39)K4pleA (). We next investigated whether the anti-APE1 antibody 1D4 recognized the N-terminus region of the protein. Cell extracts overexpressing the full-length FLAG-tagged protein or the 1–49 N-terminal region were again probed with 1D4, and results are reported in . As shown, the ectopically expressed and endogenous proteins were detected, although the endogenous APE1 was less visible. Importantly, the shortened 1–49 polypeptide was detected under the same conditions, confirming that recognition occurred on the N-terminal portion. Protein identity was confirmed by probing them with an anti-FLAG antibody. Taken together, these findings suggest that recognition of APE1 acetylated lysines by the new 1D4 antibody only occurs within the context of the protein and specifically involves the N-terminal residues of APE1.
Figure 4. (A) Western blot analysis carried out on extracts of HeLa cells overexpressing APE1 wt variant or the HeLa cell extracts knocked-down by suitable ShRNAs (CL.3).Citation18 Here are shown the entire filters for 4 mAbs in comparison with the commercial polyclonal antibody (NB 100–101, Novus Biologicals). To highlight the different affinity of antibodies the same amounts of total proteins was loaded on the different lanes. Endogenous APE1 is also stained by 1D4 and the commercial polyclonal antibody (boxed in red). (B) The selected 1D4 mAb stained in a dose-response fashion the APE1 variants. As low as 75 ng of recombinant Flag-tagged wild-type protein (indicated as WT) immunopurified from HeLa cell extractsCitation21 were clearly detected in this assay. Also the protein lacking the first 33 residues on the N-terminus (indicated as NΔ33) was similarly detected under the same conditions, suggesting that K35, still present on the protein, was acetylated and recognized by the antibody. On the same gel, the variant, indicated as K4pleA, where K27, K31, K32 and K35 were mutated to alanines was loaded. Also this mutated protein was readily detected under these conditions by 1D4. (C) Detection of full length acetylated ectopic (ecto) and endogenous (endo) APE1 protein (indicated as APE wt) by 1D4. The endogenous protein band was weak but clearly visible at the indicated MW. In the same experiment we verified that recognition was specific for the N-terminal region of APE1 protein by analyzing cell extracts expressing the 1–49 APE1 fragment (indicated as APE1(1–49)). The reduced intensity of this band could be due to reduced in cell acetylation compared to the full-length protein. Protein identity was confirmed by using an anti-FLAG antibody.
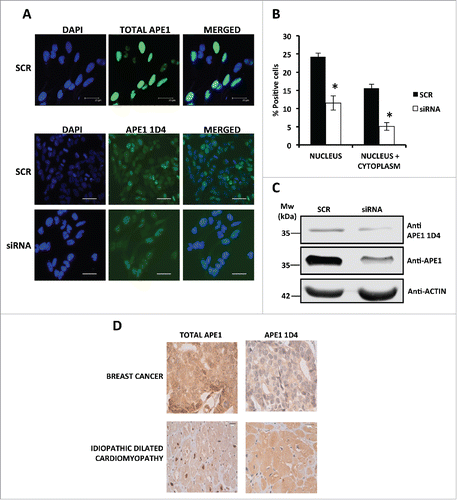
Then, we checked the efficiency of 1D4 to recognize APE1 both in paraformaldehyde fixed cells and in formalin fixed paraffin-embedded (FFPE) tissue samples. Results were compared with those obtained employing a specific mAb recognizing total APE1 protein.Citation27 Immunofluorescence analysis () was performed on HeLa cells that were either expressing endogenous APE1 (treated with a scrambled siRNA, SCR) or silenced through specific siRNA (siRNA). Data showed the ability of anti-APE1 1D4 mAb to recognize the endogenous protein. However, while total APE1 antibody gave the classic strong nuclear staining,Citation17,18 1D4 resulted again in a nuclear-cytoplasmic staining. Quantification of the staining signals (expressed as percentage of positive cells) () and Western blot analyses () in SCR and siRNA-treated cells confirmed the specificity of the recognition by the anti-APE1 1D4 antibody. Moreover, we also compared the staining pattern of the 1D4 antibody with that of the total APE1 antibody, in immunoperoxidase-stained FFPE tissue samples (). Immunohistochemistry was performed on biopsies obtained both from a breast cancer patient and from a patient affected by idiopathic dilated cardiomyopathy that underwent cardiac transplantation. The representative images demonstrate a predominantly distribution of acetylated APE1 in the cytoplasm with respect to total APE1 in accordance with the immunofluorescence data.
Figure 5. (A) Representative immunofluorescence on HeLa cells scramble and APE1 siRNA-treated cells shows the efficiency of APE1 knock-down and a distribution of AcAPE1 mainly in the cytoplasm respect to total APE1. Bars 25 μm. (B) Histogram reporting the average of positive cells for acetylated APE1 in the nucleus and in the cytoplasm. The experiment was done in triplicate counting 1000 total cells for each condition. Asterisks represent a significant difference respect to control (SCR).* P < 0.05. (C) Western blotting analysis on whole cell extracts from scramble siRNA- or APE1 siRNA-treated cells showing APE1 protein and its acetylated form. Actin was used as loading control. (D) Immuno-histochemistry on samples obtained by a patient affected by breast cancer and by a patient affected by idiopathic dilated cardiomyopathy (the study was approved by the ethics committee of Udine University). The representative images demonstrate a predominantly distribution of acetylated APE1 in the cytoplasm with respect to total APE1.
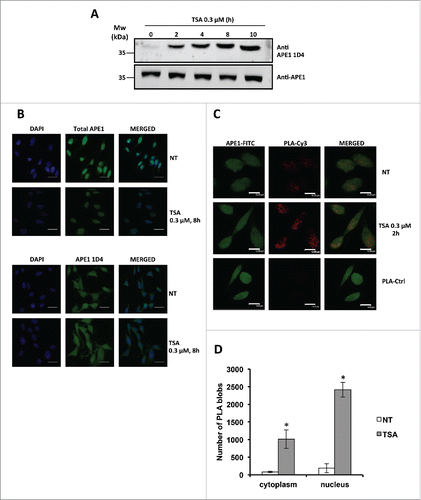
Finally, we tested the ability of the 1D4 antibody to recognize endogenous hyperacetylated APE1.Citation21 HeLa cells were treated with the deacetylases inhibitor trichostatin A (TSA) and the amount of acetylated APE1 was evaluated in Western blotting analyses performed on total cell extracts. Data clearly showed that the new mAb was indeed able to detect the increasing amounts of hyperacetylated APE1 (). Recognition was further confirmed by using 1D4 in immunofluorescence experiments showing that HeLa cells treated with TSA displayed an increased number of pancellular (nucleus+cytoplasm) AcAPE1 positive cells upon TSA treatment (). To further confirm this effect, as well as the specificity of the developed antibody against acetylated APE1, we next performed a proximity ligation assay (PLA) in HeLa cells using a commercial antibody recognizing the N-terminus (residues 1–14) of APE1 in combination with 1D4 that specifically interacts with the acetylated portion of the protein. The PLA is used to evaluate the physical proximity (<40 nm) between 2 epitopes recognized by 2 different antibodies. It is thus the ideal technology to map the proximity of epitopes in a protein, and thus the occurrence of post-translational modifications in a specific amino acid region.Citation28 The representative immunofluorescence experiment reported in and indeed shows a significant increase of positive signals in cells treated with TSA in accordance with increased acetylation of the region of interest of APE1, definitely demonstrating the specificity of recognition by the 1D4 antibody and the increased acetylation of APE1 in the lysine residues 27, 31, 32, 35 upon inhibition of histone deacetylase activity by TSA treatment.
Figure 6. (A) Western blotting analysis on whole cell extracts from HeLa cells treated with TSA 0.3 µM, showing time-dependent increase of APE1 acetylation. (B) Representative immunofluorescence on HeLa cells treated with TSA 0.3 µM for 8 h stained with total APE1 and AcAPE1 (1D4) antibody showing increased distribution of the acetylated APE1 form in the cytoplasm compared to total APE1 upon TSA treatment. NT, non-treated. Bars 50 μm. (C) Nucleoplasmic interaction between APE1 N-terminal and Ac-APE1 (1D4) antibody after TSA treatment. HeLa cells were seeded on a glass coverslip and treated with TSA 0.3 µM for 2h. PLA reaction was carried out using anti-APE1 N-terminalCitation21,27 and anti-acetylated APE1 1D4 antibodies. APE1 expression was detected by using the anti-APE1 N-terminal antibody and was used as a reference for the nuclei. PLA control was performed omitting the primary antibody for Ac-APE1 (1D4) (PLA-Ctrl). (D) Histogram accounting for the average number of PLA signals of at least 30 randomly selected cells per condition. Asterisks represent a significant difference respective to control (NT). * P < 0.05. NT, non-treated cells. Bars 14 μm.
Discussion
Proteins are reversibly and dynamically acetylated on the ε-amino group of the lysine side chain by specific acetyltransferases and deacetylases. Like other covalent post-translational modifications (such as phosphorylation of serine, threonine or tyrosine), lysine acetylation works through “loss-of-function” and “gain-of-function” mechanisms, which regulate protein activity and play a major role in most pathological and physiological conditions. While protein N-terminal acetylation generally stabilizes proteins, several proteomics/acetylomes studies suggest a much wider role for lysine acetylation, also in light of its widespread occurrence in a number of eukaryotic cellular processes, including chromatin structure and gene activity, cell growth, differentiation, and apoptosis.Citation1,29-31
Despite substantial progress, the study of acetylomes is still limited by the complexity of samples in terms of analyte concentration, and redundancy and similarity of substructures to be detected. They are also sometimes hampered by difficulties in accessing highly sensitive mass spectrometers and by the lack of suitable complementary biochemical methods that allow the detection of specific acetylated protein without pre-enrichment steps. The task is further complicated by the occurrence in the cell of other highly abundant acetylated proteins, such as histones, tubulin and mitochondrial proteins.
In this context, antibodies specific for a given acetylated protein would be valuable and cost-effective tools. While a number of antibodies against acetyl-lysine are available, no reagents able to detect a specific acetyl-lysine and the surrounding peptide structure are known. Typically, antigens used to generate antibodies to acetyl-lysine are either acetyl-lysine-containing synthetic peptides or chemically acetylated carrier proteins (http://www.antibodyresource.com/proteinsearch/Protein/?searchTerm=acetyl+lysine).Citation4-6, 8-13
Random mixtures of synthetic peptides containing acetylated lysines have been used to generate antibodies able to detect acetylated proteins in cellular extracts in a non-specific way. Likewise, immunization of mice with chemically acetylated proteins resulted in antibodies that recognized acetylated lysines independently of flanking sequences.Citation4-6, 8-13
To explore the possibility of generating antibodies specific for a given acetylated epitope, we immunized mice with a discrete pool of antigens containing all combinations of peptides, acetylated or non-acetylated on the side chains of a given set of lysines. The mAb pools have been subsequently screened to isolate some able to interact with a specific structure. Although several reports have described immunization with multiple antigens, one major problem associated with this approach could be the difference in antigenicity of the various components and the consequent unequal distribution of antibody populations. This was not expected to happen, or the problem was strongly attenuated, with the mixtures we used in this work because the peptide portion of the molecules was largely conserved and the overall antigenicity was also expectedly the same, ensuring a nearly similar representation of the induced antibody population. Data shown in and S4A–C strongly corroborate this hypothesis. The approach also ensured that the resulting antibodies were specific for the chosen sequence because the peptide portion, with its more- or less-antigenic elements, was present in all the library components. Following a subtractive screening approach, based on the exclusion of mAbs binding the non-acetylated peptide variant, we isolated a set of antibodies recognizing with comparable affinity all the library components. Remarkably, the selected antibodies were completely inactive toward the non-acetylated peptide, an observation strongly supporting the notion that they bind with high affinity to at least one acetylated lysine and a second specific site on the APE1 peptide chain. This hypothesis is further corroborated by the observation that all the 4 peptides with a single acetylation have similar binding affinities and by the lack of antibody recognition by both the non-acetylated peptide and the chemically acetylated peptides and proteins with unrelated sequences. The single tetra-acetylated peptide appeared to bind less well the 4 mAbs, also suggesting that a second free lysine, among those on positions 27, 31, 32 and 35, could be required for a more efficient interaction with antibodies.
Altogether, the data support the conclusion that mAbs specific for the singly or multi-acetylated variants of the 24–39 APE1 region can be generated with our approach, but they are not sufficiently specific to discriminate between the differently acetylated forms of the peptide and of the protein. Despite the lack of such fine specificity, the new antibodies allow very sensitive and selective detection of acetylated APE1 in cells expressing the protein and monitoring of the protein trafficking between cytoplasm and nucleus (). In addition, using in parallel antibodies recognizing the acetylated or non-acetylated protein, the relative abundance of modified versus total protein could be estimated, a factor that can help to monitor the extent of protein activation, its ability to interact with rRNA, NPM1 or other protein partnersCitation16-24 and the protein stability under diverse stimuli.Citation25,26 Furthermore, the role played by a secreted acetylated APE1 variant in tumor necrosis factor-mediated inflammation mechanisms in endothelial cellsCitation32 also suggests the potential use of these new mAbs in targeted therapeutic applications.
The ability of the new mAbs to recognize both mono- or poly-acetylated APE1 antigens suggests they have some polyclonal features, combining the advantages of targeting multiple antigenic determinants and the very high affinity and specificity. However, further refinements based on antibody sequencing, structural studies and site-directed mutagenesis may help in obtaining new antibodies with improved specificity against the single antigens. The approach reported here provides a rapid way to generate site-specific multi- and monoacetyl-lysine antibodies that are applicable to protein acetylation studies in other tissues and organisms.
Materials and methods
All protected amino acids and resins for peptide synthesis were from either Novabiochem (Laufelfingen, Switzerland) or from GL Biochem (Shanghai, PRC). Fmoc-L-Lys(Ac)-OH was from Bachem (Bubendorf, Switzerland). Solvents for peptide synthesis and purification were from LabScan (Dublin, Ireland). Other reagents for peptide synthesis, BSA, KLH, OVA and glutaraldehyde were from Sigma-Aldrich (Milan, Italy). Preparative RP-HPLC were performed on a Waters Prep 150 LC preparative system, equipped with a 2489 UV/Visible detector and with a ONYX monolithic C18 column (100 × 10 mm ID), Phenomenex, Torrance, CA, USA). The ALPA was from Millipore (Cat. # AB3879); the anti-APE1 polyclonal antibody was from Novus Biologicals (Cat. NB 100–101). All media for cell culture were from Sigma-Aldrich (Milan, Italy). The RPMI 1640 medium, containing 10% fetal bovine serum (FBS) and 1% penicillin, streptomycin and glutamine (PSG) was used for maintaining myeloma cells. The RPMI-GM medium contained RPMI 1640 with added 1% non-essential amino acids (NEAA). For the fusion of splenocytes and myeloma cells, we used an RPMI 1640 medium supplemented with 15% FBS, 2% HAT (hypoxantine-aminopterine-thymine), 1% PSG and 10% HES (hybridoma enhancing supplement). The medium for clone selection was RPMI 1640 supplemented with 15% FBS, 2% HAT, 1% PSG. BALB/c mice were from Charles River Laboratories. Animals were housed and handled according to the institutional guidelines. Recombinant human APE1 protein (rh-APE1) was expressed in E. coli. The protein was prepared as previously described.Citation18-22 Data were fitted using the software GraphPad Prism 4, ver. Four.02 (GraphPad Software, San Diego, CA, USA). Explanted human hearts, and breast cancer biopsies were collected by the Pathology Department of our institution following, respectively, cardiac transplantation or mastectomy procedures performed at our Academic Hospital. The protocol was approved by the Ethics Committee of Udine, in accordance with the Declaration of Helsinki and written informed consent was obtained from the enrolled patients. Other materials are indicated below.
Immunogen preparation
Antigen generation
Antibodies were raised against the 24–39 region of APE1.Citation20 We focused our attention on this peculiar protein to implement a novel approach for generating mAbs recognizing specific acetylated lysines within a given peptide sequence. For this purpose, we designed and prepared a mixture of 16 synthetic APE1 peptides containing all combinations of sequences, where K27, K31, K32, and K35, previously reported as potentially acetylated, were either acetylated or non-acetylated (24 = 16 combinations, ).
The following single-letter code sequence has been used to indicate the peptide mixture: KKSK*TAAK*K*NDK*EAAG. Here K* stands for either acetylated or non-acetylated lysines. Peptides were C-terminally amidated. The peptide mixture was manually prepared by Fmoc chemical synthesis by the solid phase method using a RINK amide resin with a 0.57 mmol/g substitution.Citation33,34 The synthesis scale was 0.05 mmoles. Canonical coupling and deprotection conditions were used to incorporate amino acids into the sequence, using a 5-fold excess of protected residues (0.25 mmoles). To generate mixtures, in correspondence of K27, K31, K32, and K35, we used an equimolar mixture of Fmoc-L-Lys(Boc)-OH and Fmoc-L-Lys(Ac)-OH, performing the preactivation as for the other residues. Synthesis progression was tightly monitored during chain assembly by removing small resin aliquots and characterizing the products by LC-MS, thus assessing identity and distribution of the expected peptides. These analytical controls were carried out after incorporation of the first Lys/Lys(Ac) mixture (2 compounds generated), the second Lys/Lys(Ac) incorporation (4 compounds generated), the third Lys/Lys(Ac) incorporation (8 compounds generated) and the fourth Lys/Lys(Ac) incorporation (16 compounds generated). Such analyses allowed tight control over the relative distribution of compounds within the mixtures. For this purpose we integrated and compared peak areas from UV traces at 214 nm of chromatographically resolved peaks. In complex mixtures where not all peaks were chromatographically resolved, the relative distribution of compounds was determined by integrating and comparing peaks obtained by extracted ion chromatograms. After synthesis completion, peptides were removed from the solid support using a trifluoroacetic acid-triisopropylsilane-water (TFA-TIS-H2O) mixture (90:2.5:2.5, v:v:v), precipitated in cold diethyl ether and lyophilized. To remove salts deriving from the cleavage procedure, a 10 mg aliquot of the crude material was loaded on a C18 100×10 mm ID ONYX monolithic column equilibrated at 0.5% CH3CN in H2O, 0.1% TFA. After 10 column volume washes with the same solvent at 10 mL/min, the material was eluted by changing the solvent to 95% CH3CN in H2O, 0.1% TFA, collected in a single fraction and lyophilized. This material was used for conjugation to KLH. Another aliquot of the same material was fractionated by preparative RP-HPLC using the same column. For this purpose, 14.0 mg aliquots of the crude peptide mixture were dissolved in 2% CH3CN in H2O, 0.1% TFA (2.0 mL) and injected onto a column equilibrated with the same solvent at 20 mL/min. A gradient from 2% CH3CN in H2O, 0.1% TFA to 30% CH3CN in H2O, 0.1% TFA in 8 minutes was applied to separate the components, monitoring the eluate at 214 nm. 16 fractions (#1 – #16) of 10 mL each were manually collected every 0.5 minutes. Isolated fractions were lyophilized and characterized by high-resolution LC-MS analyses using an Agilent 6230 Time-Of-Flight (TOF) LC-MS System (Agilent Technologies, Cernusco Sul Naviglio, Italy). The LC module consisted of an Agilent 1290 HPLC Infinity system equipped with a photodiode array detector, a binary solvent pump degasser, a column heater and an autosampler. Gradients were from 1% CH3CN in H2O, 0.1% TFA to 30% CH3CN in H2O, 0.1% TFA. A C18 LC-MS AERIS column (Phenomenex) was used for these analyses. The column flow rate was kept at 0.2 mL/min with the heater set at 20°C. UV spectra were monitored in the range between 200 nm and 400 nm. The mass analyzer Agilent 6230 TOF-MS was set to operate in positive ion scan mode with mass scanning from 100 to 3200 m/z. The ion source was upgraded from the original Agilent Jet Stream source to the dual-sprayer version for improved reference mass delivery. Nitrogen was used as the drying and nebulizer gas. The instrument acquired data using the following parameters: drying gas temperature, 325°C; drying gas flow, 10 L/min; nebulizer, 20 psi; sheath gas temperature, 400°C; sheath gas flow, 11 L/min; VCap. Three.500 V; nozzle, 0 V; fragmentor, 200 V; skimmer, 65 V; and octapole RF Vpp was 750. The instrument state was set to extended dynamic range mode (2 GHz). Tuning and calibration were performed before sample runs. Data collection and integration were performed using MassHunter workstation software (version B.05.00). Data were stored in both centroid and profile formats during acquisition. A constant flow of Agilent TOF reference solution through the reference nebulizer allowed the system to continuously correct for any mass drift by using 2 independent reference lock-mass ions, purine (m/z 119.03632) and HP-922 (m/z 922.000725), to ensure high mass accuracy and reproducibility. Target compounds were detected and reported from accurate mass scan data using Agilent MassHunter Qualitative software. Spectra were also acquired by direct infusion at 1 µL/min using a HCT Ultra ion trap mass spectrometer (Bruker Daltonics, Macerata, Italy), equipped with a nanoESI source maintained at 4.8 kV and 250°C. Typical spectra were recorded between m/z 200 and 2800 in the positive ion mode. Four single peptides of sequence K24KSKTAAKKNDKEAAG39, mono-acetylated on either K27, K31, K32, or K35, were prepared in a similar way by solid phase synthesis, purified to homogeneity by RP-HPLC as described for library fractionation, and characterized by LC-MS. These single peptides are here reported as: APE1(24–39)K27Ac, APE1(24–39)K31Ac, APE1(24–39)K32Ac and APE1(24–39)K35Ac, respectively. The peptide K24KSATAAAANDAEAAG39, where K27, K31, K32, or K35 were replaced by alanines, was similarly prepared and characterized by MS. The peptide is here reported as APE1(24–39)K4pleA. Also the non-acetylated peptide was prepared as single molecule and purified to homogeneity by RP-HPLC. The library of c-Myc (138–161)-derived acetylated peptides and the acetylated peptide termed N_1–13, used as controls, were prepared as described above and characterized by direct infusion MS using the ESI-Ion trap mass spectrometer described above. Protein MS analyses were performed using the LC/MS Agilent system described above and a Phenomenex AERIS WIDEPORE 3.6 µm C4 50 × 2.1 mm applying a gradient from 30% solvent B (CH3CN, 0.1% TFA) to 90% solvent B in 10 minutes.
Antigen conjugation
Conjugation reactions were performed mixing 1.0 mg peptide material with 3.0 mg of KLH protein in 1.0 mL PBS containing 0.2% v/v glutaraldehyde in water.Citation35 Mixtures were left under stirring for 3 h, then 1.0 mL of 1.0 M glycine in water was added and solutions stirred again at room temperature (RT) for 1 h. At the end, solutions were extensively dialyzed against PBS buffer, pH 7.3 and lyophilized. The amount of peptide-protein conjugate was determined by the method of Bradford.Citation36 The desalted peptide-KLH mixture was used as immunogen.
A mixture of fractions #4 through #8 (thus excluding the non-acetylated peptide, present only in fraction #1) was prepared by mixing identical amounts (0.2 mg) from each fraction. This mixture was subsequently conjugated also to BSA as described above and named Ac-APE1(24–39)-BSA. The isolated fractions #1, #4, #5, #6, #7, and #8 were also conjugated under the same conditions to BSA. BSA-conjugated fraction #1, containing only the non-acetylated peptide, was named APE1(24–39)-BSA. Ac-APE1(24–39)-BSA and APE1(24–39)-BSA were used to monitor immunization progression and to perform the first step of hybridoma screening. The library of c-Myc (138–161)-derived acetylated peptides described in the previous section was conjugated to BSA under identical experimental conditions and used as control in several ELISA experiments. BSA2, used as an additional control, was prepared in a similar way, omitting the peptide solution.
Immunogen preparation
The immunogen was prepared by mixing 500 µL of KLH-conjugated desalted peptide mixture at 4.0 mg/mL in PBS with 500 µL of complete TiterMax Gold Adjuvant (Sigma-Aldrich, Milan, Italy). Mixing was achieved using a 3-way valve connected with 2 syringes, one filled with the conjugate and the other filled with the adjuvant, and allowing the aqueous solution to flow within the oily adjuvant solution.
MAbs generation
Mice immunization
Two 5-week old Balb/c mice (from Charles River Laboratories) were immunized with 50 μL of suspension containing about 100 μg of adjuvant emulsified KLH-conjugated peptide mixtures. Two independent injections were carried out subcutaneously with 25 μL immunogen. Before immunization, 250 µL blood samples were taken from each mouse from the caudal vein and used as the pre-immune control (T0 samples). Mice were again immunized with the same amount of immunogen at day 30 after the first immunization (T = 30d), at day 40 (T = 40d) and at day 60 (T = 60d), and blood samples were taken from the caudal vein (250 µL) before every subsequent immunization and tested by ELISA to monitor the increase of antibody titer compared to the pre-immune serum (see the section on details for the ELISA test). Mice showing the highest antibody titer were sacrificed and splenectomized.
Fusion of myeloma cells and mice splenocytes to generate hybridomas
The 2 mice showing the highest titer of specific antibodies against the peptide mixture, were sacrificed by cervical disarticulation and splenectomized, placing the spleens in a 50 mL tube containing 10 mL of RPMI-GM medium (Sigma-Aldrich, Milan, Italy) pre-warmed at 37°C. Spleens were then moved to 100 mm plates containing 10 mL of RPMI-GM medium and grinded using a cell filter (BD-Falcon, Milan, Italy). The resulting suspensions were centrifuged at 800 rpm for 5 minutes at RT, removing the supernatants, then cells were suspended in 5 mL of RPMI-GM and counted in the presence of 0.4% Trypan Blue (Sigma-Aldrich, Milan, Italy) to exclude dead cells. To perform cell fusion, 1/5 of splenocytes in 10 mL RPMI-GM were mixed with 4/5 of SP2/0 myeloma cells in 5 mL RPMI-GM (ATCC), then 15 mL of RPMI-GM were further added. Cells were spun at 600 rpm for 5 minutes at RT. After discarding the supernatant, 1 mL of RPMI-GM containing PEG 1300–1600 (Hybri-Max, Sigma-Aldrich, Milan, Italy) and 75 µL DMSO (Sigma-Aldrich, Milan, Italy) were added. Cell pellets were gently disrupted with a pipette tip, adding at the same time 9 mL of RPMI-GM supplemented with 10% FBS. Cells were then centrifuged at 500 rpm for 7 minutes at RT, then the pellet was re-suspended in 200 mL of RPMI-HAT medium and cells seeded in 20 96-well plates and incubated for at least 10 d at 37°C under 5% CO2. After about 2 weeks, supernatants were assayed by ELISA using plates coated with both Ac-APE1(24–39)-BSA and APE1(24–39)-BSA to detect the presence of antibodies against either the pool of acetylated peptides or the non-acetylated variant, operating as described in the previous section. The most reactive clones, were isolated, sub-cloned by limited dilution and re-tested by ELISA. By this step, 8 clones had lost productivity; the remaining clones were submitted to a stabilization process.
Clone stabilization
Clones were stabilized by 3 sequential rounds of cloning performed by the method of limited dilution. At every round, cells were counted and diluted up to about 3 cells/mL; 20 mL of this solution were plated in 2 96-well plates, dispensing 100 µL/well, thus reaching about 0.3 cells/well. Cells were incubated for 2 weeks at 37°C under 5% CO2, then supernatants from each well were again tested for the presence of antibodies against the peptide antigen. Supernatants showing the highest antibody titer were isolated and again cloned thrice by limited dilution, always testing by ELISA for the presence of specific antibodies.
Adaptation of clones in serum-free medium
To obtain secreted mAbs in the absence of serum-derived bovine antibodies, selected clones were adapted to grow in serum-free medium. To this end, the medium was first changed to OPTIMEM (Sigma-Aldrich, Milan, Italy) supplemented with 10% FBS, then FBS was progressively reduced and finally eliminated over a period of about 4 week. Productivity was always monitored by ELISA assays.
Antibody purification
Antibodies were purified by standard procedures. After centrifugation at 12000 rpm for 20 minutes at 4°C, hybridoma cell-culture supernatants were filtered on a 0.22 μm filter and loaded onto a Protein G Sepharose column (GE, Healthcare) using PBS as loading buffer. Elution was performed using 0.1 M glycine pH 2.7. Protein G-purified mAbs were dialyzed overnight against 20 mM Tris buffer pH 7.0 at 4°C, and then concentrated using Vivaspin™ 500 centrifugal filter (30 kDa MWCO, Sartorius) by centrifugation at 4000 rpm, at 4°C. Purity of mAbs was estimated by size-exclusion chromatography using a Superdex 200 Hr 10/30 column (GE, Healthcare) and by 12% SDS-PAGE under reducing and non-reducing conditions. Concentration was determined using a BIORAD kit.Citation36
ELISA assays
ELISA assays were performed as described in detail in the Supplementary Material.
Preparation of acetylated proteins
0.5 mg of OVA (about 11 nmoles), BSA (about 7 nmoles), KLH (about 11 nmoles) and rh-APE1 (about 14 nmoles, residues 1 to 318, Uniprot P27695), produced in E. coli,Citation18,20,22 were dissolved in 0.5 mL of 0.1 M Na2CO3.Citation5 Acetylation was achieved adding drop-wise 10 μL of acetic anhydride (about 0.167 mmoles) and then 4 μL of pyridine, under constant shaking to avoid precipitation. Reaction mixtures were incubated at 30°C for 5 h, then 4 μL of 1 M Tris pH 9.0 was added to block the reaction. Proteins were extensively dialyzed against 25 mM Tris pH 7.5, 100 mM NaCl, overnight, at 4°C. Protein acetylation was assessed by SDS-PAGE analysis as reported elsewhere.Citation5 The acetylation of rh-APE1 was also assessed by LC-MS analysis using the Agilent LC-MS system described above.
Cell culture and transient transfection
The variants of human APE1 protein (Uniprot P27695) expressed in HeLa cells were prepared as previously described.Citation18-22 APE1 proteins used in this study were: WT, corresponding to the wild type protein from residues from 1 to 318; NΔ33, corresponding to the protein lacking residues from 1 to 33; K4pleA, where the K27, K31, K32, and K35 were substituted with alanines residues; the presence of alanine mimicked the constitutively acetylated protein. APE1(1–49) fragment corresponding to residues from 1 to 49 of the protein.
The endogenous protein was indicated as APE1 wt; CL3 indicated HeLa cells where silencing of endogenous human APE1 protein was performed as previously described.Citation18 For inducible shRNA experiments, doxycycline (1 μg/mL) (Sigma Aldrich, Milan, Italy) was added to the cell culture medium and cells were grown for 10 d. HeLa cells were grown in Dulbecco's Modified Eagle's Medium (Invitrogen) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin sulfate. One day before transfection, cells were seeded in 10-cm plates at a density of 3.0 × 10Citation6 cells/plate. Cells were then transiently transfected using the lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Cells were harvested either 24 or 48 h after transfection.
For siRNA experiments, HeLa cells were transfected with 100 pmol siRNA APE1 5′-UACUCCAGUCGUACCAGACCU-3′ or the scrambled control siRNA 5′-CCAUGAGGUCAUGGUCUGdTdT-3′ (Dharmacon, Lafayette, CO) using DharmaFECT reagent (Dharmacon) for 72h.
Western blotting analysis
For Western blotting analyses, 15 μg of whole cell lysate or the indicated amount of purified proteins from HeLa cellsCitation18-22 were resolved on 12% SDS-PAGE, transferred onto nitrocellulose membranes (Schleicher & Schuell Bioscience, Dassel, Germany). Membranes were blocked with 5% w/v BSA in TBS containing 0.1% v/v Tween 20 and probed with polyclonal antibody (NB 100–101, Novus Biologicals), anti-FLAG (Sigma Aldrich, Milan, Italy) or anti-APE1 mAbs. Similar procedures for the staining of chemically acetylated proteins (rh-APE1 and control proteins) were performed using anti-APE1 mAbs at the concentrations of 1 μg/mL.
Immunofluorescence confocal and Proximity Ligation analyses
For immunofluorescence analyses, HeLa cells treated with 0.3 µM TSA or silenced for APE1 were grown on glass coverslips. Cells fixed in 4% (wt/vol) paraformaldehyde for 20 min at RT were permeabilized for 5 min with PBS-0.25% (wt/vol) Triton X-100 and incubated for 30 min with 5% normal goat serum in PBS-0.1% (wt/vol) Triton X-100 (blocking solution) to block unspecific binding of the antibodies. Cells were then incubated with the mouse monoclonal anti-APE1Citation17,18,20 or anti-AcAPE1 (1D4) diluted 1:22 and 1:100, in blocking solution, for 3 h at 37°C or O/N at 4°C, respectively. After washing, cells were incubated for 2 h at RT with secondary antibody Alexa Fluor 488-conjugated goat anti-mouse IgG1 (1:600). The preparations were then washed with PBS 3 times for 5 min each in the dark. Nuclei were then stained by 5 minutes of incubation in 300 nM solution of 4, 6-diamidino-2-phenylindole dihydrochloride (Sigma-Aldrich) in PBS. The preparations were then washed 3 times in PBS for 5 min. The microscope slides were then mounted onto slides in Mowiol 4–88 supplemented with DABCO (1:5) as an anti-fade reagent.
Immunofluorescence procedures and PLA were carried out as described previously.Citation21,28 To study the interaction between total APE1 and acetylated APE1 in vivo, we used the in situ PLA technology (Duolink, Sigma-Aldrich). After incubation with polyclonal anti-APE1 (N-terminal) (NB 100–897, Novus Biologicals) (1:200) for 3 h at 37°C, cells were incubated with anti-AcAPE1 (1D4) (1:100) overnight, at 4°C. PLA was performed following manufacturer's instructions. Technical controls, represented by the omission of anti-AcAPE1 primary antibody 1D4, resulted in the complete loss of PLA signal.
Cells were examined under a Leica TCS SP laser-scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with a 488-nm argon laser, a 543-nm He-Ne laser, and a 63× oil immersion objective (HCX PL APO CS 63×/1.32–0.60; Leica). Data were acquired at RT (23°C) using the integrated Leica Confocal Software package; multicolor images were captured through sequential scanning. Determination of PLA signal was performed using the Blob Finder software (Center for Image Analysis, Uppsala University).
Immunohistochemistry
For ICH, the histological sections of breast cancer and idiopathic dilated cardiomyopathy, embedded in paraffin, were initially incubated in citrate buffer (Dako EnVision™ FLEX-Target retrieval solution- Low pH 50X) for 40 minutes at 98°C, cooled for 20 minutes at RT and washed in distilled water for 2 minutes. Then, the sections were incubated with a solution of hydrogen peroxide at 3.5% in distilled water, for blocking of the endogenous peroxidase, 10 minutes at RT, followed by a wash with distilled water 3 minutes, RT and PBS 10 minutes, RT. Once dried, the slides were incubated with mouse monoclonal anti-APE1 and with mouse anti-AcAPE1 antibodies diluted 1:100 and 1:300, respectively, for 1 hour at RT. When the primary antibody incubation finished, the slides were dried and incubated for 40 minutes at RT with the detection system EnVision (Dako Real). Subsequently, after washing with PBS, chromogen (3,3′-diaminobenzidine) was added to the slides and left for 1–4 minutes at RT. After this step, the slides were washed for 5 minutes with distilled water and counterstained with Gill's hematoxylin for 1 minute at RT, followed by washing in running water for 2 minutes and in distilled water for 1–2 minutes. At staining end, the slides were dehydrated by immersion in ascending scale of alcohol (80% to the absolute starting alcohol for at least 2 minutes each alcohol solution) and clearing by xylene for 2 times for 2 minutes each. Finally, the stained sections were mounted using the medium Bio Mount (Bio-Optica) and examined with an optical microscope (Olympus,Hicksville, NY). The study was approved by ethics committee of Udine University.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Data.docx
Download MS Word (2.5 MB)Funding
The project was partially funded by the project OPTIMA, n° PON03PE_00155_2 from MIUR. This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC - IG14038) to GT.
References
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834-40; PMID:19608861; http://dx.doi.org/10.1126/science.1175371
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 2014; 15:536-50; PMID:25053359; http://dx.doi.org/10.1038/nrm3841
- Zee BM, Garcia BA. Validation of protein acetylation by mass spectrometry. Methods Mol Biol 2013; 981:1-11; PMID:23381849; http://dx.doi.org/10.1007/978-1-62703-305-3_1
- Qiang L, Xiao H, Campos EI, Ho VC, Li G. Development of a PAN-specific, affinity-purified anti-acetylated lysine antibody for detection, identification, isolation, and intracellular localization of acetylated protein. J Immunoassay Immunochem 2005; 26:13-23; PMID:15754801; http://dx.doi.org/10.1081/IAS-200041153
- Guan KL, Yu W, Lin Y, Xiong Y, Zhao S. Generation of acetyllysine antibodies and affinity enrichment of acetylated peptides. Nat Protoc 2010; 5:1583-95; PMID:21085124; http://dx.doi.org/10.1038/nprot.2010.117
- Xu W, Zhao S. Generation and characterization of pan-specific anti-acetyllysine antibody. Methods Mol. Biol 2013; 981:137-50; PMID:23381859; http://dx.doi.org/10.1007/978-1-62703-305-3_11
- Zee BM, Garcia BA. Discovery of lysine post-translational modifications through mass spectrometric detection. Essays Biochem 2012; 52:147-63; PMID:22708569; http://dx.doi.org/10.1042/bse0520147
- Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene 2005; 24:1653-62; PMID:15744326; http://dx.doi.org/10.1038/sj.onc.1208173
- Vigushin DM, Coombes RC. Targeted histone deacetylase inhibition for cancer therapy. Curr Cancer Drug Targets 2004; 4:205-18; PMID:15032670; http://dx.doi.org/10.2174/1568009043481560
- Wu L, Ma CA, Zhao Y, Jain A. Aurora B Interacts with NIR-p53, Leading to p53. Phosphorylation in Its DNA-binding Domain and Subsequent Functional Suppression. JBC 2011; 286:2236-44; PMID:20959462; http://dx.doi.org/10.1074/jbc.M110.174755
- Kuo HP, Lee DF, Chen CT, Liu M, Chou CK, Lee HJ, Du Y, Xie X, Wei Y, Xia W, et al. ARD1 stabilization of TSC2 suppresses tumorigenesis through the mTOR signaling pathway. Sci Signal 2010; 3(108):ra9; PMID:20145209; http://dx.doi.org/10.1126/scisignal.2000590
- Wang S, Yan-Neale Y, Zeremski M, Cohen D. Transcription regulation by histone deacetylases. Novartis Found Symp 2004; 259:238-45; PMID:15171258; http://dx.doi.org/10.1002/0470862637.ch18
- VanDemark AP, Kasten MM, Ferris E, Heroux A, Hill CP, Cairns BR. Autoregulation of the rsc4 tandem bromodomain by gcn5 acetylation. Mol Cell 2007; 27:817-28; PMID:17803945; http://dx.doi.org/10.1016/j.molcel.2007.08.018
- Li M, Wilson DM. Human apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal 2014; 20:678-707; PMID:23834463; http://dx.doi.org/10.1089/ars.2013.5492
- Busso CS, Lake MW, Izumi T. Posttranslational modification of mammalian AP endonuclease (APE1). Cell Mol Life Sci 2010; 67:3609-20; PMID:20711647; http://dx.doi.org/10.1007/s00018-010-0487-3
- Antoniali G, Lirussi L, D'Ambrosio C, Dal Piaz F, Vascotto C, Casarano E, Marasco D, Scaloni A, Fogolari F, Tell G. SIRT1 gene expression upon genotoxic damage is regulated by APE1 through nCaRE-promoter elements. Mol Biol Cell 2014; 25:532-47; PMID:24356447; http://dx.doi.org/10.1091/mbc.E13-05-0286
- Vascotto C, Cesaratto L, Zeef LA, Deganuto M, D'Ambrosio C, Scaloni A, Romanello M, Damante G, Taglialatela G, Delneri D, et al. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics 2009; 9:1058-1074; PMID:19180539; http://dx.doi.org/10.1002/pmic.200800638
- Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D'Ambrosio C, Scaloni A, et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol 2009; 29:1834-54; PMID:19188445; http://dx.doi.org/10.1128/MCB.01337-08
- Tell G, Wilson DM, Lee CH. Intrusion of a DNA repair protein in the RNome world: is this the beginning of a new era. Mol Cell Biol 2010; 30:366-71; PMID:19901076; http://dx.doi.org/10.1128/MCB.01174-09
- Fantini D, Vascotto C, Marasco D, D'Ambrosio C, Romanello M. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res 2010; 38:8239-56; PMID:20699270; http://dx.doi.org/10.1093/nar/gkq691
- Lirussi L, Antoniali G, Vascotto C, D'Ambrosio C, Poletto M, Romanello M, Marasco D, Leone M, Quadrifoglio F, Bhakat KK, et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol Biol Cell 2012; 23:4079-96; PMID:22918947; http://dx.doi.org/10.1091/mbc.E12-04-0299
- Poletto M, Vascotto C, Scognamiglio PL, Lirussi L, Marasco D, Tell G. Role of the unstructured N-terminal domain of the hAPE1 (human apurinic/apyrimidinic endonuclease 1) in the modulation of its interaction with nucleic acids and NPM1 (nucleophosmin). Biochem J 2013; 452:545-57; PMID:23544830; http://dx.doi.org/10.1042/BJ20121277
- Choi S, Lee YR, Park MS, Joo HK, Cho EJ, Kim HS, Kim CS, Park JB, Irani K, Jeon BH. Histone deacetylases inhibitor trichostatin A modulates the extracellular release of APE1/Ref-1. BBRC 2013; 435:403-7; PMID:23665318; http://dx.doi.org/10.1016/j.bbrc.2013.04.101
- Lee YR, Kim KM, Jeon BH, Choi S. Extracellularly secreted APE1/Ref-1 triggers apoptosis in triple-negative breast cancer cells via RAGE binding, which is mediated through acetylation. Oncotarget 2015; 6:23383-98; PMID:26125438; http://dx.doi.org/10.18632/oncotarget.4345
- Bhakat KK, Sengupta S, Adeniyi VF, Roychoudhury S, Nath S, Bellot LJ, Feng D, Mantha AK, Sinha M, Qiu S, et al. Regulation of limited N-terminal proteolysis of APE1 in tumor via acetylation and its role in cell proliferation. Oncotarget 2016; 7:22590-604; Mar 10. PMID:26981776; http://dx.doi.org/10.18632/oncotarget.8026
- Meisenberg C, Tait PS, Dianova II, Wright K, Edelmann MJ, Ternette N, Tasaki T, Kessler BM, Parsons JL, Kwon YT, et al. Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic Acids Res 2012; 40:701-711; PMID:21933813; http://dx.doi.org/10.1093/nar/gkr744
- Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, Kelley MR, Quadrifoglio F, Tell G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res 2005; 33:4379-4394; PMID:16077024; http://dx.doi.org/10.1093/nar/gki751
- Weibrecht I, Leuchowius KJ, Clausson CM, Conze T, Jarvius MW, Howell M, Kamali-Moghaddam M, Söderberg O. Proximity ligation assays: a recent addition to the proteomics toolboxExpert. Review of Proteomics 2010; 7:401-409; PMID:20536310; http://dx.doi.org/10.1586/epr.10.10
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006; 23:607-18; PMID:16916647; http://dx.doi.org/10.1016/j.molcel.2006.06.026
- Polevoda B, Sherman F. The diversity of acetylated proteins. Genome Biol 2002; 3:reviews0006; PMID:12049668; http://dx.doi.org/10.1186/gb-2002-3-5-reviews0006
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010; 327:1000-4; PMID:20167786; http://dx.doi.org/10.1126/science.1179689
- Park MS, Choi S, Lee YR, Joo HK, Kang G, Kim CS, Kim SJ, Lee SD, Jeon BH. Secreted APE1/Ref-1 inhibits TNF-α-stimulated endothelial inflammation via thiol-disulfide exchange in TNF receptor. Sci Rep 2016; 6:23015; PMID:26964514; http://dx.doi.org/10.1038/srep23015
- Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9 fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res 1990; 35:161-214; PMID:2191922; http://dx.doi.org/10.1111/j.1399-3011.1990.tb00939.x
- Sandomenico A, Russo A, Palmieri G, Bergamo P, Gogliettino M, Falcigno L, Ruvo M. Small peptide inhibitors of acetyl-peptide hydrolase having an uncommon mechanism of inhibition and a stable bent conformation. J Med Chem 2012; 55:2102-11; PMID:22309188; http://dx.doi.org/10.1021/jm2013375
- Carter JM. Techniques for conjugation of synthetic peptides to carrier molecules. Methods Mol Biol 1994; 36:155-91; PMID:7697109; http://dx.doi.org/10.1385/0-89603-274-4:155
- Bradford MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72:248-54; PMID:942051; http://dx.doi.org/10.1016/0003-2697(76)90527-3