
ABSTRACT
We developed an IgG1 domain-tethering approach to guide the correct assembly of 2 light and 2 heavy chains, derived from 2 different antibodies, to form bispecific monovalent antibodies in IgG1 format. We show here that assembling 2 different light and heavy chains by sequentially connecting them with protease-cleavable polypeptide linkers results in the generation of monovalent bispecific antibodies that have IgG1 sequence, structure and functional properties. This approach was used to generate a bispecific monovalent antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor that: 1) can be produced and purified using standard IgG1 techniques; 2) exhibits stability and structural features comparable to IgG1; 3) binds both targets simultaneously; and 4) has potent anti-tumor activity. Our strategy provides new engineering opportunities for bispecific antibody applications, and, most importantly, overcomes some of the limitations (e.g., half-antibody and homodimer formation, light chains mispairing, multi-step purification), inherent with some of the previously described IgG1-based bispecific monovalent antibodies.
Introduction
Monoclonal antibodies are monospecific and bivalent for a single antigen. Although the bivalent binding of the antibodies to their antigens provides the immune system with many advantages, such as increasing the binding events and biologic responses, a single antibody with specificity to 2 distinct antigens is preferred for many therapeutic applications.Citation1 For example, a monovalent bispecific antibody that binds to T cells and to a target expressed on a tumor cell surface can facilitate T-cell redirected target cell killing.Citation2 Many existing bispecific antibody (BsAb) formats bind bivalently to each antigen, but their structures deviate substantially from that of a canonical IgG structure.Citation3-5 Some of these BsAbs have inferior pharmacokinetic properties, including rapid clearance and shorter half-life relative to their IgG counterparts, thus limiting their therapeutic applications.Citation6-12
Several solutions have been described for the generation of BsAb with an IgG chain-like sequence and structure (IgG-Bs).Citation13 For example, IgG-Bs have been produced by expressing the IgGs separately and subsequently mixing them,Citation14,15 but these methods require both site-specific mutations and reduction and oxidation, which complicate downstream development. Moreover, these approaches require the production of 2 separate cell lines expressing the 2 antibody pairs. Other IgG-Bs have been generated by culturing 2 different Escherichia coli, each transformed with DNA encoding one half-antibody obtained by introducing specific mutations in the CH3 domain of the heavy chain.Citation16 This approach has limitations, including mutagenesis in the CH3 domains, inability to use a single cell line for production, and, most notably, the final product does not have the conserved N-297 linked glycans in the CH2 domains that are believed to be important to maintain the structure and function of antibodies.Citation17-21
A computational design strategy has been applied to express the IgG-Bs in a single mammalian cell by identifying complementary point mutations at the heavy and light chain interfaces.Citation22 When expressed using the same mammalian cell, the heavy and light chains assemble into IgG-Bs based on their complementary mutations. However, this approach is laborious because 15 to 20 point mutations are required, and these mutations are not universally applicable to all IgG isotypes. Moreover, this method is only applicable to kappa but not lambda light chains, and only to the VH3 and VH1 antibody gene families. Lastly, a crossover approach has been developed for the production of IgG-Bs antibodies.Citation23 This approach involved the molecular exchange of heavy chain and light chain domains within the antigen-binding fragment (Fab), which deviates from the canonical domain structure of IgG, and included mutations in the CH3 domains to favor heterodimerization. Other designs of IgG-Bs have been described, but with the exception of IgG-Bs that are restricted to the same heavy chain on both binding armsCitation24 and the ‘two-in-one’ IgG,Citation25 these other IgG-Bs are either variations or optimizations of the methods described thus far.Citation26-28 Although these approaches generate IgG-Bs, the deviation from the IgG chain sequence and structure remains a substantial issue,Citation6-12 and novel antibody engineering solutions are thus still needed.
Generating IgG-Bs with an IgG-like chain sequence and structure is challenging for a 2 reasons. First, the homodimeric structure of IgG must be maintained. Second, 2 genes, one for the light chain and another for the heavy chain, are required. The 2 chains are independently translated and assembled into a homodimeric bivalent IgG due to their complementary domain interfaces. We set out to investigate if a single-chain IgG design without any domain interface mutations (herein termed ‘innovative monoclonal antibody’ (iMab)) could overcome inefficiencies inherent with the use of 2 independent genes to form a full-length IgG-Bs molecule with monovalent antigen binding and with IgG1-like sequence and structure. The key design element of the iMab is based on the hypothesis that the light and heavy chains of different antigen specificity (i.e., 2 different light and heavy chains), if in close proximity (i.e., co-transcribed, co-translated, co-folded, co-secreted), will force the monovalent bispecific formation of the iMab.
The iMab design offers a unique engineering solution to overcome some of the limitations of the described previously IgG-Bs,Citation13-28 such as the potential formation of half-antibodies and homodimers, which can be difficult to eliminate and require complex multi-step purification methods.
Results
iMab design
The iMab design uses a single-chain DNA expression cassette () where the IgG1 domains of the 2 different antibodies are connected by protease-cleavable polypeptide linkers (). The polypeptide linkers are designed to be unstructured (i.e., rich in glycine-serine, ) to provide structural flexibility during the assembly of the iMab.
Figure 1. Molecular design, amino acid sequence of the linkers, cartoon representation and protease processing of the iMab, and frequency of the thrombin recognition site in the human CDR-H3. (A) Schematic representation of the tethered single-chain iMab design. The different domains of the iMab are schematically labeled. The linkers are shown in red as dotted lines and the protease cleavage sites are shown by red filled diamonds. (B) Amino acid sequence of the linkers. The thrombin recognition sites are underlined and the arrows represent the cleavage sites. In italics and bold text are the sequences remaining with the iMab after thrombin cleavage. (C) Cartoon representation of the iMab with linkers and (D) iMab with the linkers removed after thrombin treatment. (E) Number of unique CDR-H3 sequences from MedImmune and from abYsis databases with the number of hits found for the thrombin recognition sequence and its frequency.
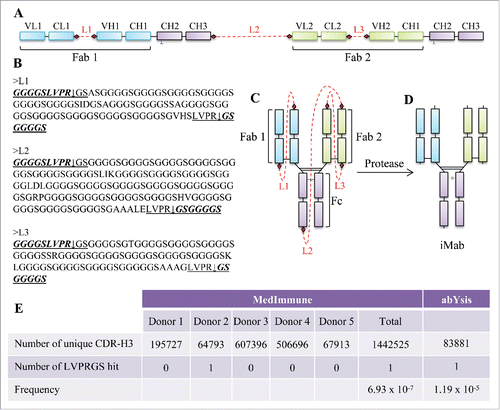
Our selection of the linker length was informed by published studies. Prior work aimed at improving the correct formation of single-chain antibodies by Lee et alCitation29 and Schirrmann et al,Citation30 in which the C-terminus of the light chain was linked to the N-terminus of the heavy chain, resulted in the correct expression of the single-chain IgG, but with the presence of dimers and oligomers. Lee et al and Schirrmann et al used 30 and 34 glycine-serine linkers, respectively, to connect the C-terminus of the light chain to the N-terminus of the heavy chain. A similar linker length was also used by Hust to generate single-chain Fabs.Citation31 This single-chain Fab format required removal of the interchain disulfide bond between the light and heavy chain to achieve appreciable display on phage and expression in Escherichia coli, but expression resulted in high levels of aggregates. Further work on linker length optimization by KeorberCitation32 suggested that the linker lengths used by Lee, Schirrmann and Hust were likely too short to connect the C-terminus of the light chain to the N-terminus of the heavy chain. In fact, modeling of glycine/serine-rich linkers using PEP-FOLDCitation33 suggested that linkers between 50 to 80 amino acids in length may enhance expression, folding and stability of single-chain Fabs.Citation32 To improve molecular assembly, expression and monomeric content, we hypothesized that longer linkers could be beneficial to the iMab design. To this end, the linker we used to connect the C-terminus of the 2 light chains to the N-terminus of their respective heavy chains (linker L1 and L3 in ) was ∼3X longer than that used by Lee,Citation29 SchirrmannCitation30 and Hust,Citation31 and about 1.5X longer than that used by Koerber,Citation32 while the linker we used to connect the C-terminus of the first heavy chain to the N-terminus of the second light chain (linker L2 in ) was ∼4.5X longer than the linker used by Lee,Citation24 SchirrmannCitation25 and Hust,Citation31 and 2.5X longer than that used by Koerber.Citation32
The linkers also contain protease cleavage sites (diamond symbols in ; underlined sequence in ) at their ends to facilitate cleavage upon protease treatment (). We chose to use thrombin as a protease to process the linkers because sequence analysis of V, D and J gene segments of the human antibody heavy chains, and the V and J segments of the kappa and lambda light chains, compiled from the international ImMunoGeneTics information system® (IMGT; www.imgt.org),Citation34 revealed no matched hit of thrombin recognition sequence (). Given that the insertions and deletions at VDJ junctions would generate complementarity-determining region (CDR)-H3 sequences that could not be found in human antibody germline sequences, we searched for the thrombin recognition sequence in 2 additional databases: 1) a MedImmune proprietary next-generation sequencing database of over 1.4 million unique CDR-H3 sequences obtained from primary B cells from 5 healthy human donors, and 2) sequences retrieved from the abYsis database.Citation35 Both database searches revealed a very low frequency for the thrombin recognition sequence for the CDR-H3 (). Cleavage with thrombin resulted in 9 residues (i.e., GGGGSLVPR) to remain at the C-terminus of the 2 light chains and C-terminus of the first heavy chain, and 7 residues (i.e., GSGGGGS) to remain at the N-terminus of the 2 heavy chains and at the N-terminus of the second light chain ().
Design of an iMab targeting EGFR and IGF1R (iMab-EI)
To demonstrate feasibility of the iMab, we designed, expressed and characterized iMab-EI, a bispecific monovalent iMab that binds the epidermal growth factor receptor (EGFR) and the type I insulin-like growth factor receptor (IGF1R) (Fig. S1). EGFR and IGF1R are cell surface receptor tyrosine kinases known to cooperate to promote tumor growth and progression.Citation36 Dual inhibitions of EGFR and IGF1R have been shown to improve anti-tumor activity and to overcome resistance to therapy against a single receptor inhibition in preclinical models.Citation36 In iMab-EI, the anti-EGFR light chain is connected to its heavy chain by a 104-residue peptide linker (linker L1 in ; ; Fig. S1). This half-antibody generates the anti-EGFR monovalent binding arm of iMab-EI. The monovalent anti-EGFR arm is connected to the anti-IGF1R light chain by a 165-residue peptide linker also rich in glycine-serine (linker L2 in ; ; Fig. S1), which is then connected to the anti-IGF1R heavy chain by a 103-residue glycine-serine linker (linker L3 in ; ; Fig. S1), thus generating the anti-IGF1R monovalent binding arm.
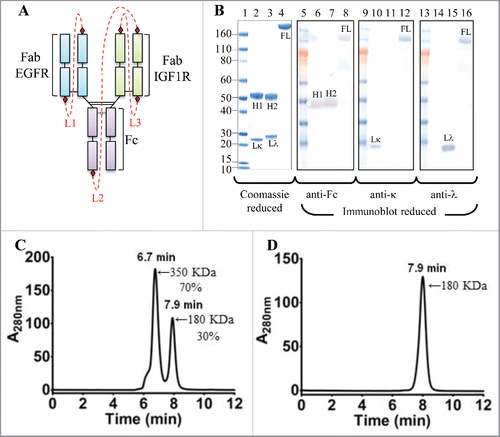
Figure 2. Cartoon representation, SDS-PAGE, reduced western blot and SEC-MALS analyses of the iMab-EI. (A) Cartoon representation of the iMab-EI with linkers connecting the antibody domains shown as red dotted lines. (B) Reduced SDS-PAGE (lanes 1 to 4) and reduced western blot analysis (lanes 5 to 16) of iMab-EI, anti-EGFR and anti-IGF1R antibodies. Molecular mass standards are schematically shown. Lκ, Lλ, H1 and H2 denote anti-EGFR kappa light chain, anti-IGF1R lambda light chain, anti-EGFR heavy chain, anti-IGF1R heavy chain, respectively. FL denotes the full-length iMab-EI. Lane 2 is the anti-EGFR antibody, lane 3 is the anti-IGF1R antibody and lane 4 is the full-length iMab-EI. Lanes 6, 7 and 8 is the reduced western blot probed with an anti-human Fc antibody. Lanes 10, 11 and 12 is the reduced western blot probed with an anti-human kappa antibody. Lane 14, 15 and 16 is the reduced western blot probed with an anti-human lambda antibody. (C) SEC-MALS of the iMab-EI after protein A purification. (D) SEC-MALS of monomeric iMab-EI. The molecular weights in KDa were obtained using SEC-MALS.
Expression and purification of the iMab-EI
The iMab-EI single-chain gene was codon optimized for optimal expression in mammalian cells (Fig. S2). The iMab-EI gene was cloned into a mammalian IgG1 expression vector that has a cytomegalovirus promoter and the native light chain kappa leader sequence (Fig. S1).Citation4 The iMab-EI expression vector was transfected into HEK293 cells. The production yield of iMab-EI was 10 mg/L as determined using a quantitative protein A binding method.Citation4 This expression yield is similar to the single-chain IgG1 reported by Schirrmann et al.Citation30 However, the expression level of the iMab-EI was improved by using 2 engineering strategies. First, by cloning the iMab-EI expression cassette into a vector containing UCOEs elements,Citation37 which maintains the chromatin in an open state and facilitates transcription, we obtained a 3-fold improvement (30 mg/L) in the expression level (Fig. S3). Second, by engineering endogenous protease cleavage sites instead of thrombin recognition sites (i.e., furin cleavage sites replaced those of thrombin) (Fig. S4), we observed a transient expression of 45 mg/L, with about 70% of the iMab-EI fully processed in the culture media (Fig. S4). This expression level corresponds to a 4-fold improvement of that obtained when the thrombin cleavage site was used. The iMab-EI was purified using protein A chromatography following methods commonly used for the purification of IgG1. The ability to purify iMab-EI using a protein A column indicates that the iMab-EI has a domain structure similar to that of a conventional IgG1, and that the connecting linkers do not block the binding of iMab-EI to protein A.
Analytical characterization of the iMab-EI
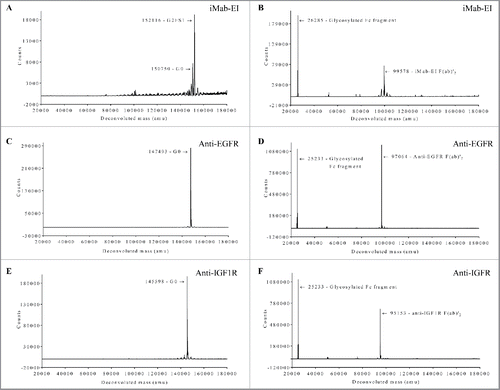
When the protein A purified iMab-EI was analyzed using reduced SDS-PAGE (), a single polypeptide chain was observed, while 2 distinct heavy and light chains were observed for the 2 parental antibodies ( and ). To verify identity of the polypeptide chains, anti-human-Fc, anti-human-kappa and anti-human-lambda monoclonal antibodies were used for reduced western-blot analysis (). The anti-human-Fc antibody identified the heavy chains of the 2 parental antibodies () at a molecular weight of about 50 KDa, and the iMab-EI polypeptide chain at a molecular weight of about 180 KDa (). When the reduced western blots were processed using the anti-human-kappa and anti-human-lambda antibodies, the light chains of the 2 parental antibodies were identified as the anti-EGFR (), the anti-IGF1R (), while both anti-light chain antibodies identified the polypeptide chain of the iMab-EI because it contains both light chain isotypes ().
The iMab-EI was subsequently analyzed using size-exclusion chromatography multi-angle light scattering (SEC-MALS) and shown to be composed of 2 peaks (). The 2 peaks have an estimated molecular weight of 180 KDa with a retention time of 7.9 min and 350 KDa with a retention time of 6.9 min, corresponding to 30% and 70%, respectively. These molecular weights likely correspond to monomer and dimer. Before performing thrombin digestion, we proceeded with the purification of the monomeric species using preparative gel-filtration. The monomeric peak was successful purified from the dimeric peak, and, when re-analyzed by SEC-MALS, the purified monomeric peak maintained its retention time of 7.9 min and apparent molecular weight of 180 KDa (). Moreover, the monomeric peak remained monomer (i.e., no formation of dimer, aggregates or fragments) upon storage at 4°C for over 3 months (data not shown).
Thrombin cleavage and analytical characterization of the thrombin-processed iMab-EI
We next performed thrombin digestion to cleave the polypeptide linkers. Digestion was performed in solution and the cleaved peptide linkers and the thrombin were removed by centrifugation using a 50,000 molecular weight cut-off membrane filter. The reduced SDS-PAGE analysis of the thrombin-treated iMab-EI showed the presence of 4 polypeptides corresponding to the 2 light chains and the 2 heavy chains, respectively (). In this analysis, the parental anti-EGFR and anti-IGF1R antibodies were used for identification of the light and heavy chains ( for the anti-EGFR and anti-IGF1R, respectively). Reduced western blot was then used for identification of the 4 polypeptide chains in the iMab-EI. As shown in , the anti-Fc antibody identified the 2 heavy chains of the iMab-EI, and the heavy chains of the 2 parental antibodies (). The anti-lambda and anti-kappa light chains specific antibodies revealed that, as expected, the iMab-EI possesses both light chain isotypes (). SEC-MALS analysis () of the iMab-EI showed that it is 99% monomer, with a retention time of 8.3 min and a molecular mass of 154 KDa similar to the 2 parental antibodies (data not shown).
Figure 3. Reduced SDS-PAGE, reduced western-blot, SEC-MALS, rRP-HPLC and rLCMS of the iMab-EI after thrombin cleavage. (A) Reduced SDS-PAGE (lanes 1 to 4) and western blot analysis (lanes 5 to 16) of iMab-EI, anti-EGFR and anti-IGF1R antibodies. Molecular mass standards are schematically shown. Lκ, Lλ, H1 and H2 denote anti-EGFR kappa light chain, anti-IGF1R lambda light chain, anti-EGFR heavy chain, anti-IGF1R heavy chain, respectively. Lκ*, Lλ*, H1* and H2* denote the iMab-EI anti-EGFR kappa light chain, anti-IGF1R lambda light chain, anti-EGFR heavy chain, and anti-IGF1R heavy chain, respectively. Lane 2 is the anti-EGFR antibody, lane 3 is the anti-IGF1R antibody and lane 4 is the iMab-EI. Lanes 6, 7 and 8 is the reduced western blot probed with an anti-human Fc antibody. Lanes 10, 11 and 12 is the reduced western blot probed with an anti-human kappa antibody. Lanes 14, 15 and 16 is the reduced western blot probed with an anti-human lambda antibody. The chains of the iMab-EI are migrating slower compared to the chains from the 2 parental antibodies due their slightly higher molecular weight. (B) SEC-MALS analysis of the iMab-EI after thrombin cleavage showed that the iMab-EI is 99% monomer and has a molecular weight resembling that of an intact IgG1. (C) rRP-HPLC of the iMab-EI, (D) anti-IGF1R, and (E) anti-EGFR. The identity of each peak is schematically labeled as shown in panel A. (F) rLCMS of iMab-EI. The chains of the iMab-EI are labeled as shown in the panel A and as in panel C. The molecular masses of the 4 chains are shown in Daltons. The rLCMS was performed after Endo S treatment to trim the N-glycan found at the conserved N297 site in iMab-EI CH2 domain.
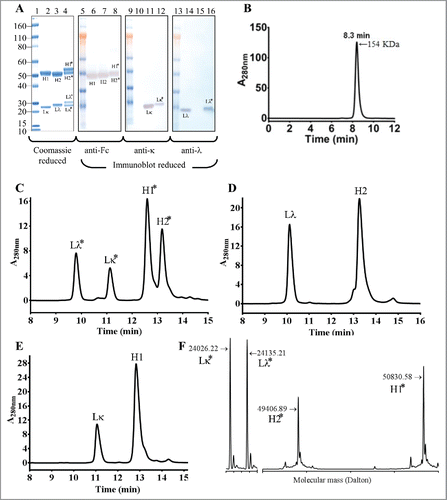
To further confirm that the iMab-EI is composed of 2 light chains and 2 heavy chains, we used reduced reverse-phase liquid chromatography and mass spectrometry (rRP-HPLC and rLCMS). The rRP-HPLC analysis revealed that the iMab-EI has 4 peaks (), corresponding to the 2 light and the 2 heavy chains of the anti-IGF1R and anti-EGFR arms, respectively (). and show the rRP-HPLC of the 2 parental antibodies. The 4 chains of the iMab-EI were further identified based on their molecular masses using rLCMS (). These analyses confirmed the efficient cleavage of the connecting linkers, and that the iMab-EI possesses 4 polypeptide chains.
To confirm that the iMab-EI has disulfide bonds at the hinge and at the heavy light chains similar to those of an IgG1, we used non-reduced SDS-PAGE and non-reduced LCMS. Non-reduced SDS-PAGE shows that iMab-EI has correctly formed the interchain disulfide bonds at the hinge and at the heavy and light chains (), similar to the 2 parental antibodies (and). Non-reduced LCMS of the intact iMab-EI and the iMab-EI treated with IdeS, which cleaves IgG1 at the lower hinge and releases the Fc fragment and the F(ab)2, clearly confirmed that iMab-EI has interchain disulfide bonds at the hinge and at the heavy and light chains similar to the 2 IgG1 parental antibodies ().
Figure 4. iMab-EI has native interchain disulfide bonds at the hinge and at the heavy-light chains as demonstrated using non-reduced SDS-PAGE. Lane 1 is the molecular weight standards; lane 2 is the anti-IGF1R, lane 3 is the anti-EGFR.
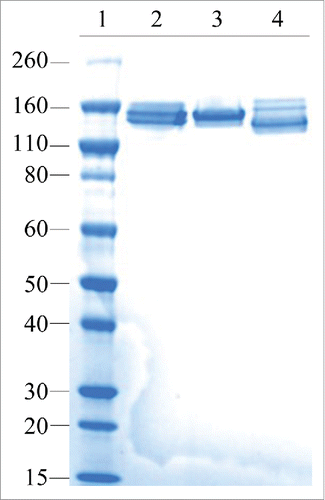
Figure 5. Non-reduced LCMS of the iMab-EI and the 2 parental antibodies intact glycosylated and after treatment with IdeS, which release the Fc fragment from the F(ab)’2. (A) Intact glycosylated iMab-EI; (B) IdeS-treated iMab-EI; (C) Intact glycosylated anti-EGFR; (D) IdeS-treated anti-EGFR; (E) Intact glycosylated anti-IGF1R; (F) IdeS-treated anti-IGF1R; The deconvoluted mass in Dalton is shown on the x-axis and the ions counts are shown on the y-axis. The molecular weight of each peak is schematically shown with the corresponding glycoform. LCMS show the iMab-EI has correctly formed the interchain disulfide bonds at the hinge and at the heavy and light chains similar to IgG1 parental antibodies.
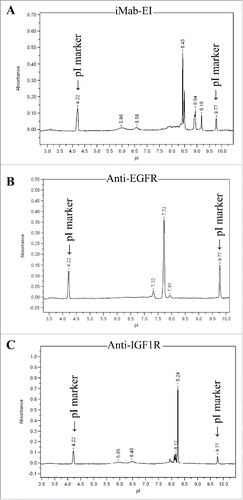
To demonstrate that the iMab-EI is homogeneous (i.e., does not contain a mixture of the 2 parental antibodies), we performed isoelectric point determination for iMab-EI and the 2 parental antibodies. Our rationale was that each protein has its unique isoelectric point and charge characteristic, and therefore it would be a simple and efficient approach to detect the presence of the 2 parental antibodies in the iMab-EI preparation. As shown in , iMab-EI's charge characteristics and its isoelectric point are distinct from those of the 2 parental antibodies ().
Figure 6. Isoelectric point (pI) of the iMab-EI and the 2 parental antibodies. (A) iMab-EI pI main peak is 8.43; (B) anti-EGFR antibody pI main peak is 7.72; (C) anti-IGF1R pI main peak is 8.24. pI marker peaks 4.22 and 9.77 are schematically shown.
Biophysical characterization of the iMab-EI
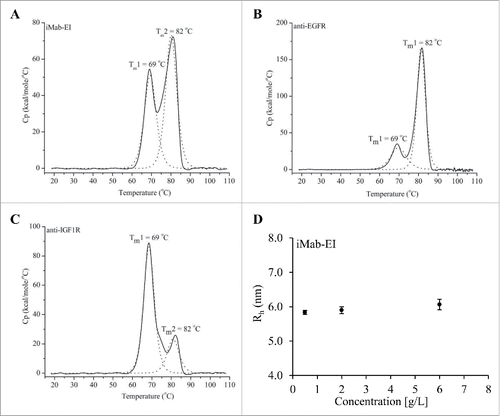
Transition temperatures (Tm) and hydrodynamic radius (Rh) at 3 different concentrations of the iMab-EI were determined using differential scanning calorimetry (DSC) and SEC-MALS. DSC analysis showed that the iMab-EI has 2 transition temperatures, 69°C and 82°C (), which are same as the 2 transition temperatures observed for the 2 parental antibodies ( and ). This indicates the iMab-EI has a structure similar to the IgG1 parental antibodies. In addition, the Rh of the iMab-EI remained similar when analyzed at 0.5, 2 and 6 mg/mL (). Rh data also confirmed that the Rh of the iMab-EI is similar to that of IgG1.Citation4
Figure 7. Differential scanning calorimetry (DCS) of the iMab-EI (A), the anti-EGFR (B) and anti-IGF1R (C) antibodies. Transition temperatures are shown as Tm in°C. DSC analysis shows that the iMab-EI has transition temperatures similar to the transition temperatures of the anti-EGFR and anti-IGF1R antibodies, indicating the iMab-EI has a structure similar to the anti-EGFR and anti-IGF1R antibodies. (D) Hydrodynamic radios (Rh) determined using SEC-MALS of the iMab-EI at 0.5, 2 and 6 mg/mL. The Rh of the iMab-EI remains similar at the 3 concentrations tested, which is an indication of structural stability of the iMab-EI.
Concurrent binding and kinetics of binding of the iMab-EI to EGFR and IGF1R
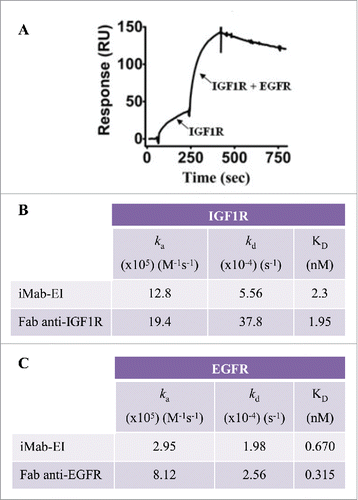
To study concurrent binding to EGFR and IGF1R, the iMab-EI was immobilized on a BIAcore CM5 sensor chip. Concurrent binding to IGF1R and EGFR was demonstrated by first injecting IGF1R, followed by a mixture of EGFR and IGF1R (). Having demonstrated concurrent binding to EGFR and IGF1R, we proceeded to determine the kinetics of binding of the iMab-EI to EGFR and IGF1R using BIAcore. The KD of the iMab-EI for EGFR and IGF1R are similar to the KD determined using the Fabs prepared from the anti-EGFR and anti-IGF1R antibodies ( and ).
Figure 8. Binding of iMab-EI to EGFR and IGF1R using BIAcore. (A) Concurrent binding to antigens using BIAcore. iMab-EI was immobilized on the BIAcore sensor chip and dual binding was determined by first injecting IGF1R, followed by co-injection of IGF1R and EGFR. (B) Binding kinetics to IGF1R of the iMab-EI and the Fab prepared from the anti-IGF1R antibody. (C) Binding kinetics to EGFR of the iMab-EI and the Fab prepared from the anti-EGFR antibody.
Assessment of iMab-EI homogeneity
Non-reduced LCMS and pI studies confirmed that the iMab-EI is homogeneous and does not contain light and heavy chains mispaired iMabs ( and ). To further demonstrate that the iMab-EI preparation is homogeneous, we developed an ELISA transfer assay. This assay consisted of incubating iMab-EI in 2 distinct ELISA plates, one with coated EGFR and one with coated IGF1R, at a pre-determined binding saturation concentration of 0.02 μg/mL (data not shown), followed by transferring the supernatant from the EGFR-coated plate to the IGF1R-coated ELISA, and the supernatant from the IGF1R-coated plate to the EGFR-coated ELISA plate. If iMab-EI is homogeneous, we expect no binding signal when the well supernatants are swapped. In fact, and showed that the iMab-EI supernatant taken from the EGFR-coated ELISA well did not result in binding when transferred to the IGF1R-coated ELISA plate () and vice versa ().
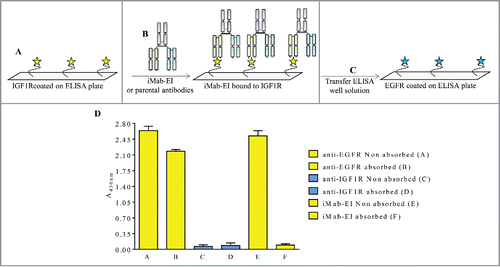
Figure 9. Schematic representation and data of the EGFR ELISA transfer assay. (A) EGFR was immobilized on the ELISA plate, (B) and incubated with iMab-EI or the 2 parental antibodies. The well volume of the iMab-EI or the parental antibodies pre-absorbed on an EGFR-coated ELISA plates (C) were transferred to an ELISA plate with immobilized IGF1R, (D) followed by detection of binding using an anti-human-lambda-HRP labeled antibody.
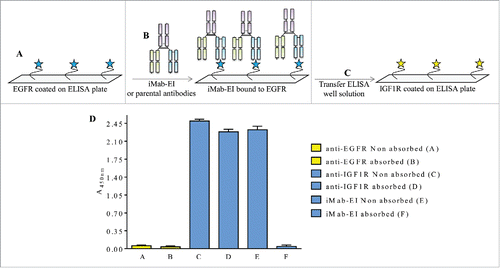
Figure 10. Schematic representation and data of the IGF1R ELISA transfer assay. (A) IGF1R was immobilized on the ELISA plate, (B) and incubated with iMab-EI or the 2 parental antibodies. The well volume of the iMab-EI or the parental antibodies pre-absorbed on an IGF1R-coated ELISA plates (C) were transferred to an ELISA plate with immobilized EGFR, (D) followed by detection of binding using an anti-human-kappa-HRP labeled antibody.
Binding of iMab-EI to FcRn and C1q
To confirm that iMab-EI has an intact and functional Fc similar to IgG1, we verified that binding to FcRn and C1q is similar to that of the parental anti-IGF1R and anti-EGFR antibodies. Binding to FcRn is important in maintaining IgG1 pharmacokinetics, and binding to C1q is important to preserve complement-dependent cytotoxicity during the humoral immune response against pathogens. iMab-EI was able to bind to human FcRn (Fig. S5A) and human C1q (Fig. S5B) with similar EC50 as the parental anti-IGF1R and anti-EGFR antibodies, indicating iMab-EI maintains a functional Fc.
In vivo tumor growth inhibition of iMab-EI
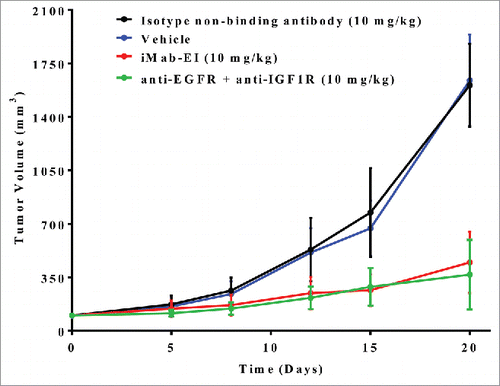
Supported by the successful preparation of the iMab-EI and its IgG1-like analytical and functional properties, we tested in vivo tumor growth inhibition. Patient-derived stage IV recurrent metastatic non-small cell lung cancerCitation38 was implanted subcutaneously as a xenograft into RAG2 knockout (KO) mice. The non-small cell lung tumor was shown to express EGFR and IGF1R (Fig. S6). As shown in , treatment of the mice twice weekly for 2 weeks with iMab-EI resulted in a 73% reduction in tumor growth, indicating that the iMab-EI has potent in vivo tumor growth inhibition. The in vivo efficacy of the iMab-EI matched that of the mixture of the anti-EGFR and anti-IGF1R antibodies (), despite the fact that iMab-EI is monovalent for each target, while the anti-EGFR and anti-IGF1R binds their respective targets bivalently.
Figure 11. In vivo efficacy of iMab-EI using a xenograft mouse model of a patient derived non-small cell lung cancer. iMab-EI (red curve), isotype non-binding antibody (black curve) and combination of the anti-EGFR and anti-IGF1R antibodies (green curve) were dosed at 10 mg/kg 2 times weekly for 2 weeks. Vehicle-treated mice (blue curve) were used as negative control.
Discussion
A major impediment to the generation of bispecific monovalent antibodies with IgG1-like chain sequence and structure is that current methods require the use of 2 independent expression cassettes, one for the light and one for the heavy chains, and subsequent assembly of these 2 chains in a single cell. This approach results in the production of mixtures of mispaired, nonfunctional products that need to be separated from the correctly paired product. This process is inefficient and requires multi-step purifications. To overcome these technical difficulties, we designed and characterized a single-chain IgG1 by tethering 2 IgG1 of different specificity with unstructured and protease-cleavable linkers. The experimental evidence showed that our engineering approach to prepare a monovalent bispecific antibody with IgG1-like chain sequence and structure, which we named iMab, was successful. Our single-chain design approach has the potential for wide use as an antibody engineering tool to generate bispecific monovalent constructs for research purposes.
Key design elements of the iMab are the linkers connecting the light chains to their respective heavy chains and the linker connecting the 2 half antibodies. Previous studies using peptide linkers to connect different protein domains have shown that linkers lacking secondary structure and displaying a high degree of flexibility offer many advantages compared with rigid and structured linkers, such as increasing expression yield and improved folding.Citation39,40 Seminal work performed by Argos,Citation39 and George and HeringaCitation40 suggested that pentapeptides consisting of glycine, serine and threonine would make the best modular linkers because these residues are most frequently present in naturally occurring linkers, provide high flexibility due to their small sizes, and, importantly, they are hydrophilic, which help stabilize their conformation in aqueous solvent through formation of hydrogens bonds. For these reasons, linkers rich in glycine and serine have been widely used for the construction of stable, bioactive fusion proteins, including antibodies.Citation3-5,41 The linkers we used for the iMab are also rich in glycine and serine and have been designed to have a longer length than previous described linkers.Citation3-5, 29-32 Our hypothesis is that longer linkers would facilitate the correct assembly of the different domains of the iMab.
We engineered the iMab linkers to include protease-cleavable sites at their respective ends. This design feature allows cleavage of the linkers upon protease treatment, thereby removing the linkers from the iMab after purification. We selected thrombin as the protease because sequence analyses revealed a very low frequency of thrombin recognition sequence in antibodies. Moreover, the thrombin catalytic activity is highly efficient, and can be used in solution or immobilized on agarose beads, thus facilitating downstream purification. The amino acid sequence that remains after thrombin cleavage is minimal, thus limiting potential sources of immunogenicity. The linkers were also engineered to contain unique DNA restriction site sequences at their ends to facilitate cloning of different antibody pairs.
To test our antibody engineering approach, we developed a monovalent bispecific antibody with an IgG1-like chain sequence and structure that concurrently binds to EGFR and IGF1R (i.e., iMab-EI). iMab-EI can be expressed in mammalian cells using standard antibody expression methods.
The iMab-EI with the intact linkers was purified using protein A affinity chromatography. Successful purification of iMab-EI using protein A indicates the connecting linkers do not block the binding to protein A and do not destabilize the structure of the iMab-EI. Subsequently the monomeric iMab-EI was purified and the linkers were efficiently cleaved with thrombin.
Results from the analytical characterization provided highly convincing evidences of the correct formation of the iMab-EI. In fact, results from the reduced SDS-PAGE, reduced RP-HPLC and reduced LCMS showed the iMab-EI is composed of 4 distinct polypeptide chains corresponding to the light and heavy chains of the anti-IGF1R and EGFR antibodies. The same analytical assays under non-reducing conditions confirmed the correct formation of the hinge and light heavy chains disulfides.
Correct paring of the light and heavy chains was further confirmed by the ELISA transfer assay, which showed the iMab-EI preparation was homogeneous (i.e., the binding was not due to the presence of a mixture of the 2 parental antibodies). The iMab-EI retained similar kinetics of binding to the 2 antigens as the Fab prepared using the 2 parental antibodies. Moreover, the iMab-EI was able to concurrently bind both antigens and had potent in vivo efficacy, which is an indication of in vivo stability.
The iMab-EI has a functional Fc as demonstrated by binding to FcRn and C1q, which is similar to the binding of the parental antibodies.
DSC studies showed that the iMab-EI has transition temperatures similar to the 2 parental antibodies, further confirming the antibody-like stability of iMab-EI. Recent reports have shown that antibodies with a lambda light chain are not as stable as those with a kappa light chain, mainly due to a high propensity of disulfide bond reduction between the light and heavy chains in antibodies with the lambda light chain.Citation42 LCMS analysis revealed that the last serine residue of the lambda light chains contribute to the susceptibility of the interchain disulfide bond reduction.Citation43 One of the light chains in iMab-EI is lambda and its last serine residue is followed by the 4 residues LVPR, which remain at the N-terminus of the light chain after thrombin cleavage. These residues do not have a negative effect on the stability of the iMab, as determined by DSC, and do not have any effect on the formation of the light and heavy chain disulfide bonds, as determined by non-reduced LCMS and non-reduced SDS-PAGE.
Thrombin cleavage of the linkers results in GGGGSLVPR sequence to remain at the C-terminus of the 2 light chains, a single GGGGSLVPR sequence to remain at the C-terminus of one heavy chain and GGGGSGS sequence to remain at the N-terminus of the 2 light chains and one heavy chain. The BIAcore binding experiments demonstrated that the GGGGSGS sequences remaining at the N-terminus of the iMab do not have any negative effect on the kinetics of binding and affinities for EGFR and IGF1R.
As is the case for other bispecific antibodies that contains linkers,Citation4,5 immunogenicity of the linker sequences in the iMab is a potential clinical risk. However, the iMab was designed as a tool for research, where the bispecific monovalent antibody format and the single-chain tethered approach may facilitate discovery of cognate targets that cannot be identified by using conventional monospecific antibodies.
We demonstrated that thrombin is an ideal protease to process the linkers because its sequence recognition motif has a very low frequency in antibody repertoires and its catalytic activity is highly efficient. However, the use of thrombin may have some drawbacks. For example, it requires an additional purification step during the iMab preparation to remove the thrombin and the linkers, which can complicate preparation of the iMabs. A potential solution to eliminate this additional purification step is to engineer the linkers with protease motifs that are recognized by endogenous proteases (i.e., proteases that are ubiquitously expressed in the host used to express the iMab). To this end, we engineered furin cleavage sites to replace the thrombin cleavage sites and observed a transient expression 4-fold higher than the one obtained when the thrombin cleavage site was used.
Furin could be an ideal enzyme for the intracellular cleavage of the linkers because it is enriched in the Golgi, which is a translocation route during the iMab intracellular processing. Additional studies that examine furin iMab technology to improve processing are warranted. For example, engineering the mammalian expression host to overexpress the furin protease, which should improve yields of fully processed iMab, may be possible.
Another option would be to link a protease at the C-terminus of the iMab. In this arrangement, the protease will cleave itself from the polypeptide (i.e., the protease is linked at the C-terminus of the iMab with the same protease cleavage sequence used in the linkers), and thereafter could cleave the linkers in the iMab.
The iMab design also allows each of the 2 Fc chain to have different mutations, such as mutations in one chain to modulate Fc effector functions and mutations in the other chain for conjugation of cytotoxic drugs.Citation44-46 Finally, the iMab technology may be applied to mammalian, yeast and ribosomal display systems, which use a single polypeptide for selection. Moreover, the tethered single-chain polypeptide approach can be applied to conventional IgG to facilitate in vivo gene transfection.Citation47
In conclusion, the innovative design of the iMab offers an antibody engineering framework for future applications and paves the way for use of a “tethered and cleavable connecting linker” approach to the engineering of monovalent multispecific biologics for preclinical research applications.
Materials and methods
Cloning
The amino acid sequences of the anti-EGFR antibody variable domains were obtained using SciFinder database with entry code Panitumumab (CAS Registry Number 339177-26-3) in the substance identifier search box. The amino acid sequences of the panitumumab variable domains were back-translated to DNA and the sequences were optimized for optimal expression in mammalian cells. The panitumumab variable domains were cloned into a human IgG1κ mammalian expression vector.Citation4 The anti-IGF1R antibody was identified at MedImmune from an antibody phage display library. The DNA sequences of the variable domains of the anti-IGF1R antibody were synthetized and optimized for optimal protein expression in mammalian cells and cloned into a human IgG1λ mammalian expression vector.Citation4 The iMab-EI DNA sequence, which consisted of the variable domains from the 2 parental antibodies, the light and heavy chain constant domains, and the linkers, was assembled using synthetic oligonucleotides (Thermo Fisher Scientific). The iMab-EI DNA and amino acid sequences are shown in Fig. S2. The iMab-EI with the furin cleavage site consisted of the same iMab-EI DNA sequence, but with the thrombin cleavage site changed to the furin cleavage site. UCOEs-containing vector was supplied by Millipore Corporation. Key elements of the UCOEs expression vector are shown in Fig. S3.
Western blot of endogenous furin expressed in Chinese hamster ovary cells
1×106 cell/mL Chinese hamster ovary cells were centrifuged at 1,500 g for 10 min and the pellet suspended in 30 mL of phosphate-buffered saline (PBS) pH 7.2. As a positive control, 0.3 μg of recombinant furin (R&D System, cat. No. 1503-SE-010) was prepared in 30 μL of PBS pH 7.2. Ten μL of 4X protein sample buffer supplemented with NuPAGE reducing agent (Thermo Fisher Scientific) was added to the culture media, cell pellet and to the recombinant furin. Samples were incubated at 70°C for 10 min, briefly spun down using a benchtop centrifuge at 13,500 rpm for 30 sec and loaded on a Nupage 10% SDS-PAGE and electrophoresed at 200 V in 3-(N-morpholino) propanesulfonic acid buffer (Thermo Fisher Scientific). SeeBlue Plus2 proteins molecular weight standard was used (Thermo Fisher Scientific). The SDS-PAGE was transferred to PVDF membranes (Thermo Fisher Scientific) using iBlot2 Dry Blotting System (Thermo Fisher Scientific). After the proteins were transferred, the PVDF membranes were blocked with 3% on non-fat dry milk in PBS (blocking buffer) for one hour at room temperature. The membranes were washed 3 times with PBS and incubated for one hour at room temperature with 0.1 μg/mL anti-human furin antibody (R& D System, cat. No. MAB15032-SP) prepared in blocking buffer. The membranes were washed 3 times with PBS and 1:10.000 of anti-mouse anti-goat Fc antibody HRP conjugated (R& D System, cat. No. G-102-C) prepared in blocking buffer was added for one hour at room temperature. After 3 washes with PBS, the furin protein was visualized using 1 mL of 3,3′,5,5′-tetramethylbenzidine substrate (KPL) for 5 min at room temperature. The membranes were then washed with water and stored dried.
Transfection, expression and quantification
Transient antibody expression was performed in HEK239F suspension cells. Cells were cultured in FreeStyle™ serum-free media (Thermo Fisher Scientific) at 120 rpm, 37°C and 8% CO2. Twenty-four hours before transfection, the cells were split to 7 × 105 cells and cultured in FreeStyle™. Two hundred μg of vector DNA and 300 μL of 293fectin™ transfection reagent (Thermo Fisher Scientific) were independently mixed with 10 mL of Opti-MEM® I Reduced Serum Medium (Thermo Fisher Scientific) and incubated for 5 min at room temperature. Next, the 2 mixtures were mixed together, incubated for 25 min at room temperature, and then added to 300 mL of 1×106 HEK239F cells. The cells were grown for 11 days with addition of 300 mL of FreeStyle™ every 3 days. HEK239F cells transfection and expression of iMab-EI-Enterokinase was performed as described for the iMab-EI with the furin cleavage sites. The antibody expression level during cell culture was determined using a protein A binding method. One hundred μL of culture media was filtered using a 0.2 μm Eppendorf filter (Eppendorf), and loaded onto a protein A column (POROS® A 20 µm Column, 4.6 × 50 mm, 0.8 mL) using an HPLC system (Agilent 1100 Capillary LC System). Antibodies were detected using absorbance at 280 nm. The column with antibody bound was washed with a solution of PBS pH 7.2, and antibodies were eluted with 0.1% phosphoric acid (pH 1.8). The area corresponding to the eluted peak was integrated and antibody concentration was determined by comparing the area under the peak obtained using an IgG at known concentration.
Purification
Antibodies were purified using protein A affinity chromatography. The culture media was collected after 11 days post transfection and cells were removed using centrifugation at 1500 rpm for 5 min. The media was then filtered using a 0.2 μM vacuum filter (Nalgene). HiTrap 5 mL protein A columns (GE Healthcare) were used for the purification with an AKTA Explorer (GE Healthcare). The protein A column was equilibrated with 20 column volumes of PBS pH 7.2 using a flow rate of 5 mL/min and the culture media was loaded also using a flow rate of 5 mL/min. Unbound material was removed by using 20 column volumes with PBS pH 7.2. Elution of the antibodies was performed using 5 column volumes of 0.1 M glycine, 150 mM sodium chloride pH 3.2, and the elution was monitored using an absorbance of 280 nm. The protein A eluted antibodies were immediately neutralized by using 1/10 of volume of 1 M Tris-HCl pH 7.0. The antibodies were then filtered using a 0.22 μM syringe filter (Nalgene) and dialyzed overnight at 4°C in PBS pH7.2. The concentration of the purified antibodies was determined by reading the absorbance at 280 nm using a NanoDrop (Thermo Fisher Scientific) and an extinction coefficient of 1.4 M−1cm−1. Preparative gel-filtration chromatography was performed using HiLoad Superdex 200 PG (GE Healthcare) at 1 mL/min.
Analytical characterization
Reduced SDS-PAGE was performed using 5 μL of SDS-PAGE loading buffer (Thermo Fisher Scientific) with the addition of NuPAGE reducing reagent (Thermo Fisher Scientific) and 2 μg of antibodies for the Coomassie Blue staining, and 0.5 μg for the western blot. Ten µL of the Novex Sharp Pre-Stained Protein Standard (Thermo Fisher Scientific) was used as a marker. The samples were heated at 70°C for 10 min, centrifuged at 13,500 rpm using a benchtop centrifuge and loaded onto 4-12% Bolt gel (Thermo Fisher Scientific). Electrophoresis was performed in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer at 150 V for one and half hours. The SDS-PAGE gels were stained with SimplyBlue™ SafeStain (Thermo Fisher Scientific) and distained in water overnight.
For the western blot analysis, the antibodies separated by the SDS-PAGE were transferred to PVDF membranes (Thermo Fisher Scientific) using the iBlot 2 Dry Blotting System (Thermo Fisher Scientific) following manufacturer's instructions. After transfer, the membranes were blocked using 3% of non-fat dry milk in PBS for one hour at room temperature. The membranes were washed 3 times with PBS. Horseradish peroxidase (HRP)-labeled anti-human-Fc (Thermo Fisher Scientific; cat. No. 31413), anti-human-kappa (SouthernBiotech; cat. No. 2060-05) and anti-human-lambda (SouthernBiotech; cat. No. 2070-05), were diluted 1:3000 in blocking buffer and added to the membranes for one hour at room temperature under gentle shaking. The membranes were washed 3 times with PBS and proteins were visualized using 1 mL of TMB substrate (KPL) for 5 min and stopped with 1 mL of TMB stop solution (KPL) for 5 min. The membranes were then washed with water and stored dried.
Analytical SEC-HPLC was performed using an Agilent 1100 HPLC (Agilent) with a TSK GEL G3000SWXL column (Tosoh Bioscience). One hundred μg of antibodies were used for the analysis. The mobile phase used was 0.1 M sodium sulfate, 0.1 M sodium phosphate pH 6.8, and antibodies were monitored using absorbance at 280 nm. Chemstation software (Agilent) was used for the analysis and the figures were prepared using Prism5 software (GraphPad). rRP-HPLC was performed using an Agilent 1100 HPLC (Agilent) with a PLRP-S, 2.1 × 50 mm, 1000 Å column (Agilent). 10 μg of antibodies in PBS pH 7.2 were reduced with 42 mM dithiothreitol at 37°C for 20 min. rRP-HPLC was performed at 80°C, 1 mL/minute flow rate. The mobile phase A used was 0.1% trifluoroacetic acid in water (J.T. Baker); while the mobile phase B was 0.1% trifluoroacetic acid in acetonitrile (J.T. Baker). The antibodies were monitored using an absorbance of 280 nm. Before analysis, the column was equilibrated for 20 min using a linear gradient of 80% of mobile phase A. The antibodies were analyzed using an isocratic gradient from 20% to 50% mobile phase B for 20 min. Chemstation software (Agilent) was used for the analysis and the figures were prepared using Prism5 software (GraphPad).
SEC-MALS was performed in-line with the SEC-HPLC, as described above, using a Dawn Heleos II fitted with a solid-state laser operating at 658 nm in series with an Optilab rEX refractive index detector (both from Wyatt Technologies). Data were analyzed using Astra software version 5.3.2 (Wyatt Technologies). rLCMS was performed using an Agilent 1290 HPLC system coupled to an Agilent 6230 TOF equipped with an electrospray ionization source. IdeS digestion of the iMab-EI and the 2 parental antibodies was performed using fabRICATOR as recommended by the manufacturer (Genovis). iMab-EI was deglycosylated using Remove-iT endoglycosidase S as recommended by the manufacturer (New England Biolabs). Briefly, iMab-EI was diluted to 1 mg/mL in PBS pH 7.2 and 90 µL of sample were mixed with 10 µl of 10X Glyco Buffer 1 (New England Biolabs), 1 µL Remove-iT, and incubated at 37°C for 2 hours. Five µL of sample was loaded onto a Poroshell 300SB-C3 column (Agilent) and eluted using a flow rate of 0.5 mL/min and a linear gradient of 1% of B for 2 min, followed by elution with a linear gradient of 80% of B for additional 2 min. The mobile phase A was 0.1% formic acid in water and the mobile phase B was 0.1% formic acid in acetonitrile. Agilent MassHunter was used for data acquisition and processing. Peaks were assigned based on their molecular weight determined using the primary amino acid sequence and the figure was prepared using Prism 5 (GraphPad).
Imaged capillary isoelectric focusing was performed using an iCE analyzer (ProteinSimple). Pharmalytes pH 3-10 and 8-10.5 were purchased from Sigma. The FC cartridge and the performance evaluation chemical kit, including anolyte (80 mM phosphoric acid in 0.1% methyl cellulose), catholyte (100 mM sodium hydroxide in 0.1% methyl cellulose), 0.5% methylcellulose, ampholytes and pI markers were purchased from ProteinSimple. Deionized water ≥ 18 MΩcm was generated using a Milli-Q water purification system (Millipore). pI marker 4.22 and 9.77 were purchased from ProteinSimple. Antibodies used for the analysis were formulated at 1 mg/mL in deionized water and were 99% monomer as determined by SEC-HPLC. Sample were prepared before the isoelectric focusing experiments by adding 50 μl of 1 mg/mL of antibody, 1 μl of 4.22 pI marker, 1 μl of 9.77 pI marker, 140 μl of 0.5% methylcellulose, 2 μl of 3-10 and 6 μl of 8-10.5 pharmalytes, respectively. The mixture was vortexed for 45 sec and centrifuged at 10,000 rpm for 3 min. An autosampler (ProteinSimple) was used to load the samples into the capillaries. Sample separation was performed by a pre-focus at 1000 kV for 1 min/sec, followed by 3000 kV for 7 min. Detection was performed using an absorbance of 280 nm. Data were analyzed using the iCE analyzer software.
ELISA transfer assay
iMab-EI and parental antibodies ELISA binding to EGFR and IGF1R was used to determine the lowest antibody concentration to achieve maximum binding signal, which was 0.2 µg/mL. Therefore, 0.2 µg/mL of iMab-EI, anti-EGFR and anti-IGF1R antibodies were used for the ELISA transfer assay. Two µg/mL (30 µL/well) of EGFR (R&D system, cat. No. 236-EG-200) or IGF1R (R&D System, cat. No. 391-GR-050) prepared in PBS, pH 7.2, were coated on ELISA plates overnight at 4°C. The ELISA plates were blocked with 150 µL of 3% non-fat dry milk in PBST (blocking solution) for one hour at room temperature, followed by 5 washes with PBST. Thirty µL of 0.2 µg/mL of the antibodies prepared in blocking buffer were added to the EGFR and IGF1R plates and incubated for one hour at room temperature under shaking. Twenty-five µL of the antibodies pre-incubated with their respective ELISA plates were carefully removed and transferred to newly prepared EGFR- and IGF1R-coated ELISA plates, so that the solution from the EGFR plates was transferred to newly prepared IGF1R plates and vice versa. The ELISA plates were incubated for one hour at room temperature under shaking, followed by 5 washes with PBST. A 1:3000 dilution of goat anti-human-kappa-HRP (Southern Biotech cat. No. 2060-05) for the EGFR plates or 1:3000 dilution of goat anti-human-lambda-HRP (Southern Biotech; cat. No. 2070-05) for IGF1R plates was added (30 µL/well) for one hour at room temperature. The plates were washed 5 times with PBST and developed with 30 µL/well of 3,3′,5,5′-tetramethylbenzidine substrate (KPL). Thirty µL/well of 0.2 N HCl was added to stop the enzymatic reaction. The absorbance at 405 nm was measured using an ELISA plate reader (Perkin Elmer). EC50 were determined using non-linear regression analysis (log dose-response) and binding curves were plotted using Prism5 (GraphPad).
FcRn ELISA
Five µg/mL (30 µL/well) of the antibodies were coated on ELISA plates (Corning) overnight in PBS pH7.2. The ELISA plates were washed 5 times with PBST pH 5.8 and blocked for 1 hour at room temperature with blocking buffer composed of 10% fish gelatin, 50% Superblock (Pierce) in PBST pH 5.8. A 2-fold dilution of biotinylated FcRn (MedImmune) in blocking buffer from 100 µg/mL to 0.19 µg/mL was added to the ELISA plates (30 µL/well) and incubated at room temperature for one hour under shaking. The plates were washed 5 times with PBST pH 5.8 and a 1:12500 dilution of streptavidin-HRP (Pierce) in blocking buffer was added and incubated for one hour at room temperature. The plates were washed 5 times with PBST pH 5.8, and 30 µL/well of 3,3′,5,5′-tetramethylbenzidine substrate (KPL) was added and the plates were incubated for 5 min at room temperature under shaking. The enzymatic reaction was stopped using 30 µL/well of 0.2 N HCl. The absorbance at 450 nm was measured using an ELISA plate reader (Perkin Elmer) and data were plotted using Prism5 (GraphPad).
C1q ELISA
Five µg/mL (30 µL/well) of the antibodies were coated on ELISA plates (Corning) overnight in PBS pH 7.2. The ELISA plates were washed 5 times with PBST and blocked with a blocking buffer composed of 3% BSA, 50% Superblock (Pierce) in PBST for one hour at 37°C. A 2-fold dilution of human C1q (Cell Sciences, cat. No. CRC 162B) from 100 µg/ml to 0.19 µg/mL prepared in blocking buffer was added (30 µL/well) and incubated at room temperature for one hour. The ELISA plates were washed 5 times with PBST, and 1 µg/mL of sheep anti-human C1q (Immunology Consultants Laboratory, cat. No. SC1Q-80A) in blocking buffer was added to the ELISA plates and incubated for one hour at room temperature under shaking. The ELISA plates were washed 5 times with PBST and a 1:2500 dilution of donkey anti-sheep/goat-HRP (Biorad, cat. No. STAR88P) in blocking buffer was added, and incubated for one hour at room temperature under shaking. The ELISA plates were washed 5 times with PBST, and 30 µL/well of 3,3′,5,5′-tetramethylbenzidine substrate (KPL) was added and the plates were incubated for 5 min at room temperature under shaking. The enzymatic reaction was stopped using 30 µL/well of 0.2 N HCl. The absorbance at 450 nm was measured using an ELISA plate reader (Perkin Elmer). EC50 were determined using non-linear regression analysis (log dose-response) and binding curves were plotted using Prism5 (GraphPad).
Frequency of thrombin recognition sequence in human antibodies
The sequences for the V, D and J genes of the human heavy chain antibody and the V and J genes of the kappa and lambda light chains were retrieved form the IMGT database.Citation34 For the abYsis database, the thrombin recognition sequence was searched online using Homo sapiens as organism.Citation35 Sequence repertoires for the CDR-H3 were also obtained from human blood samples. One million B cells from 5 healthy donors were isolated and used to generate VH and VL amplicons that were sequenced using Illumina MiSeq 2×300 bp (SeqMagic). Raw Fastq sequences were qualify-filtered using FastQC (Babraham Bioinformatics), aligned to IMGT V and J genes, and annotated according to Kabat definitions to extract CDR-H3 information. A custom Python script was developed to search for the thrombin recognition sequence.
Thrombin digestion
Thrombin (GE Healthcare) was prepared in PBS pH 7.2. Five thrombin units per mg of iMab-EI in PBS pH 7.2 were used and cleavage was performed for 30 min at room temperature. Following cleavage, the thrombin was inactivated using 1 mM phenylmethylsulfonyl fluoride and thrombin and linkers were removed using ultracentrifugation with 50,000 Da cutoff membrane filters.
Fab preparation
Fab from the anti-EGFR and anti-IFG1R antibodies were prepared using papain (Thermo Fisher Scientific). Antibodies were prepared at 1 mg/mL in papain digestion buffer (0.1 M Tris-HCl, 4 mM EDTA, 1 mM cysteine, pH 7.4). A papain solution of 0.1 mg/mL was prepared prior the digestion in papain digestion buffer. The antibody to papain molar ratio used was 100:1 in a volume of 1.5 mL of papain digestion buffer. The digestion was performed for 2 hours at 37°C and then the reaction mixture was buffer exchanged into 2 ml of PBS pH 7.2 using PD-10 columns (GE Healthcare). The Fabs were purified using gel filtration chromatography with a Superdex 200 column (GE Healthcare). Purification was performed at room temperature in PBS. An absorbance of 280 nm was used to monitor the proteins during the purification. Fabs were dialyzed overnight at 4°C in PBS pH 7.2 and were >95% monomer as determined using analytical SEC-HPLC. Analytical SEC-HPLC was performed using 100 μg of Fabs as described for the SEC-HPLC of antibodies.
Binding to EGFR and IGF1R using BIAcore
Human EGFR and IGF1R were obtained from R&D Systems. BIAcore 3000 (GE Healthcare) was used to carry out binding. The EGFR and IGF1R proteins were diluted to 0.5 µg/mL in 10 mM acetate buffer, pH 5.0 and immobilized on a CM5 chip surface (2000 and 3000 resonance units, respectively) using standard amine coupling protocols. The anti-EGFR Fab, the anti-IGF1R Fab and the iMab-EI were diluted in HEPES-buffered saline (HBS; GE Healthcare) at 100 nM, 33.3 nM, 11.11 nM, 3.7 nM, 1.23 nM, 0.41 nM, 0.137 nM and 0.046 nM and flowed over the EGFR or IGF1R surfaces at 5 µL/min for 200 sec. Dissociation was allowed to occur for 350 sec. Surfaces were regenerated by a double injection of 15 μL of glycine pH 2.0 at 30 µL/min flow rate. Binding Curves were fit to a 1:1 Langmuir binding model using BIAevaluation (GE Healthcare) to obtain binding kinetics and equilibrium KD. All experiments were performed at 25°C using HBS-EP (GE Healthcare) as the running buffer.
Concurrent binding to EGFR and IGF1R using BIAcore
Concurrent binding experiments were performed on a BIAcore 3000 (GE Healthcare) at 25°C. iMab-EI was diluted to 75 µg/mL in 10 mM acetate buffer, pH 5 and immobilized on separate CM5 sensor chip surfaces at 150 resonance units, using standard amine coupling protocols provided by the manufacturer (GE Healthcare). Binding was performed using HBS-EP buffer (GE Healthcare). IGF1R and a mixture of IGFR and EGF1R were prepared in HBS buffer (GE Healthcare) at 330 nM, and 125 nM IGF1R, respectively. IGF1R and the mixture of IGF1R and EGFR were injected at a flow rate of 5 µL/min for 4 min. The mixture of IGF1R and EGFR was injected after IGF1R. The BIAcore chip surfaces were regenerated by a double injection of 15 µL of 0.1 M glycine, pH 2 at a flow rate of 30 µL/min. The data were analyzed using BIAevaluation (GE healthcare) and the figure was prepared using Prism 5 (Graph Pad).
Expression of EGFR and IGF1R on human-derived small cell lung tumors
The human-derived small cell lung tumor was grown in RAG2KO mice. When the tumor reached a volume of 800 mm3, the mouse was sacrificed and tumor removed, washed 3 times with PBS pH 7.4, and minced using razor blades. The tumor was digested using 1.5 µg/mL of Dispase (Stem Cell Technologies) in RPMI (Thermo Fisher Scientific) for 2 hours at 37°C, with gentle agitation every 15 min. Digested tumor was passed over a 70 µm syringe filter and rinsed with RPMI containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific). The cells were then washed with RPMI, 10% FBS and collected by centrifugation at 1000 rpm for 5 min at room temperature. Aliquots of 5 × 106 cells/mL were prepared in cell freezing medium (Thermo Fisher Scientific) and stored at −80°C. Cells were thawed and transferred to a 15 ml conical tube containing 5 mL of FACS Staining Buffer (FSB; PBS + 3% FBS). The cells were washed twice by centrifugation at 1500 rpm for, 5 min at room temperature. Cells were resuspended in FSB containing Fc block (R&D System) and incubated for 15 min at 4°C. Cells were added at 1 × 105 cells per well into a 96-well plate (Corning). Cells were incubated with the anti-IGF1R or anti-EGFR antibodies (R&D Systems) for 30 min at 4°C. Cells were washed twice with FSB and collected by centrifugation at 1500 rpm for 3 min after each wash. Cells were incubated with a goat anti-human Alexa Fluor 647 conjugated antibody (Thermo Fisher Scientific) for 30 min at 4°C. Cells were washed 3 times as described above and then resuspended in FSB containing DAPI (KBL). IGFIR and EGFR expression was analyzed using LSR II flow cytometer (BD Biosciences). Data were collected and analyzed using FlowJo software (FlowJo).
In vivo tumor growth inhibition
Animal experiments were performed under MedImmune Institutional Animal Care and Use Committee according to established guidelines. RAG2KO female mice (Taconic Biosciences Inc.) at 5 to 7 weeks of age were implanted subcutaneously, using a trocar, with 30 mm3 tumor fragments of patient-derived xenograft of a stage IV recurrent metastatic non-small cell lung tumor cancer, which had been previously in vivo passaged in RAG2 KO mice. When mean tumor volume reached 90 mm3 as measured by calipers, tumor-bearing mice were randomized into groups (n = 8). Antibodies were prepared in PBS, pH 7.2 and were endotoxin free as determined using the Endosafe PTS testing system (Charles River Laboratories). The mice were then injected intravenously with iMab-EI, a combination of the anti-EGFR and anti-IGF1R antibodies, or a negative isotype-matched control antibody at 10 mg/kg, 2 times weekly, for 2 weeks. Vehicle-treated mice were included as a control. Tumors were measured twice weekly using caliper and data were analyzed using Prism5 (GraphPad). Animals were sacrificed when tumors reached 2000 mm3. Tumor volume (TV) was calculated using the following formula: (tumor length × (tumor width)Citation2) / 2. Tumor growth inhibition was calculated as follows: 1 - ((iMab-EI mean TV) / (control mean TV)) × 100.
Disclosure of potential conflicts of interest
Authors are shareholders of AstraZeneca and are used by MedImmune.
Author contributions
ND designed the iMab, coordinated the studies, and wrote the manuscript. RF, BB performed protein expression and analytical characterization. ES, CH and KS performed FACS binding and tumor growth inhibition experiments. LZ and MK performed protein expression using UCOE technology. PC and AB identified the anti-IGF1R antibody. SR and JW contributed the human antibody repertoire sequencing and analysis, respectively. CG and HW provided scientific support.
Supplemental_Figures_1-6.zip
Download Zip (1.6 MB)Acknowledgments
We thank Jie Zhu for help with preparation of the transient expression vector containing the UCOE sequence.
References
- Grandjenette C, Dicato M, Diederich M. Bispecific antibodies: an innovative arsenal to hunt, grab and destroy cancer cells. Curr Pharm Biotechnol 2015; 16:670-683; PMID:25941884; http://dx.doi.org/10.2174/1389201016666150505124037
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 2009; 69:4941-4944; PMID:19509221; http://dx.doi.org/10.1158/0008-5472.CAN-09-0547
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67:95-106; PMID:25637431; http://dx.doi.org/10.1016/j.molimm.2015.01.003
- Dimasi N, Gao C, Fleming R, Woods RM, Yao XT, Shirinian L, Kiener PA, Wu H. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J Mol Biol 2009; 393:672-692; PMID:19699208; http://dx.doi.org/10.1016/j.jmb.2009.08.032
- Bezabeh B, Fleming R, Fazenbaker C, Zhong H, Coffman K, Yu XQ, Leow CC, Gibson N, Wilson S, Stover CK, Wu H, Gao C, Dimasi N. Insertion of scFv into the hinge domain of full-length IgG1 monoclonal antibody results in tetravalent bispecific molecule with robust properties. MAbs. 2016; PMID:27981887
- Dela Cruz JS, Trinh KR, Morrison SL, Penichet ML. Recombinant anti-human HER2/neu IgG3-(GM-CSF) fusion protein retains antigen specificity and cytokine function and demonstrates antitumor activity. J Immunol 2000; 165:5112-5121; PMID:11046042; http://dx.doi.org/10.4049/jimmunol.165.9.5112
- Dong J, Sereno A, Aivazian D, Langley E, Miller BR, Snyder WB, Chan E, Cantele M, Morena R, Joseph IB, et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. MAbs 2011; 3:273-88; PMID:21393993; http://dx.doi.org/10.4161/mabs.3.3.15188
- Rossi EA, Chang CH, Cardillo TM, Goldenberg DM. Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties. Bioconjugate Chem 2013; 24:63-71; PMID:23116517; http://dx.doi.org/10.1021/bc300488f
- Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 2009; 113:6161-6171; PMID:19372261; http://dx.doi.org/10.1182/blood-2008-10-187138
- Croasdale R, Wartha K, Schanzer JM, Kuenkele KP, Ries C, Mayer K, Gassner C, Wagner M, Dimoudis N, Herter S, et al. Development of tetravalent IgG1 dual targeting IGF-1R-EGFR antibodies with potenttumor inhibition. Arch Biochem Biophys 2012; 526:206-218; PMID:22464987; http://dx.doi.org/10.1016/j.abb.2012.03.016
- Yazaki PJ, Lee B, Channappa D, Cheung CW, Crow D, Chea J, Poku E, Li L, Andersen JT, Sandlie I, et al. A series of anti-CEA/anti-DOTA bispecific antibody formats evaluated for pre-targeting: comparison of tumor uptake and blood clearance. Protein Eng Des Sel 2013; 26:187-193; PMID:23175797; http://dx.doi.org/10.1093/protein/gzs096
- Datta-Mannan A, Croy JE, Schirtzinger L, Torgerson S, Breyer M, Wroblewski VJ. Aberrant bispecific antibody pharmacokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. MAbs 2016; 8:969-982; PMID:27111637; http://dx.doi.org/10.1080/19420862.2016.1178435
- Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, Schaefer W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. MAbs 2012; 6:653-63; PMID:22925968;http://dx.doi.org/10.4161/mabs.21379
- Strop P, F Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol 2012; 420:204-219; PMID:22543237; http://dx.doi.org/10.1016/j.jmb.2012.04.020
- Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A 2013; 110:5145-5150; PMID:23479652; http://dx.doi.org/10.1073/pnas.1220145110
- Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol 2013; 8:753-758; PMID:23831709;http://dx.doi.org/10.1038/nbt.2621
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21:11-16; PMID:15903235; http://dx.doi.org/10.1021/bp040016j
- Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7:1401-1413; PMID:17727329; http://dx.doi.org/10.1517/14712598.7.9.1401
- Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, Tsumoto K, Kumagai I, Shitara K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J Mol Biol 2004; 336:1239-1249; PMID:15037082; http://dx.doi.org/10.1016/j.jmb.2004.01.007
- Mimura Y, Church S, Ghirlando R, Ashton PR, Dong S, Goodall M, Lund J, Jefferis R. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol Immunol 2000; 37:697-706; PMID:11275255; http://dx.doi.org/10.1016/S0161-5890(00)00105-X
- Subedi GP, Barb AW. The structural role of antibody N-Glycosylation in receptor interactions. Structure 2015; 23:1573-1583; PMID:26211613; http://dx.doi.org/10.1016/j.str.2015.06.015
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol 2014; 32:191-198; PMID:24463572; http://dx.doi.org/10.1038/nbt.2797
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci USA 2011; 108:11187-11192; PMID:21690412; http://dx.doi.org/10.1073/pnas.1019002108
- Fischer N, Elson G, Magistrelli G, Dheilly E, Fouque N, Laurendon A, Gueneau F, Ravn U, Depoisier JF, Moine V, et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat Commun 2015; 6:6113; PMID:25672245; http://dx.doi.org/10.1038/ncomms7113
- Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 2009; 323:1610-1614; PMID:19299620; http://dx.doi.org/10.1126/science.1165480
- Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs 2015; 7:377-389; PMID:25621507; http://dx.doi.org/10.1080/19420862.2015.1007816
- Liu Z, Leng EC, Gunasekaran K, Pentony M, Shen M, Howard M, Stoops J, Manchulenko K, Razinkov V, Liu H, et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. J Biol Chem 2015; 290:7535-7562; PMID:25583986; http://dx.doi.org/10.1074/jbc.M114.620260
- Schanzer JM, Wartha K, Moessner E, Hosse RJ, Moser S, Croasdale R, Trochanowska H, Shao C, Wang P, Shi L, et al. XGFR, a novel affinity-matured bispecific antibody targeting IGF-1R and EGFR with combined signaling inhibition and enhanced immune activation for the treatment of pancreatic cancer. MAbs 2016; 4:811-827; PMID:26984378; http://dx.doi.org/10.1080/19420862.2016.1160989
- Lee HS, Shu L, De Pascalis R, Giuliano M, Zhu M, Padlan EA, Hand PH, Schlom J, Hong HJ, Kashmiri SV. Generation and characterization of a novel single-gene-encoded single-chain immunoglobulin molecule with antigen binding activity and effector functions. Mol Immunol 1999; 36:61-71; PMID:10369421; http://dx.doi.org/10.1016/S0161-5890(98)00109-6
- Schirrmann T, Menzel C, Hust M, Prilop J, Jostock T, Dübel S. Oligomeric forms of single chain immunoglobulin (scIgG). MAbs 2010; 1:73-76; PMID:20081378; http://dx.doi.org/10.4161/mabs.2.1.10784
- Hust M, Jostock T, Menzel C, Voedisch B, Mohr A, Brenneis M, Kirsch MI, Meier D, Dübel S. Single chain Fab (scFab) fragment. BMC Biotechnol 2007; 7:14; PMID:17346344; http://dx.doi.org/10.1186/1472-6750-7-14
- Koerber JT, Hornsby MJ, Wells JA. An improved single-chain Fab platform for efficient display and recombinant expression. J Mol Biol 2015; 427:576-586; PMID:25481745; http://dx.doi.org/10.1016/j.jmb.2014.11.017
- Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res 2012; 40:W288-93; PMID:22581768; http://dx.doi.org/10.1093/nar/gks419
- Lefranc MP, Giudicelli V, Ginestoux C, Bodmer J, Müller W, Bontrop R, Lemaitre M, Malik A, Barbié V, Chaume D. IMGT, the international ImMunoGeneTics database. Nucl Acids Res 1999; 1:209-212; PMID:9847182; http://dx.doi.org/10.1093/nar/27.1.209
- Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin AC. abYsis: integrated antibody sequence and structure-management, analysis, and prediction. J Mol Biol 2016; in press; PMID:27561707
- Dong J, Sereno A, Aivazian D, Langley E, Miller BR, Snyder WB, Chan E, Cantele M, Morena R, Joseph IB, et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. MAbs 2011; 3:273-288; PMID:21393993; http://dx.doi.org/10.4161/mabs.3.3.15188
- Hou JJC, Hughes BS, Smede M, Leung KM, Levine K, Rigby S, Gray PP, Munro TP. High-throughput ClonePix FL analysis of mAb-expressing clones using the UCOE expression system. New Biotechnol 2014; 31:214-220; PMID:24518824; http://dx.doi.org/10.1016/j.nbt.2014.02.002
- Sandercock AM, Rust S, Guillard S, Sachsenmeier KF, Holoweckyj N, Hay C, Flynn M, Huang Q, Yan K, Herpers B, et al. Identification of anti-tumour biologics using primary tumour models, 3-D phenotypic screening and image-based multi-parametric profiling. Mol Cancer 2014; 14:147; PMID:26227951; http://dx.doi.org/10.1186/s12943-015-0415-0
- Argos P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J Mol Biol 1990; 211:943-958; PMID:2313701; http://dx.doi.org/10.1016/0022-2836(90)90085-Z
- George R, Heringa J. An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng 2002; 15:871-879; PMID:12538906; http://dx.doi.org/10.1093/protein/15.11.871
- Chen X, Zaro J, Shen WC. Fusion protein linkers: Property, design and functionality. Adv Drug Deliv Rev 2013; 65:1357-1369; PMID:23026637; http://dx.doi.org/10.1016/j.addr.2012.09.039
- Shen Y, Zeng L, Zhu A, Blanc T, Patel D, Pennello A, Bari A, Ng S, Persaud K, Kang YK, et al. Removal of a C-terminal serine residue proximal to the inter-chain disulfide bond of a human IgG1 lambda light chain mediates enhanced antibody stability and antibody dependent cell-mediated cytotoxicity. MAbs 2013; 5:418-431; PMID:23567210; http://dx.doi.org/10.4161/mabs.24291
- Liu H, Zhong S, Chumsae C, Radziejewski C, Hsieh CM. Effect of the light chain C-terminal serine residue on disulfide bond susceptibility of human immunoglobulin G1λ. Anal Biochem 2011; 408:277-283; PMID:20869344; http://dx.doi.org/10.1016/j.ab.2010.09.025
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-2324; PMID:16793771; http://dx.doi.org/10.1074/jbc.M604292200
- Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall'acqua WF. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol 2009; 46:1750-1755; PMID:19250681; http://dx.doi.org/10.1016/j.molimm.2009.01.026
- Thompson P, Fleming R, Bezabeh B, Huang F, Mao S, Chen C, Harper J, Zhong H, Gao X, Yu XQ, et al. Rational design, biophysical and biological characterization of site-specific antibody-tubulysin conjugates with improved stability, efficacy and pharmacokinetics. J Control Release 2016; 236:100-116; PMID:27327768; http://dx.doi.org/10.1016/j.jconrel.2016.06.025
- Prasad GL, Lee HS, Iwahashi M, Milenic DE, Abrams S, Schlom J, Kashmiri SV. In vivo gene inoculation of a recombinant single-chain antitumor antibody induces anti-immunoglobulin response. Cancer Gene Ther 1997; 4:253-259; PMID:9253511