
ABSTRACT
Junctional adhesion molecule-A (JAM-A) is an adherens and tight junction protein expressed by endothelial and epithelial cells and associated with cancer progression. We present here the extensive characterization of immune complexes involving JAM-A antigen and three monoclonal antibodies (mAbs), including hz6F4-2, a humanized version of anti-tumoral 6F4 mAb identified by a functional and proteomic approach in our laboratory. A specific workflow that combines orthogonal approaches has been designed to determine binding stoichiometries along with JAM-A epitope mapping determination at high resolution for these three mAbs. Native mass spectrometry experiments revealed different binding stoichiometries and affinities, with two molecules of JAM-A being able to bind to hz6F4-2 and F11 Fab, while only one JAM-A was bound to J10.4. Surface plasmon resonance indirect competitive binding assays suggested epitopes located in close proximity for hz6F4-2 and F11. Finally, hydrogen-deuterium exchange mass spectrometry was used to precisely identify epitopes for all mAbs. The results obtained by orthogonal biophysical approaches showed a clear correlation between the determined epitopes and JAM-A binding characteristics, allowing the basis for molecular recognition of JAM-A by hz6F4-2 to be definitively established for the first time. Taken together, our results highlight the power of MS-based structural approaches for epitope mapping and mAb conformational characterization.
Abbreviations
| Ag | = | antigen |
| ADC | = | antibody-drug conjugate |
| D2O | = | deuterium oxide |
| Fab | = | Fragment antigen-binding |
| GuHCl | = | guanidine hydrochloride |
| HCl | = | hydrochloric acid |
| HDX-MS | = | H/D exchange followed by MS |
| IgG | = | immunoglobulin G |
| JAM-A | = | Junctional adhesion molecule-A |
| K2HPO4 | = | dibasic potassium phosphate |
| KH2PO4 | = | monobasic potassium phosphate |
| LC-MS(/MS) | = | liquid chromatography coupled to MS or tandem mass spectrometry |
| mAb | = | monoclonal antibody |
| MS | = | mass spectrometry |
| MS/MS | = | tandem mass spectrometry |
| NH4Ac | = | ammonium acetate |
| SPR | = | Surface plasmon resonance |
| TCEP | = | tris(2-carboxyethyl)phosphine |
Introduction
As class of therapeutic agents, monoclonal antibodies (mAbs) and related products have shown increasing growth for over a decade.Citation1,2 More than 70 mAbs and derivatives are currently approved by the US Food and Drug Administration and the European Medicines Agency, and indicated for the treatment of multiple pathologies such as cancer, inflammation, transplantation or infectious diseases.Citation3 Unfavorable pharmaceutic or physicochemical properties can severely limit their potency and delay their clinical development. In this context, it is of paramount importance to precisely determine the sequence, post-translational modifications or glycoform profile to name a few critical quality attributes.Citation4,5 Another key issue for mAb design and characterization is the determination of binding regions between mAbs and their target antigens (Ag), which drives their function and efficacy. The determination of the interacting areas on the antigen side (epitope) is thus essential to understand associated biologic mechanisms,Citation6-9 but also for antibody design and intellectual property.
The gold standard for protein/protein interacting region mapping remains X-ray crystallography, which provides atomic resolution.Citation10 However, mAbs and mAb:Ag complexes are difficult to crystallize due to the flexibility of given regions. X-ray crystallography also has drawbacks because it provides only a static picture of the protein in its thermodynamic stable state. Therefore, it is thus of utmost interest to use and develop new structural and analytical tools to gain insight into structure-function relationships of mAb:Ag complexes.Citation11-13 Mass spectrometry (MS) techniques already play a dominant role in mAb characterization.Citation14,15 In the past decade, several MS-based approaches have been reported for the characterization of mAb:Ag complexes. Among those, native MS, which aims at analyzing intact noncovalent complexes in the gas phase of the mass spectrometer, allows direct determination of mAbs:antigen binding stoichiometry.Citation16-19 Several MS footprinting strategies comprising chemical crosslinking,Citation20-22 hydroxyradicalCitation23,24 or carboxyradical footprinting,Citation25 chemical painting/footprintingCitation26,27 or hydrogen/deuterium exchange (HDX-MS)Citation28-30 have been developed for protein/protein interface determination. All those approaches are based on protein labeling before enzymatic digestion into peptides and further LC-MS(/MS) analysis. The structure of the protein is then directly encoded on the amino acid sequence, the structure being further inferred through the MS identification of solvent accessible versus non-accessible peptides.
Among these, HDX-MS analysis has moved to the forefront, especially for epitope mapping applicationsCitation27,31-35 and mAb comparability studies.Citation36,37-40 It consists of the reversible exchange of amidic hydrogens of the backbone upon deuterated solvent exposure, revealing less exposed peptides when interacting region are concerned. For epitope mapping purposes, the HDX-MS approach is a differential approach that compares deuterium uptake kinetics of the antigen in the presence or absence of the mAb. Of note, HDX-MS has been shown to be particularly valuable for conformational (non-linear) epitope mapping.Citation9,41,42 Due to extensive automation and progress in bioinformatics, HDX-MS is now amenable to routine high throughput analysis for mAb characterization.Citation43-46 Even if HDX-MS cannot claim to reach an atomic spatial resolutions comparable to the one obtained by crystallography,Citation47 it is less hindered by experimental limitations.Citation48 Also, several studies have shown its potential for the mapping of mAbs binding areas compared to classical methodologies.Citation49-51
In a previous work, we selected murine mAb 6F4 as a mAb of interest based on MCF-7 tumor cells growth inhibition screen.Citation52 To identify the antigen recognized by 6F4, immunopurification was performed using HT-29 cells expressing high amount of JAM-A. After extraction and solubilization with detergents, membrane proteins were incubated in the presence of the 6F4 mAb immobilized on Sepharose beads. The 6F4 antigen target was identified by proteomics approach as JAM-A, and hz6F4-2, a humanized version, was produced.Citation16 In addition, a thorough characterization of hz6F4-2:JAM-A immune complexes using native MS and ion mobility-MS revealed for the first time an unexpected binding stoichiometry of the hz6F4-2 mAb (hinge-stabilized IgG4) with recombinant JAM-A through the formation of specific 1:4 mAb:Ag complexes. Moreover, a bispecific antibody formed by one heavy chain and one light chain of the wild type IgG4 variant of hz6F4-2 and by one heavy chain and one light chain of natalizumab still binds two JAM-A moleculesCitation53 and confirms the stoichiometry of the binding of 2 JAM-A molecule per antigen-binding fragment (Fab). Altogether, our previous native MS results prompted us to propose a selective binding of JAM-A dimers to hz6F4-2. To provide additional insight into the hz6F4-2:JAM-A complex, we present here a differential analysis of JAM-A binding to either hz6F4-2 or two commercially available mAbs, F11 and J10.4, which are also described in the literature to target JAM-A,Citation54,55 using orthogonal biophysical approaches. For this, we combine three methods: 1) native MS to determine binding characteristics of JAM-A with the three different antibodies, 2) surface plasmon resonance (SPR) to evaluate similarity between epitopes and 3) HDX-MS for precise epitope mapping. Altogether, our results give a definite explanation for the selective interaction between JAM-A dimers and hz6F4-2, and highlight the benefits of combining structural MS approaches to more classical biophysical techniques for mAb:Ag immune complex characterization.
Results
Native MS reveals different stoichiometries for mAb:JAM-A complexes
Native MS was used to rapidly and precisely determine the binding stoichiometries of each mAb:JAM-A complex. For this, the Fabs of each mAb were produced (see Materials and Methods section) and incubated with increasing concentrations of JAM-A. Prior to native MS analysis of Fab:Ag complexes, JAM-A and Fabs were first analyzed alone in native conditions (See Supplementary Information S1 and ). As already reported, 16 JAM-A is detected as both monomers and dimers, in a concentration-dependent manner, the dimers being favored at higher concentrations (Supplementary Information S2, ). The presence of dimers is in agreement with crystallographic data of JAM-A, which describe two JAM-A molecules interacting through expanding ionic and hydrophobic areas.Citation56 Regarding Fabs, most of them revealed a significant heterogeneity coming from the papain cleavage ().
Table 1. Masses measured by native mass spectrometry for JAM-A antigen, the three monoclonal antibodies (hz6F4-2, F11 and J10.4) and related mAb:JAM-A complexes.
Native MS titration experiments were next performed in order to determine Fab:Ag binding stoichiometries. For these, a fixed concentration of Fab (5 µM) was incubated with increasing concentrations of JAM-A. presents the relative proportions of Fab:JAM-A complexes as deduced from native mass spectra peak intensities (see Materials and Methods section). With a four-fold excess of JAM-A, both hz6F4-2 and F11 Fabs were able to bind up to two JAM-A molecules, while J10.4 only binds one (, Supplementary Information S3). The 1:1 and 1:2 hz6F4-2:JAM-A complexes were detected with masses of 73033 ± 1 Da and 97587 ± 4 Da, respectively (), in agreement with previously published data,Citation16 and providing a first strong hint for JAM-A binding as a dimer to hz6F4-2. From native MS experiments, it seems that F11 has a similar binding behavior as hz6F4-2, up to two JAM-A molecules being detected on the F11 Fab with measured masses of 71778 ± 5 Da and 96358 ± 10 Da for 1:1 and 1:2 F11:JAM-A complexes, respectively (). No higher-order binding stoichiometries were detected for JAM-A binding to either hz6F4-2 or F11, which confirms the specificity of the detected interaction and the probable binding of one dimer of JAM-A per Fab for these two mAbs. In addition, 70% of 1:2 hz6F4-2:JAM-A complex is detected compared to 52% for 1:2 F11:JAM-A species, which suggests that the binding of hz6F4-2 is more effective in stabilizing the JAM-A dimer than F11. Conversely, even in the presence of a large excess of JAM-A, only 1:1 J10.4:JAM-A complexes (73311 ± 1 Da) were detected, suggesting that only monomeric JAM-A can bind to J10.4 Fab.
Figure 1. Fab:JAM-A binding stoichiometries as deduced from native mass spectrometry. Relative proportions of each species were deduced from native MS experiments in large excess of JAM-A (≥ 4 molar equivalents).
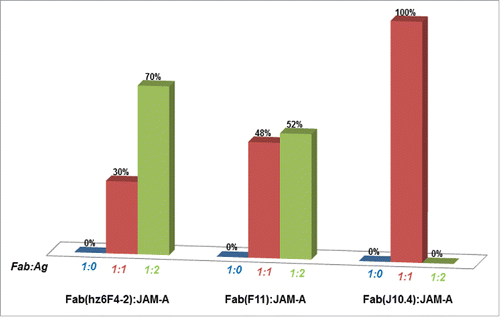
These different binding stoichiometries reflect the specificity of each mAb to bind a given target with a specific recognition mode. We thus hypothesized that these different stoichiometries might be related to the different epitopes.
Label-free interaction analysis using SPR defines overlapping epitopes for hz6F4-2 and F11
Label-free interaction analysis using SPR was next used to eva-luate overlapping epitopes between the three different mAbs. For this, mAb indirect and sequential competition experiments were designed (see ). JAM-A was first immobilized on the SPR sensor chip using an anti-6His mouse mAb and the binding of a first full-length mAb (either hz6F4-2, F11 or J10.4) was monitored. This step leads to almost quantitative saturation of the JAM-A binding site by any mAb, leading to stable mAb:JAM-A complexes. An injection of a second mAb (either hz6F4-2, F11 or J10.4) was next performed in order to distinguish overlapping epitopes (no or at least a weak binding level of the second antibody) from distant epitopes (high binding level of the second antibody). Three experiments (one for each of the three antibodies used in the second injection) were run for each antibody injected first. The three sensorgrams are superposed in each figure. In , hz6F4-2 was first bound to the immobilized JAM-A antigen (the capture level of the antigen and the binding level of the antibody are very reproducible). When hz6F4-2 is re-injected as the second mAb, the SPR signal still increases, suggesting incomplete saturation of JAM-A by hz6F4-2 (red line) after the first injection. When F11 (green line) is used in the second step, the SPR signals only slightly increase, suggesting that F11 binding is hindered by the binding of hz6F4-2. Conversely when J10.4 is injected as the second mAb, the SPR signal significantly increases, which demonstrates that J10.4 binds to JAM-A even when hz6F4-2 is already bound. Control experiments in which F11 is injected in the first step (), followed by either F11 (green line), hz6F4-2 (red line) or J10.4 (blue line) showed that only J10.4 can still bind the preformed F11/JAM-A complexes, while hz6F4-2 and F11 only induce a slight SPR signal increase, suggesting again that F11 binding hinders hz6F4-2 simultaneous binding. Finally, the complexes formed between JAM-A and J10.4 () do not prevent the binding of hz6F2 or F11.
Figure 2. Label-free interaction analysis monitored by surface plasmon resonance. Workflow of indirect and sequential competition experiments designed for the SPR study (a). Parts 2b-c shows the superposition of three sensorgrams starting with the capture of JAM-A by an anti-6His mouse monoclonal antibody chemically grafted on a CM5 sensor chip followed by the injection of a 50 µg/ml solution of antibody either hz6F4-2 (b), F11 (c) and J10.4 (d). A second solution of antibody (50 µg/ml) is then injected at each cycle either hz6F4-2 (red), F11 (green) or J10.4 (blue). The regeneration step at the end of each cycle is not shown.
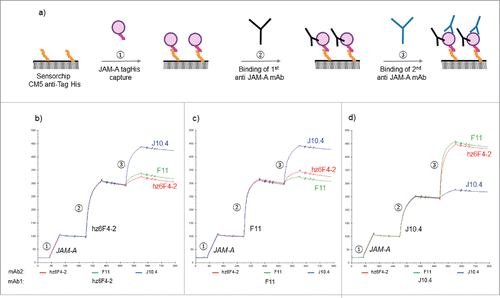
Taken together, the SPR results suggest that hz6F4-2 and F11 might share closely located epitope areas while J10.4 clearly binds to a distant region on JAM-A.
HDX-MS for epitope mapping
In order to confirm the results of the SPR experiments and precisely locate JAM-A epitopes, we next performed HDX-MS experiments. This method allows the identification of regions that either are protected from the solvent upon complex formation or undergo conformational changes resulting in differences in solvent accessibility. We thus compared the deuterium incorporation rates of free JAM-A versus mAb:JAM-A complexes for all three full-length mAbs. HDX-MS experiments were performed with the mAb:JAM-A ratio that ensures the antigen is saturated with mAb according to previous native MS analysis (see Material and Methods).
Pepsin digestion generated numerous peptides for JAM-A, of which 40 could be monitored upon deuteration, resulting in >95% sequence coverages and redundancy index >2.0 in presence of the mAbs (Supplementary Information S4). Almost total linear sequence coverage was still obtained for JAM-A after mAb binding, except for F11 for which the region 40–59 could not be recovered (Supplementary Information S4).
presents the differential HDX heat map of JAM-A in the presence of either hz6F4-2 (), F11 () or J10.4 (). Deuterium incorporation plotted as a function of time revealed that most peptides showed no difference in exchange rate between JAM-A alone or in complex with any of the mAbs ().
Figure 3. HDX difference plots and heat maps for free JAM-A compared to hz6F4-2:JAM-A (a), F11:JAM-A (b) and J10.4:JAM-A (c) complexes. The difference plots illustrate differential deuterium uptake for each identified peptide at different exposure times 0.5 (orange), 2 (grey), 10 (yellow), 30 (blue) and 60 min (green). Differences are summed into striped histograms which highlight localized variations. Significant differences in deuterium uptake are highlighted by red horizontal dotted line. Heat maps are colored from −3% (blue) to 9% (red) difference of deuterium uptake after 60 min. Color coding for relative deuterium uptake difference is shown at the bottom right (cold colors correspond to areas that incorporate more deuterium in the complexed state, and so become more exposed to deuterated buffer; while hot colors are associated with areas that incorporate less deuterium in the complexed state, and so are protected from labelling buffer).
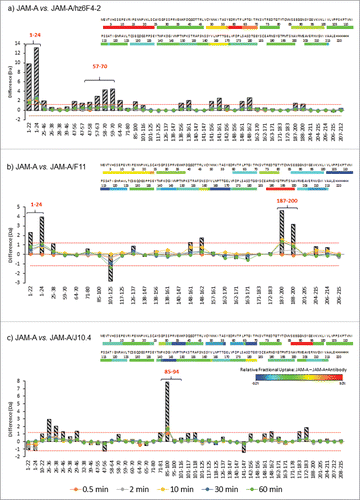
For the binding of hz6F4-2, the deuterium uptake is notably reduced for regions 1–24 and 59–70 (), corresponding to areas protected from deuterium incorporation in the presence of hz6F4-2. Residues 1–24 displayed strong protection in the presence of hz6F4-2 (>2 Da mass shift), while residues 59–70 displayed modest protection (>0.5-Da mass shift) (, ). Changes in these regions were reproducible in replicate experiments, and overlapping peptides displayed similar behavior, giving us high confidence that the changes observed were real and significant. No other part of the antigen seems to be strongly and significantly affected upon hz6F4-2 binding. JAM-A peptides affected upon hz6F4-2 binding after 60 min of labelling were next highlighted on JAM-A crystal structure (PDB: 1NBQ, ). Even if the regions revealed by HDX-MS are very close on the amino acid sequence, they are discontinuous polypeptide segments that seem to be brought into spatial proximity by the folding of the protein in its native structure, which is in favor of a conformational epitope.
Figure 4. HDX-MS epitope mapping results plotted on JAM-A crystal structure (PDB: 1NBQ). Relative deuterium uptake differences were plotted on JAM-A crystal structure (PDB: 1NBQ) for hz6F4-2:JAM-A (a), F11:JAM-A (b) and J10.4:JAM-A (c). Deuterium uptake graphs of peptides showing significant differences in deuterium uptake are provided in panels (b), (d) and (f). Red lines stand for JAM-A alone while mAb:JAM-A complex are represented in blue curves.
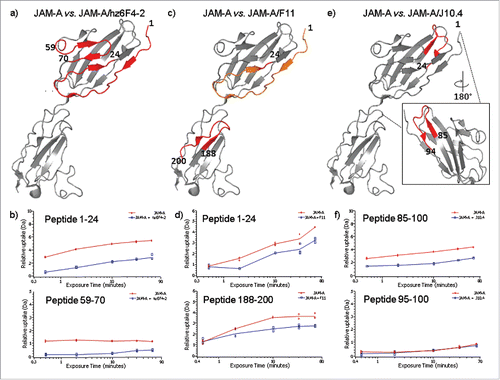
For F11 binding to JAM-A, the most significant differential reduction of deuterium uptake happens in the C-terminal domain, more precisely for the peptide 188–200 (), which displayed a strong protection in the presence of F11 (>1 Da mass shift, ). However, the N-terminus peptide 1–24 also presents a decrease in deuterium incorporation, even if the difference is less marked than for hz6F4-2 (>0.5 Da mass shift, ). Of note, no conclusion could be drawn on the role of the region 40–59 of F11 because no peptide could be generated upon biological duplicates of HDX-MS experiments.
For J10.4 binding to JAM-A, the peptide redundancy allows to show a significant reduction in deuterium uptake only for residues 85–94 (), with a strong decrease (>2 Da mass shift, ). This region is located on the opposite side of the segment 1–24 previously involved in the interaction with hz6F4-2 and F11 (). The binding of J10.4 mAb induces other slight conformational changes, including the regions of residues 34–38.
Altogether, HDX-MS results allowed us to unambiguously assess different epitopes on JAM-A for either hz6F4-2, F11 or J10.4 binding. Two discontinuous regions (residues 1–24 and 59–70) were identified on JAM-A upon hz6F4-2 binding, suggesting a conformational epitope. The binding of hz6F4-2 also mainly affects two areas: one located at the N-terminus part of JAM-A and the other one more central. HDX-MS experiments also revealed that F11 and hz6F4-2 share a common binding region on JAM-A (residues 1–24), which confirms the hypothesis of overlapping epitopes suggested by the SPR data. Conversely, HDX-MS results obtained for J10.4 binding to JAM-A unambiguously confirmed the existence of a different and distant epitope located around residues 85–94.
Rationale for different binding modes for anti-JAM-A antibodies binding to JAM-A
summarizes the results obtained by HDX-MS, by mapping the epitope residues that are protected from H/D exchange for each mAb binding on the crystal structure of JAM-A. For hz6F4-2 and F11 binding, two discontinuous regions were affected upon binding to JAM-A: residues 1–24 and 59–70 for hz6F4-2; and residues 1–24 and 188–200 for F11, allowing conclusions to be made regarding conformational epitopes for both mAb binding. Our work unambiguously highlights for the first time that hz6F4-2 and F11 share a common epitope region consisting of the N-terminal region of JAM-A. From a dynamic point of view, the most significant reduction in deuterium uptake is attributable to hz6F4-2, for which the difference between the free and complexed state is marked at short time points (), while this trend is observed only after longer time points for F11 (). In addition, a second nonconse-cutive peptide is specific to each mAb: for hz6F4-2, the specific area consists of residues 59–70 located in the N-terminal domain of JAM-A while for F11, the specific region of the conformational epitope is located in the C-terminal domain (residues 188–200).
Figure 5. JAM-A epitopes superimposition for hz6F4-2, F11 and J10.4 binding. JAM-A epitopes were superimposed on monomeric (a) and dimeric (b) structures of JAM-A (PDB: 1NBQ). Blue: common area to hz6F4-2 and F11 epitope; Cyan: specific hz6F4-2 epitope; Magenta: specific F11 epitope; Red: J10.4 epitope.
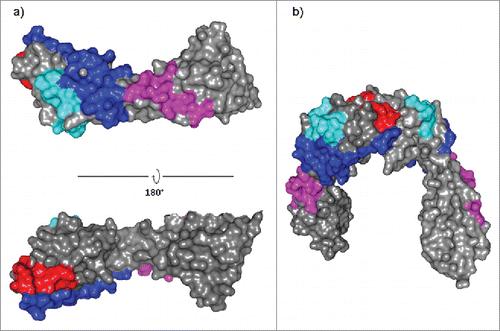
Of note, even if both epitopes for F11 binding (1-24 and 188–200) are located in two distinct domains, these are positioned on the same antigen face (), which can explain similar binding behavior for hz6F4-2 and F11. Furthermore, the epitopes for hz6F4-2 or F11 binding do not overlap with the JAM-A dimerization interface (), which rationalizes the binding of up to 4 JAM-A molecules per mAb.
Conversely, HDX-MS demonstrated that the J10.4 epitope consists of one linear epitope (residues 84–95) and is located at the dimerization interface of JAM-A. These results are in good correlation with phage display epitope determination already reported in literature (residues 84–96).Citation55 The N-terminal region of JAM-A is thus not affected upon J10.4 binding. However, this region overlaps with the JAM-A dimerization interface. Thus, J10.4 binding to JAM-A disrupts or prevents antigen dimerization, which results in the formation of 1:2 J10.4:JAM-A complexes.
By comparing HDX-MS data obtained for hz6F4-2/F11 and J10.4, definitive reasons for specific molecular recognition between JAM-A and the three different mAbs could be deduced. A coherent explanation for the unexpected binding stoichiometry of JAM-A and hz6F4-2 already reported can be provided: the J10.4 epitope overlaps with the dimerization interface of JAM-A, which prevents JAM-A dimerization and its further binding to J10.4 as a dimer. Subsequently, only JAM-A monomers can bind to J10.4, leading to the detection of 1:1 complexes between the Fab of J10.4 and JAM-A. In contrast, hz6F4-2 or F11 binding do not occur at JAM-A dimerization interface. As a consequence, JAM-A, mostly dimeric at high concentrations, is able to bind as a dimer to hz6F4-2 or F11 Fabs.
Discussion
The determination of binding stoichiometries and epitope mapping is particularly important for in-depth understanding of molecular recognition mechanisms associated to immunological responses, and thus can guide specific design of bio-therapeutic agents. Different binding stoichiometry and sizes of soluble tumor necrosis factor-antagonist complexes for adalimumab, infliximab, and etanercept in human serum and in phosphate-buffered saline, for example, have been recently published based on analytical ultracentrifugation with fluorescence detection system.Citation57
Here, we presented the combination of orthogonal biophysical techniques, namely MS-based approaches and SPR, to perform structural characterization of the antigenic JAM-A target involved in immune complexes. Because co-crystallization attempts for JAM-A extra-cellular-domain with hz6F4 Fab failed, we sought to determine the epitope of anti-JAM-A antibodies to gain insight into JAM-A mechanism of binding using three complementary approaches: native MS, HDX-MS and SPR.
An extended MS-based structural study of immune complexes involving JAM-A antigen and three different mAbs allowed us to highlight two different behaviors as a function of their binding properties. Native MS revealed that both Fabs of hz6F4-2 and F11 can bind the monomers and dimers of JAM-A molecules while J10.4 Fab only binds the monomer form. Next, SPR sequential competition experiments suggested that hz6F4-2 and F11 might have overlapping epitopes, as binding of one mAb almost completely blocks the quantitative binding of the second mAb. Conversely, it could be concluded from SPR that J10.4 epitope is distant from hz6F4-2/F11 binding regions. Finally, HDX-MS experiments revealed not only that hz6F4-2 and F11 epitopes are conformational epitopes formed by two nonconsecutive sequence of JAM-A, but also that both hz6F4-2 and F11 share the N-terminal 1–24 region as epitope. The linear epitope determined with J10.4 explains the different binding behaviors for this mAb compared to hz6F4-2 or F11.
Collectively, our data demonstrate that the combination of native MS, HDX-MS and SPR provides a valuable approach for epitope mapping of antigen–antibody interactions. The workflow can be generically applied to other systems for which a crystal structure of the interaction complex is not attainable, and was applied here to elucidate for the first time the epitope of hz6F4-2 targeting JAM-A.
Materials and methods
Materials
Ammonium acetate (NH4Ac), tris(2-carboxyethyl)phosphine (TCEP), dibasic potassium phosphate (K2HPO4), monobasic potassium phosphate (KH2PO4), guanidine hydrochloride (GuHCl), hydrochloric acid (HCl) were purchased from Sigma (St. Louis, MO, USA). Deuterium oxide (D2O) and deuterium chloride were products of Euriso-top (Saarbrücken, Germany). NAP-5 columns were obtained from GE Healthcare (Little Chalfont, UK), Zeba column (Thermoscientific, Rockford, IL, USA), Glu-fibrinogen peptide (GFP) from ERA (Golden, CO, USA). Papain came from Roche Diagnostics (Meylan, France) and pepsin-immobilized cartridge from Applied Biosystems (Forster city, CA, USA). F11 (sc-52690) and J10.4 (sc-53623) mAbs were purchased from Santa Cruz Biotechnology (Heidelberg, Germany), whereas 6F4-2 mAb and JAM-A antigen were produced in-house as previously described.Citation16
Fab preparation
The Fabs were generated as described previously.Citation58
Native mass spectrometry analysis
Sample preparation for native mass spectrometry
Prior to native MS experiments, the antigen was buffer exchanged against 150 mM NH4Ac, pH 7.5 by two cycles of gel filtration on NAP-5 columns. Fabs were exchanged twice against a 150 mM NH4Ac buffer using Zeba microcentrifuge gel filtration columns. The protein concentrations were determined by UV absorbance using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, France).
Binding stoichiometries experiments
The determination of binding stoichiometries was carried out by incubating 5 µM of Fabs with 1, 2, 4 and 6 equivalents of JAM-A for 10 minutes. Each titration point was analyzed by native mass spectrometry.
Native mass spectrometry
Mass spectrometry experiments were carried out on a hybrid electrospray quadrupole time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Manchester, UK) coupled to an automated chip-based nanoelectrospray device (Triversa Nanomate, Advion Biosciences, Ithaca, U.S.A.) operating in the positive ion mode. Denatured MS analysis was performed on the Synapt G2 HDMS instrument with external calibration using the multiply charged ions produced by 2 µM horse heart myoglobin solution diluted in water/acetonitrile/formic acid (50v/50v/1v) and classical interface tuning parameters of the mass spectrometer (Vc = 40 V, Pi = 2.1 mbar). For native MS experiments, external calibration was performed using singly charged ions produced by a 2 mg/mL solution of cesium iodide in 2-propanol/water (1v/1v). The MS parameters were carefully optimized to improve desolvation and sensitivity as well as maintaining non covalent interactions. Particularly, the sample cone voltage Vc was set to 120 V and the backing pressure Pi was increased to 7 mbar using throttling valve. Native MS data interpretation was performed using MassLynx 4.1 (Waters, Manchester, UK.).
Hydrogen/Deuterium Exchange Mass Spectrometry
JAM-A free and JAM-A/mAb complexes (for JAM-A/hz6F4-2 and JAM-A/F11 complexes, the concentration ratios were 2:1; whereas for JAM-A/J10.4 complex, the ratio was 1:1.5; all incubations were performed during 10 min prior to D20 labeling and JAM-A concentration was 40 µM) were diluted 20-fold with a 10 mM potassium phosphate equilibrium buffer at pH 7.0, or a 10 mM potassium phosphate deuterated buffer at pD 7.0. After dilution, the samples were incubated at 20°C for several kinetic times (0 s for undeuterated experiments, and 30 s, 2, 10, 30 and 60 min for deuterated experiments), then quenched by adding an equal volume of quench solution (100 mM potassium phosphate, 100 mM TCEP, 2 M GdHCl, pH 2.4) at 1°C during 30 s. When the quench solution and blank or deuteration solution were mixed at a 1:1 ratio (v/v), the solution was at pH 2.5. The quenched samples (100 pmol) were digested through pepsin-immobilized cartridge for 3 min in 0.1% aqueous formic acid solution at a 200 µL/min. The digestion occurs at 20°C in a controlled temperature compartment. The digested peptides were first trapped and desalted on UPLC pre-column (ACQUITY UPLC BEH C18 VanGuard pre-column, 2.1 mm I.D. × 5 mm, 1.7 µM particle diameter, Waters) and separated on UPLC column (ACQUITY UPLC BEH C18, 1.0 mm I.D. × 100 mm, 1.7 µM particle diameter, Waters) at 0°C with a gradient elution of solvent A (0.1% aqueous formic acid) and solvent B (acetonitrile with 0.1% formic acid) [2%−40% B (7 min), 40%−85% B (0.5 min), and 85% B (1 min)] at a flow rate of 40 µL/min. Sample dilutions were conducted with the CTC PAL robot (Leap Technologies, Zwingen, Switzerland), chromatographic step was carried out on Acquity UPLC system with HDX technology (Waters, Milford, MA, USA) and the mass spectrometry measurements were performed on Synapt G2 HDMS with electrospray ionization and using GFP as lock-mass correction. Peptic peptides were sequenced by MS/MS analysis in data-independent acquisition mode (MSE, Waters). Ion fragments identification and deuterium uptake monitoring were respectively performed using ProteinLynx Global Server 2.5.2 (Waters) and DynamX 3.0 (Waters). Acquisitions were realized in triplicate for each time point and only peptides identified in all replicates were kept, with a minimum fragment per amino acid of 0.3. Deuterium uptake for all identified peptides was manually checked and validated. The results were also statistically validated with the open source MEMHDX softwareCitation59 with a p-value fixed at 0.05 (see Supplementary Information S5).
Epitope mapping experiment using SPR
Analysis were performed at 25°C using a BX100 instrument (GE Healthcare).The mouse anti-His Tag mAb (Mab050, R&D systems) was diluted to 50 µg/ml in 10 mM sodium acetate pH 5.0 buffer and immobilized on a CM5 sensor chip (GE Healthcare) using the amine coupling kit and the HBS-EP+: 10 mM Hepes, 150 mM, NaCl, 3 mM EDTA, 0.05% (v/v) surfactant P20, pH 7.4 (GE Healthcare) as the running buffer. More than 10 000 RU of anti-His Tag antibodies were chemically linked by their amine functions to the carboxymetyl dextran matrix of both flowcells.
A recombinant form of the extracellular domains of the human JAM-A expressed with a C-terminal 6 His Tag was diluted to 2 µg/ml in the running buffer and injected during 60 s on the flowcell 2. The 3 anti-JAM-A antibodies hz6F4-2, F11 and J10.4 were diluted to 50 µg/ml in the running buffer. At each cycle, one of the 3 antibody was injected during 90 s on both flowcells. Then, one of the three antibodies was injected during 90 s on both flow cells. At the end of each cycle the regeneration of the surface was achieved by a 30 s injection of 10 mM Glycine, HCl pH 1.5 buffer (GE Healthcare).
3D Structure determination of the dimer formed by the extra cellular domains of JAM-A
The 3D structure of human extracellular domains of JAM-A is available in the Protein Data Bank under the reference 1NBQ. This PDB file contains the structure of two chains of JAM-A (chain A and B). The analysis of the crystallographic data using the open access PISA tools (www.ebi.ac.uk/pdbe/pisa) predict that each chain forms a homodimer (AA and BB). The structure used in this study corresponds to the BB homodimer.
Disclosure of interest
The authors report no conflict of interest.
supplementary_Material_SPR_and_HDX-MS_for_JAM-A_epitope_mapping.docx
Download MS Word (466 KB)Acknowledgments
The authors acknowledge L. Morel-Chevillet and M. Excoffier (CIPF) for their contribution in chromatographic and electrophoretic characterization of mAbs. This work was supported by the OptimAbs network bioclusters (LyonBiopole and Alsace Biovalley) and sponsors (DGCIS, Oséo, Feder, Régions Rhône- Alpes and Alsace, Communauté Urbaine de Strasbourg). This work was also supported by the CNRS, the Université de Strasbourg, the Agence Nationale de la Recherche (ANR) and the French Proteomic Infrastructure (ProFI; ANR-10-INBS-08-03). We thank the GIS IBiSA for financial support of a Synapt G2 HDMS mass spectrometer. G. Terral and M. Bourguet acknowledge Novalix and the Région Alsace for funding their PhD fellowships.
References
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345-52. doi:10.1038/nri2747. PMID:20414207
- Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337. doi:10.1038/nrd.2016.268. PMID:28303026
- Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9(2):167-181. doi:10.1080/19420862.2016.1269580. PMID:27960628
- Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Analytical Chem. 2012;85:715-36. doi:10.1021/ac3032355.
- Kaltashov IA, Bobst CE, Abzalimov RR, Wang G, Baykal B, Wang S. Advances and challenges in analytical characterization of biotechnology products: mass spectrometry-based approaches to study properties and behavior of protein therapeutics. Biotechnol Adv. 2012;30:210-22. doi:10.1016/j.biotechadv.2011.05.006. PMID:21619926
- Peng L, Oganesyan V, Damschroder MM, Wu H, Dall'Acqua WF. Structural and functional characterization of an agonistic anti-human EphA2 monoclonal antibody. J Mol Biol. 2011;413:390-405. doi:10.1016/j.jmb.2011.08.018. PMID:21867711
- Ahn J, Engen JR. The use of hydrogen/deuterium exchange mass spectrometry in epitope mapping. Chem Today. 2013;31:25-8
- Ultsch M, Bevers J, Nakamura G, Vandlen R, Kelley RF, Wu LC, Eigenbrot C. Structural basis of signaling blockade by anti-IL-13 antibody lebrikizumab. J Mol Biol. 2013;425:1330-9. doi:10.1016/j.jmb.2013.01.024. PMID:23357170
- Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discovery Today. 2014;19:95-102. doi:10.1016/j.drudis.2013.07.019. PMID:23928097
- Renaud JP, Chung CW, Danielson UH, Egner U, Hennig M, Hubbard RE, Nar H. Biophysics in drug discovery: impact, challenges and opportunities. Nat Rev Drug Discovery. 2016;15:679-98. doi:10.1038/nrd.2016.123. PMID:27516170
- Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys. 2012;526:159-66. doi:10.1016/j.abb.2012.03.021. PMID:22465822
- Zhang Y, Cui W, Wecksler AT, Zhang H, Molina P, Deperalta G, Gross ML. Native MS and ECD characterization of a fab–antigen complex may facilitate crystallization for X-ray diffraction. J Am Society Mass Spectrometry. 2016;27:1139-42. doi:10.1007/s13361-016-1398-9.
- Donnarumma D, Faleri A, Costantino P, Rappuoli R, Norais N. The role of structural proteomics in vaccine development: recent advances and future prospects. Expert Rev Proteomics. 2016;13:55-68. doi:10.1586/14789450.2016.1121113. PMID:26714563
- Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discovery. 2012;11:527-40. doi:10.1038/nrd3746. PMID:22743980
- Beck A, Debaene F, Diemer H, Wagner‐Rousset E, Colas O, Dorsselaer AV, Cianférani S. Cutting‐edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. J Mass Spectrometry. 2015;50:285-97. doi:10.1002/jms.3554.
- Atmanene C, Wagner-Rousset E, Malissard M, Chol B, Robert A, Corvaía N, Van Dorsselaer A, Beck A, Sanglier-Cianférani S. Extending mass spectrometry contribution to therapeutic monoclonal antibody lead optimization: characterization of immune complexes using noncovalent ESI-MS. Analytical chemistry 2009;81:6364-73. doi:10.1021/ac9007557. PMID:19588976
- Arthur KK, Gabrielson JP, Hawkins N, Anafi D, Wypych J, Nagi A, Sullivan JK, Bondarenko PV. In vitro stoichiometry of complexes between the soluble RANK ligand and the monoclonal antibody denosumab. Biochemistry. 2012;51:795-806. doi:10.1021/bi2007806. PMID:22242921
- Dyachenko A, Wang G, Belov M, Makarov A, De Jong RN, Van Den Bremer ET, Parren PW, Heck AJ. Tandem native mass-spectrometry on antibody–drug conjugates and submillion Da antibody–antigen protein assemblies on an Orbitrap EMR equipped with a High-Mass Quadrupole Mass Selector. Anal Chem. 2015;87:6095-102. doi:10.1021/acs.analchem.5b00788. PMID:25978613
- Wang G, de Jong RN, van den Bremer ET, Parren PW, Heck AJ. Enhancing accuracy in molecular weight determination of highly heterogeneously glycosylated proteins by native tandem mass spectrometry. Anal Chem. 2017;89(9):4793-4797. doi:10.1021/acs.analchem.6b05129. PMID:28383250
- Leitner A, Joachimiak LA, Bracher A, Mönkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure. 2012;20:814-25. doi:10.1016/j.str.2012.03.007. PMID:22503819
- Hall Z, Schmidt C, Politis A. Uncovering the early assembly mechanism for amyloidogenic β2-microglobulin using cross-linking and native mass spectrometry. J Biol Chem. 2016;291(9):4626-37. doi:10.1074/jbc.M115.691063.
- Lössl P, Sinz A. Combining amine-reactive cross-linkers and photo-reactive amino acids for 3D-structure analysis of proteins and protein complexes. Methods Mol Biol. 2016;1394:109-27. doi:10.1007/978-1-4939-3341-9_9. PMID:26700045
- Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev. 2007;107:3514-43. doi:10.1021/cr0682047. PMID:17683160
- Kiselar JG, Chance MR. Future directions of structural mass spectrometry using hydroxyl radical footprinting. J Mass Spectrometry. 2010;45:1373-82. doi:10.1002/jms.1808.
- Wecksler AT, Kalo MS, Deperalta G. Mapping of fab-1: VEGF interface using carboxyl group footprinting mass spectrometry. J Am Society Mass Spectrometry. 2015;26:2077-80. doi:10.1007/s13361-015-1273-0.
- Luchini A, Espina V, Liotta LA. Protein painting reveals solvent-excluded drug targets hidden within native protein–protein interfaces. Nat Communications. 2014;5:4413. doi:10.1038/ncomms5413.
- Li J, Wei H, Krystek SR Jr, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RY, et al. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal Chem. 2017;89(4):2250-2258. doi:10.1021/acs.analchem.6b03058.
- Snijder J, Burnley RJ, Wiegard A, Melquiond AS, Bonvin AM, Axmann IM, Heck AJ. Insight into cyanobacterial circadian timing from structural details of the KaiB–KaiC interaction. Proc Natl Acad Sci. 2014;111:1379-84. doi:10.1073/pnas.1314326111. PMID:24474762
- Premchandar A, Kupniewska A, Tarnowski K, Mücke N, Mauermann M, Kaus-Drobek M, Edelman A, Herrmann H, Dadlez M. Analysis of distinct molecular assembly complexes of keratin K8 and K18 by hydrogen–deuterium exchange. J Structural Biol. 2015;192:426-40. doi:10.1016/j.jsb.2015.10.001.
- Chiu P-L, Bou-Assaf GM, Chhabra ES, Chambers MG, Peters RT, Kulman JD, Walz T. Mapping the interaction between factor VIII and von Willebrand factor by electron microscopy and mass spectrometry. Blood. 2015;126:935-8. doi:10.1182/blood-2015-04-641688. PMID:26065652
- Coales SJ, Tuske SJ, Tomasso JC, Hamuro Y. Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on‐line proteolysis, liquid chromatography and mass spectrometry. Rapid Communications Mass Spectrometry. 2009;23:639-47. doi:10.1002/rcm.3921.
- Zhang Q, Willison LN, Tripathi P, Sathe SK, Roux KH, Emmett MR, Blakney GT, Zhang HM, Marshall AG. Epitope mapping of a 95 kDa antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2011;83:7129-36. doi:10.1021/ac201501z. PMID:21861454
- Faleri A, Santini L, Brier S, Pansegrau W, Surdo PL, Scarselli M, Buricchi F, Volpini G, Genovese A, van der Veen S, et al. Two cross-reactive monoclonal antibodies recognize overlapping epitopes on Neisseria meningitidis factor H binding protein but have different functional properties. FASEB J. 2014;28:1644-53. doi:10.1096/fj.13-239012. PMID:24371123
- Casina VC, Hu W, Mao J-H, Lu R-N, Hanby HA, Pickens B, Kan ZY, Lim WK, Mayne L, Ostertag EM, et al. High-resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome. Proc Natl Acad Sci. 2015;112:9620-5. doi:10.1073/pnas.1512561112. PMID:26203127
- Bertoldi I, Faleri A, Galli B, Surdo PL, Liguori A, Norais N, Santini L, Masignani V, Pizza M, Giuliani MM. Exploiting chimeric human antibodies to characterize a protective epitope of Neisseria adhesin A, one of the Bexsero vaccine components. FASEB J. 2016;30:93-101. doi:10.1096/fj.15-273813. PMID:26304221
- Zhang A, Hu P, MacGregor P, Xue Y, Fan H, Suchecki P, Olszewski L, Liu A. Understanding the conformational impact of chemical modifications on monoclonal antibodies with diverse sequence variation using hydrogen/deuterium exchange mass spectrometry and structural modeling. Anal Chem. 2014;86:3468-75. doi:10.1021/ac404130a. PMID:24597564
- Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs. 2013;27:495-507. doi:10.1007/s40259-013-0036-3. PMID:23649935
- Rose RJ, van Berkel PH, van den Bremer ET, Labrijn AF, Vink T, Schuurman J, Heck AJ, Parren PW. Mutation of Y407 in the CH3 domain dramatically alters glycosylation and structure of human IgG. MAbs. 2013;5:219-28. doi:10.4161/mabs.23532. PMID:23406897
- Zhang A, Fang J, Chou RY-T, Bondarenko PV, Zhang Z. Conformational difference in human IgG2 disulfide isoforms revealed by hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2015;54:1956-62. doi:10.1021/bi5015216. PMID:25730439
- Yan Y, Wei H, Fu Y, Jusuf S, Zeng M, Ludwig R, Krystek SR Jr, Chen G, Tao L, Das TK. Isomerization and oxidation in the complementarity-determining regions of a monoclonal antibody: a study of the modification–structure–function correlations by hydrogen–deuterium exchange mass spectrometry. Analytical Chem. 2016;88:2041-50. doi:10.1021/acs.analchem.5b02800.
- Pandit D, Tuske SJ, Coales SJ, Liu A, Lee JE, Morrow JA, Nemeth JF, Hamuro Y. Mapping of discontinuous conformational epitopes by amide hydrogen/deuterium exchange mass spectrometry and computational docking. J Mol Recognition. 2012;25:114-24. doi:10.1002/jmr.1169.
- Gribenko AV, Parris K, Mosyak L, Li S, Handke L, Hawkins JC, Severina E, Matsuka YV, Anderson AS. High Resolution Mapping of Bactericidal Monoclonal Antibody Binding Epitopes on Staphylococcus aureus Antigen MntC. PLoS Pathog. 2016;12:e1005908. doi:10.1371/journal.ppat.1005908. PMID:27689696
- Zhang H, Cui W, Gross ML. Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. FEBS Letters. 2014;588:308-17. doi:10.1016/j.febslet.2013.11.027. PMID:24291257
- Iacob RE, Engen JR. Hydrogen exchange mass spectrometry: are we out of the quicksand? J Am Society Mass Spectrometry. 2012;23:1003-10. doi:10.1007/s13361-012-0377-z.
- Pirrone GF, Iacob RE, Engen JR. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Analytical Chem. 2014;87:99-118. doi:10.1021/ac5040242.
- Majumdar R, Middaugh CR, Weis DD, Volkin DB. Hydrogen–deuterium exchange mass spectrometry as an emerging analytical tool for stabilization and formulation development of therapeutic monoclonal antibodies. J Pharmaceutical Sci. 2015;104:327-45. doi:10.1002/jps.24224.
- Malito E, Faleri A, Surdo PL, Veggi D, Maruggi G, Grassi E, Cartocci E, Bertoldi I, Genovese A, Santini L, et al. Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc Natl Acad Sci. 2013;110:3304-9. doi:10.1073/pnas.1222845110. PMID:23396847
- Abbott WM, Damschroder MM, Lowe DC. Current approaches to fine mapping of antigen–antibody interactions. Immunology. 2014;142:526-35. doi:10.1111/imm.12284. PMID:24635566
- Clementi N, Mancini N, Criscuolo E, Cappelletti F, Clementi M, Burioni R. Epitope mapping by epitope excision, hydrogen/deuterium exchange, and peptide-panning techniques combined with in silico analysis. Methods Mol Biol. 2014;1131:427-46. doi:10.1007/978-1-62703-992-5_26.
- Hao G, Wesolowski JS, Jiang X, Lauder S, Sood VD. Epitope characterization of an anti‐PD‐L1 antibody using orthogonal approaches. J Mol Recognition. 2015;28:269-76. doi:10.1002/jmr.2418.
- Prądzińska M, Behrendt I, Astorga-Wells J, Manoilov A, Zubarev RA, Kołodziejczyk AS, Rodziewicz-Motowidło S, Czaplewska P. Application of amide hydrogen/deuterium exchange mass spectrometry for epitope mapping in human cystatin C. Amino Acids. 2016;48:2809-20. doi:10.1007/s00726-016-2316-y. PMID:27573935
- Goetsch L, Haeuw JF, Beau‐Larvor C, Gonzalez A, Zanna L, Malissard M, Lepecquet AM, Robert A, Bailly C, Broussas M, et al. A novel role for junctional adhesion molecule‐A in tumor proliferation: Modulation by an anti‐JAM‐A monoclonal antibody. Int J Cancer. 2013;132:1463-74. doi:10.1002/ijc.27772. PMID:22886345
- Debaene F, Wagner-Rousset E, Colas O, Ayoub D, Corvaïa N, Van Dorsselaer A, Beck A, Cianférani S. Time resolved native ion-mobility mass spectrometry to monitor dynamics of IgG4 Fab arm exchange and “bispecific” monoclonal antibody formation. Anal Chem. 2013;85:9785-92. doi:10.1021/ac402237v. PMID:24007193
- Babinska A, Kedees MH, Athar H, Sobocki T, Sobocka MB, Ahmed T, Ehrlich YH, Hussain MM, Kornecki E. Two Regions of the Human Platelet F11-Receptor (F11R) Are Critical for Platelet Aggregation, Potentiation and Adhesion*. Thrombosis Haemostasis. 2002;87:712-21. PMID:12008956
- Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem. 2004;279:16254-62. doi:10.1074/jbc.M309483200. PMID:14749337
- Prota AE, Campbell JA, Schelling P, Forrest JC, Watson MJ, Peters TR, Aurrand-Lions M, Imhof BA, Dermody TS, Stehle T. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc Natl Acad Sci. 2003;100:5366-71. doi:10.1073/pnas.0937718100. PMID:12697893
- Krayukhina E, Noda M, Ishii K, Maruno T, Wakabayashi H, Tada M, Suzuki T, Ishii-Watabe A, Kato M, Uchiyama S. Analytical ultracentrifugation with fluorescence detection system reveals differences in complex formation between recombinant human TNF and different biological TNF antagonists in various environments. MAbs. 2017;9(4):664-679. 1-16. doi:10.1080/19420862.2017.1297909. PMID:28387583
- Beck A, Bussat M-C, Zorn N, Robillard V, Klinguer-Hamour C, Chenu S, Goetsch L, Corvaïa N, Van Dorsselaer A, Haeuw JF. Characterization by liquid chromatography combined with mass spectrometry of monoclonal anti-IGF-1 receptor antibodies produced in CHO and NS0 cells. J Chromatography B. 2005;819:203-18. doi:10.1016/j.jchromb.2004.06.052.
- Hourdel V, Volant S, O'Brien DP, Chenal A, Chamot-Rooke J, Dillies M-A, Brier S. MEMHDX: an interactive tool to expedite the statistical validation and visualization of large HDX-MS datasets. Bioinformatics. 2016;32:3413-9. PMID:27412089