
ABSTRACT
Gel microdroplet – fluorescence activated cell sorting (GMD-FACS) is an innovative high throughput screening platform for recombinant protein libraries, and we show here that GMD-FACS can overcome many of the limitations associated with conventional screening methods for antibody libraries. For example, phage and cell surface display benefit from exceptionally high throughput, but generally require high quality, soluble antigen target and necessitate the use of anchored antibody fragments. In contrast, the GMD-FACS assay can screen for soluble, secreted, full-length IgGs at rates of several thousand clones per second, and the technique enables direct screening against membrane protein targets in their native cellular context. In proof-of-concept experiments, rare anti-EGFR antibody clones were efficiently enriched from a 10,000-fold excess of anti-CCR5 clones in just three days. Looking forward, GMD-FACS has the potential to contribute to antibody discovery and engineering for difficult targets, such as ion channels and G protein-coupled receptors.
Article
The capacity to rapidly develop monoclonal antibodies (mAbs) against virtually any target has led to their dominant position among biologics.Citation1,2 Among the ever expanding toolbox of mAb engineering technologies, high throughput screening of large antibody repertoires is perhaps the most powerful discovery and development engine. Prominent screening technologies employ surface display of single-chain variable fragments (scFvs) or antigen-binding fragments (Fabs) on phage,Citation3-6 bacteria,Citation7,8 or yeast.Citation9,10 While surface display of antibody fragments facilitates ultra-high throughput screening via biopanning, fluorescence-activated cell sorting (FACS), or other innovative technologies,Citation11 the physicochemical properties, effector functions, and other key attributes of scFvs and Fabs are different from those of full-length IgGs, and therefore downstream applications often require reformatting into full-length antibodies.Citation12-19 Such late stage modification of antibody architecture bears an element of risk because fragments of different antibodies can exhibit wide variability in the efficiency with which they reformat.
More recently, surface display of full-length IgG antibodies has been demonstrated in bacteria,Citation8,20 mammalian cells,Citation21 and yeast.Citation22-24 While circumventing the reformatting issue, these systems have their own limitations. For example, conventional bacterial hosts are unable to perform key post-translational modifications, such as glycosylation. Yeast display of full-length IgGs typically requires a secretion-capture process, which can result in “crosstalk” among library members.Citation22-24 Mammalian systems suffer from limited library size, and multiple vector integration can also complicate library display and screening in mammalian cells.Citation21 A near universal limitation for all cell display platforms is the nature of the screening antigen, which is typically purified recombinant protein. Soluble antigen screens often neglect the context of intended applications and thereby create barriers to subsequent in vivo translation. Additionally, access to high quality recombinant antigen has proven intractable for many integral membrane targets such as ion channelsCitation25,26 and various G protein-coupled receptors (GPCRs).Citation27,28 While phage and cell surface-displayed libraries have been successfully panned against whole cell targets,Citation29-31 these efforts are typically accomplished with antibody fragment libraries, which necessitate subcloning, reformatting, and dealing with the associated issues, as described above.
In cases where the ultimate application requires a full-length IgG antibody, an ideal platform would enable direct high throughput screening of soluble secreted IgGs. Progress towards this goal includes microengraving to create arrays of individual antibody-secreting cells.Citation32,33 Within these arrays, identification of desirable clones is accomplished via microscopy, which can set upper bounds that limit screening to smaller library populations (up to 105 per device).Citation34 Others have leveraged microfluidic in vitro compartmentalization to encapsulate individual antibody-secreting clones, and these picoliter compartments can be sorted on chips using customized devices.Citation35 The nature of inverted emulsions, however, precludes washing steps, rendering this screen most relevant to antibodies that inhibit or activate enzymes for which there are fluorescent reporter systems. Related strategies have employed hydrogel microdroplets for cellular encapsulation.Citation36 The hydrogel matrix permits post-production manipulation of the encapsulated cells (e.g., washing steps), but early application of this technology to antibody library screens was done only with fluorophore-conjugated recombinant antigensCitation36 or antigens captured within the hydrogel matrix by complex sandwich schemes.Citation37,38
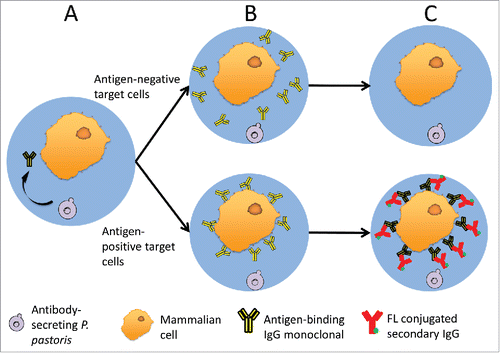
Co-encapsulation of mixed cell types in gel microdroplets (GMDs) has been used to study paracrine signalingCitation39 and as a platform for ultra-high throughput screening of antibacterial enzyme libraries.Citation40,41 Recently, GMD technology has also been adapted to screening antibody libraries against whole-cell targets.Citation42,43 In this latter work, splenocytes from immunized chickens were co-encapsulated with target cells, and B cells secreting antibody able to bind target cell antigens were identified by fluorescence microscopy. Motivated by these GMD co-encapsulation studies, we envisioned that GMDs could enable sophisticated antibody library screens in which soluble IgGs are evaluated for binding to whole cell targets using high speed flow cytometry. Specifically, libraries of recombinant mAb-producing cells are co-encapsulated with target cells that bear an antigen of interest (). Secreted antibody diffuses throughout the GMD matrix, and antigen-specific antibodies bind to cognate target cells (). Non-specific and unbound antibody is removed by washing steps, and antigen-bound IgG is detected using exogenously applied, fluorescently labeled, secondary antibodies (). Antigen specific clones are then identified and isolated by FACS screening of the fluorescently labeled GMDs.
Figure 1. A schematic of GMD-FACS antibody screening. (A) P. pastoris and mammalian target cells are co-encapsulated in GMDs. During induction, P. pastoris secretes full-length mAb, which diffuses throughout the GMD matrix. (B) Secreted full-length mAbs can bind antigen targets on the surface of antigen-positive mammalian cells (lower) but not negative cells (upper), which lack the antigen. (C) Unbound antibody is removed from the GMDs by washing, and fluorophore conjugated secondary antibodies are added to selectively detect antigen-positive target cells (lower).
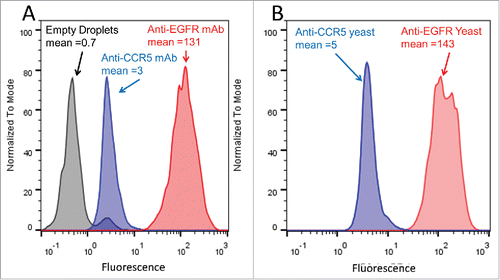
To evaluate the feasibility of selectively staining GMD-encapsulated target cells, epidermal growth factor receptor (EGFR)-expressing A431 cancer cellsCitation44 were used as targets, and anti-EGFR and anti-CCR5 mAbs were used as positive and negative control mAbs, respectively (). A431 target cells were encapsulated in agarose GMDs using a bulk stirred tank emulsification strategy. After cooling and breaking the inverted emulsion, gelled GMDs in the 40–70 µm diameter size range were selected by filtration, incubated with 20 µg/mL of purified anti-EGFR mAb or purified anti-CCR5 mAb primary, followed by staining with secondary goat anti-human IgG-PE conjugate. Stained GMDs were analyzed by FACS, and the anti-EGFR mAb was found to yield a 40-fold higher mean fluorescence signal compared to the anti-CCR5 mAb (). These control studies demonstrated that primary and secondary IgG antibodies readily diffuse into and out of the GMD matrix, and that antigen-specific primary antibodies can selectively stain encapsulated target cells expressing an antigen of interest.
Table 1. Protein sequences and estimated affinities of anti-EGFR mAb and anti-CCR5 mAb.
Figure 2. Preliminary FACS analysis of antibody staining in GMDs. (A) A431 cells were encapsulated in GMDs and labeled with purified positive control anti-EGFR (red) and negative control anti-CCR5 (blue) antibodies, respectively. Empty GMDs (grey) were treated in an analogous fashion. FACS histograms following staining with secondary antibody are shown. (B) A431 cells were co-encapsulated with yeast expressing either anti-EGFR mAb (positive control, red) or anti-CCR5 mAb (negative control, blue), respectively. FACS histograms following staining with secondary antibody are shown. Results are representative of three independent experiments.
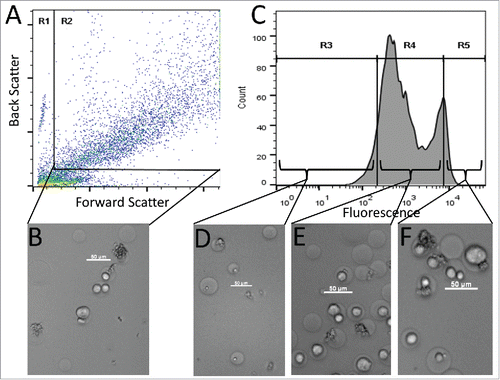
As a more practical feasibility test, antigen-targeting antibodies were next generated in situ using co-encapsulated recombinant expression hosts. First, a gene encoding the anti-EGFR IgG antibody (positive control) was cloned into Pichia pastoris yeast cells using an integrating vector. Positive control yeast were then mixed with A431 cancer cells, and the cell mixture was co-encapsulated in agarose GMDs for subsequent analysis by FACS and light microscopy (). Phase contrast microscopy of the GMD preparation revealed that it was composed of four different subpopulations: empty GMDs, GMDs containing only A431 cells, GMDs containing only P. pastoris cells, and GMDs containing both P. pastoris and A431 cells (). Additionally, following yeast cell induction, staining with secondary antibody, and FACS analysis, the positive control GMDs exhibited a multimodal fluorescence signal (). To correlate this complex fluorescence profile with the heterogeneous makeup of the GMD population as seen by microscopy, a sample of positive control GMDs was sorted into low, moderate, and high fluorescence intensity fractions. The lowest fluorescence intensity population was composed of empty GMDs or GMDs containing only yeast (). The majority of moderate fluorescence GMDs contained A431 cells, but no yeast (). The highest fluorescence GMDs contained the largest fraction co-encapsulated yeast and A431cells (). The somewhat elevated fluorescence of GMDs containing only A431 cells () was likely the result of bulk phase antibody diffusion from GMDs that contained yeast. As shown elsewhere, such crosstalk can be mitigated by employing high viscosity medium that slows antibody diffusion, by simple dilution of GMD induction samples, or by induction of GMDs in-emulsion, such that secreted proteins remain compartmentalized.Citation40,41,45
Figure 3. FACS sorting of GMD populations. (A) FACS dot plot showing forward scatter (FSC) and back scatter (BSC) signals of GMD samples. The left and bottom populations in gate R1 were a mixture of free A431 cells, GMD debris, and free P. pastoris cells. The right top population in rectangular gate R2 was intact GMDs that were either empty or contained A431 cells, P. pastoris cells, or both. Subsequent FACS sorts were gated on the R2 GMD population. (B) Micrograph image of representative GMDs before sorting. (C) PE signal of the GMDs from gate R2. Three fluorescence gates were used to sort GMDs based on PE signal intensity: the lowest 10% (R3), the central 80% (R4), and the highest 10% (R5). (D) Image of representative GMDs from the R3 sort gate. Most GMDs were empty or contained yeast cells only. (E) Image of representative GMDs from the R4 sort gate. Samples contained a large proportion of GMDs with only A431 cells. (F) Image of representative GMDs from the R5 sort gate. Samples contained a large proportion of GMD with both yeast and A431 cells. Scale bars are 50 µm. Results are representative of three independent experiments.
Subsequently, negative control yeast were constructed by inserting a gene encoding the anti-CCR5 IgG antibody into P. pastoris in a manner analogous to that used for constructing the positive control yeast. The two yeast strains (anti-EGFR = positive control; anti-CCR5 = negative control) were mixed separately with A431 cells, and the cell mixtures were each co-encapsulated in agarose GMDs. Following induction of antibody expression and staining with PE-conjugated secondary antibody, co-encapsulated GMD populations were analyzed by FACS. GMDs containing positive control yeast and A431 cells exhibited a 30-fold higher fluorescence signal than those containing negative control yeast and A431 cells (). Thus, when producing antibodies in situ with recombinant yeast, positive and negative control samples were again readily differentiated based on FACS signal.
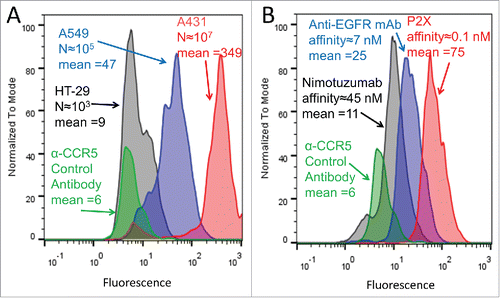
We next sought to evaluate the effect of both antigen expression level and antibody affinity on GMD-FACS signal. P. pastoris expressing the positive control anti-EGFR mAb were co-encapsulated with three different cell lines: HT29 with low level EGFR expression (103 EGFR per cellCitation46), A549 with moderate levels of EGFR expression (104-105 EGFR per cellCitation47), and A431 with high levels of EGFR expression (106-107 EGFR per cellCitation44,48). Following induction and staining with secondary antibody, the three GMD samples exhibited weak, moderate, and strong FACS fluorescence signals that correlated with the EGFR expression level of the target cells (). In a separate experiment, A431 cells, expressing high levels of EGFR, were co-encapsulated with P. pastoris expressing the original anti-EGFR mAb (KD≈1 nM) or yeast expressing anti-EGFR mAbs P2XCitation49 (KD = 0.07 nM) or nimotuzumabCitation50,51 (KD = 45 nM), respectively (). Again, following induction and immunostaining, these three GMDs exhibited weak, moderate, and strong fluorescent signals, which correlated with the EGFR binding affinities of the respective antibodies (). These findings demonstrate that the GMD-FACS assay has sufficient sensitivity to distinguish between target cells that differ in antigen expression by one to two orders of magnitude and similarly to distinguish between antibodies having one to two orders of magnitude difference in binding affinity.
Figure 4. GMD-FACS signal as a function of antigen expression level and antibody affinity. (A) Yeast expressing anti-EGFR mAb were co-encapsulated with HT29 cells (low EGFR expression, black), A549 cells (moderate EGFR expression, blue), and A431 cells (high EGFR expression, red), respectively. Following an 18 hour mAb induction, staining with secondary antibody-PE conjugate, and FACS analysis, the fluorescence intensities (mean values provided) of the three GMD preparations correlated well with target cell EGFR expression level (approximate EGFR copy per cell provided as “N”). Yeast producing an anti-CCR5 antibody co-encapsulated with A431 cells (green) are shown as a negative control. (B) A431 cells, expressing high levels of EGFR, were co-encapsulated with yeast expressing anti-EGFR antibodies of different affinities: Nimotuzumab (low affinity, black), anti-EGFR mAb (moderate affinity, blue), and P2X (high affinity, red). Following a 12 hour mAb induction, staining with secondary antibody-PE conjugate, and FACS analysis, the fluorescence intensities of the three GMD preparations correlated well with antibody affinity to the EGFR target (approximate values provided in nM). Yeast producing an anti-CCR5 antibody (green) are shown as a negative control. Results are representative of two independent experiments.
A related but as yet unexplored question is how variable antibody expression levels might affect the GMD-FACS signal. We speculate that large differences in expression level alone could alter signal when incubating GMDs in bulk, as done here. This scenario would be somewhat analogous to other work in which a cis-cell-surface capture screen (as opposed to our GMD-FACS trans-cell-surface capture screen) was developed for engineering improved protein secretion in Saccharomyces cerevisiae.Citation45 In that work, differences in antibody expression level as small as 3-fold enabled enrichment by FACS. In contrast, if GMDs were incubated in emulsion, as described elsewhere,Citation40 we speculate that the rapid accumulation of antibody to high concentrations would negate signal differences resulting from variable expression, i.e., we anticipate that even moderate affinity antibodies would saturate their target receptors in emulsified GMDs. (Note that the volume of a 50 µm GMD is ∼65 femtoliters, and yeast can secrete approximately 10,000 antibody molecules per cell per hour,Citation45 yielding 250 nM antibody in a GMD after only 1 hour incubation with a single yeast cell.)
As a proof of concept for GMD-FACS screening of antibody libraries, rare positive clones were screened from a mock “spike-in” library (). Positive control yeast, expressing the original anti-EGFR mAb, were mixed at a 1:10,000 ratio with negative control yeast, expressing the anti-CCR5 mAb. The mixed yeast population was co-encapsulated with A431 cells, antibody expression was induced for 14 hours, GMDs were stained with a secondary antibody-PE conjugate, and the stained GMDs were screened by FACS. Parallel sorts were performed using PE gates of higher stringency (HS1, top 0.05% of GMD) and lower stringency (LS1, top 0.5% of GMD). The HS1 sort was designed to assess the efficiency of single-round enrichment, and the LS1 sort was used in a follow-on round of FACS screening (). Yeast from both sorts were recovered by overnight outgrowth, and those from the LS1 sort were re-encapsulated with A431 target cells for a second round of screening. After induction and staining, this second round GMD population was FACS screened again using both higher and lower stringency sort gates (samples LS1-HS2 and LS1-LS2, respectively; ). Twenty-four clones from each of the four sort populations were selected at random, induced in deep-well microtiter plates, and the proportion of positive yeast clones from each sort was determined by FACS analysis of A431 cells stained with the respective culture supernatants (). The single higher stringency HS1 sort yielded 1 in 24 positive yeast clones, indicating an approximate 400-fold single-round enrichment relative to the initial 1:10,000 mixture. The single lower stringency LS1 sort yielded no positive yeast clones, but the sequential LS1-LS2 sort yielded 1 in 24 positive yeast clones, suggesting progressive enrichment. More importantly, the sequential LS1-HS2 sort yielded an impressive 10 in 24 positive yeast clones, which represents greater than 4000-fold enrichment in just three days. As a whole, these proof-of-concept library sorts demonstrated that GMD-FACS has the capacity to rapidly and efficiently enrich rare antibody clones from a large background of non-binding clones.
Figure 5. FACS sorting of a mock antibody library population. Positive control yeast (expressing anti-EGFR mAb) were mixed at a 1:10,000 ratio with negative control yeast (expressing anti-CCR5 mAb) followed by co-encapsulation with A431 target cells. (A) Schematic of library sorting strategy. (B) Population composition after each sort. Solid bars indicate the proportion of negative control yeast and checkered bars indicate the proportion of positive control yeast. Fold enrichment [(% anti-EGFR post sort)/(% anti-EGFR presort)] is shown.
![Figure 5. FACS sorting of a mock antibody library population. Positive control yeast (expressing anti-EGFR mAb) were mixed at a 1:10,000 ratio with negative control yeast (expressing anti-CCR5 mAb) followed by co-encapsulation with A431 target cells. (A) Schematic of library sorting strategy. (B) Population composition after each sort. Solid bars indicate the proportion of negative control yeast and checkered bars indicate the proportion of positive control yeast. Fold enrichment [(% anti-EGFR post sort)/(% anti-EGFR presort)] is shown.](/cms/asset/9b644d9b-345c-47a0-9d45-e774f73ebfea/kmab_a_1381812_f0005_oc.gif)
The GMD-FACS screening strategy described here benefits from several important advantages. First, the GMD matrix provides a robust genotype-phenotype linkage that enables ultra-high throughput screening of IgG antibody libraries in secreted soluble format. As a result, isolated binders require no reformatting or shuttling to alternative expression hosts, in turn simplifying and accelerating downstream characterization and applications. Second, GMDs are permeable at the molecular level and can be readily manipulated at any stage after production. One can therefore envision a multitude of sophisticated, multicolor, competition and blocking screens designed to isolate antagonistic or epitope-specific antibodies. Third, while yeast expression hosts were employed here, the GMD encapsulation strategy is also amenable to bacterial and mammalian expression hosts.Citation36-41 This flexibility will serve to broaden the range of antibody library screens that might be adapted to this platform. Finally, it should be noted that the strategy is not limited to screening IgG antibodies; any antibody format or various non-immunoglobulin scaffolds are likely to be amenable to GMD-FACS screening.
Perhaps the most powerful advantage of GMD-FACS screening is the capacity to use whole cells as antigenic targets. Screening against whole cells places the molecular antigen in its native context, thereby eliminating issues associated with recombinant antigens of variable or unknown quality. Moreover, the use of whole-cell targets enables facile screening of antibodies against integral membrane proteins. Thus, GMD-FACS could facilitate antibody engineering for difficult therapeutic targets such as GPCRs and ion channels. Conventional technology for screening soluble IgG antibodies against targets in their native cellular contexts requires clonal preparation of antibody producers in microtiter plates, transfer of supernatants to target cell samples, and subsequent analysis by FACS or other means. While robotic liquid handling can streamline this process, the throughput of GMD-FACS (at >1000 GMD per second) is orders of magnitude greater. Moreover, the methodology itself is simple and requires minimal resources other than access to FACS instrumentation (e.g., through a core facility).
It bears noting that a closely related GMD screening technology, termed gel encapsulated microenvironment and screening by microscopy (GEM-Microscopy), has been employed to successfully screen immune antibody libraries against whole cell targets, yielding a variety of novel antibody binders to various antigens of interest.Citation42,43 While conceptually similar to GMD-FACS, GEM-Microscopy is based on manual/visual screening of gel microdroplets on a fluorescent microscope, enabling qualitative identification of cells secreting antibodies that bind the antigen of interest. One powerful advantage of GEM-Microscopy is the potential to multiplex binding readouts to different antigens within the same GMD. However, the throughput of GEM-Microscopy is benchmarked at approximately 300,000 antibody-secreting cells per experiment (with an approximate 300-fold excess of bystander immune cells, i.e., cells not secreting sufficient antibody for detection), and, due to the qualitative nature of the primary screen, stratification of antibody candidates based on affinity requires additional culturing of clonal isolates and follow-on binding experiments.Citation43 Thus, practical throughput and quantitative discrimination during the screening process are two differentiating factors for the GMD-FACS screen described here.
One limitation of GMD-FACS for IgG antibody screens relates to screening for antigen-specific antibodies using whole cells as targets. In the context of naïve libraries, binding to proteins other than the target antigen would complicate screening. To address this issue, one might employ alternating positive and negative screens, wherein the library is first encapsulated with an isogenic cell line negative for the target receptor (e.g., via genetic knockout or RNAi knockdown) and sorted for low signal, followed by encapsulation with a cell line positive for the receptor and sorting for high signal. This strategy is analogous to negative selections in biopanning methods that employ whole cell targets.Citation29-31 A second limitation is the performance of the antibody expression host, with the current study focused specifically on yeast. Saccharomyces cerevisiae exhibits high transformation efficiency and is therefore amenable to making large recombinant libraries, but in general antibody secretion is poor in this host. Conversely P. pastoris is capable of secreting high levels of folded and functional antibody, but the transformation efficiency of this host is limited. Additionally, both of these yeast attach non-human glycoforms during post-translational modification of antibodies, which in many cases is undesirable. Having acknowledged these limitations, however, it bears noting that advances in yeast vector and host engineering are improving the performance of both yeast strains,Citation52-54 and as noted above, GMD-FACS is also amenable to other expression hosts.
The conclusions drawn here are based on successful isolation of positive clones from spike-in library sorts, and further validation of GMD-FACS for antibody screening will require application to experimental library populations, as has been done for soluble, secreted antibacterial enzymes.Citation40,41 However, we anticipate that GMD-FACS will ultimately prove useful in the screening of antibody libraries against a variety of targets, including complex cell surface proteins.
Materials and methods
Reagents, media, and cell culture
SeaPlaque Agarose (low melting temperature agarose) was purchased from Lonza. Restriction endonucleases were ordered from New England BioLabs. P. pastoris cells were grown in YPD media (1% yeast extract, 2% peptone, and 2% dextrose) and induced in BMDY media (1% yeast extract, 2% peptone, 2% dextrose, 100 mM potassium phosphate buffer pH 6.0, 1.34% yeast nitrogen base (w/o AA), and 0.8 µg/mL Biotin). Yeast extract, peptone, dextrose, and yeast nitrogen base were purchased from Research Products International Corporation. 4% paraformaldehyde in phosphate-buffered saline (PBS, 137 mM NaCl, 2.6 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, pH 7.4) was purchased from USB Corporate. Biotin was ordered from Sigma-Aldrich. Primary antibodies and secondary antibodies (goat anti-human IgG-PE) used in this study were kind gifts from Adimab, LLC. All other reagents were ordered from VWR Scientific.
A431 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS). A431 cells were fixed with 4% paraformaldehyde in PBS before encapsulation or FACS analysis.
Strains, vectors, and P. pastoris transformation
P. pastoris CBS7435 and the GAP-mAb expression vector were kind gifts from Adimab, LLC. The vector was linearized with Pme-I first and then transformed into P. pastoris by electroporation. Transformants were selected on YPD agar (1% yeast extract, 2% peptone, 2% dextrose, and 1.5% agar) plates containing 500 μg/mL Zeocin.
Expression and purification of mAbs in P. pastoris
A single colony of transformed P. pastoris was picked, inoculated in 5 mL of YPD media and grown at 250 rpm and 30°C for 24 hours. The cells were then pelleted by centrifugation, resuspended in 50 mL of BMDY media, and induced by shaking at 250 rpm and 30°C for 48 hours. Antibody purification was performed with an ÄKTA explorer 100 system following manufacture's protocol. Briefly, the supernatant was loaded on a 1 mL HiTrap Protein A FF column (from GE Healthcare Life Science) and washed with 0.02 M sodium phosphate buffer, pH 7.0. Antibodies were eluted with 0.1 M sodium citrate buffer, pH 3.0. Eluates were immediately neutralized with 1 M Tris-HCl, pH 9.0 and dialyzed in PBS.
Antibody affinity estimation
ForteBio affinity measurements for the anti-EGFR mAb were performed generally as previously described.Citation55 Briefly, ForteBio affinity measurements were performed by loading IgGs on-line onto AHQ sensors. Sensors were equilibrated off-line in assay buffer for 30 min, and then monitored on-line for 60 seconds for baseline establishment. Sensors with loaded IgGs were exposed to 100 nM EGFR-ECD for 3 min, and afterwards they were transferred to assay buffer for 3 min for off-rate measurement. Kinetic data were fit using a 1:1 binding model in the data analysis software provided by ForteBio. Results suggested a KD of 980 pM, with kon of 3.5ECitation5 M−1s−1 and koff of 3.4E−4 s−1.
Equilibrium affinity measurement for the anti-CCR5 mAb was conducted by on-cell IgG titrations. Briefly, 12 × 3-fold serial dilutions of IgG ranging between 1.8 uM and 30 pM were mixed with 105 CCR5 overexpressing cells for 30 mins at 4°C. Following two centrifugal washes in PBSF (PBS with 0.1% bovine serum albumin), cells were incubated with goat anti-human IgG R-PE detection reagent (Southern Biotech) for 15 mins at 4°C. Cells were then washed as before and resuspended in 100 µL PBSF for analysis on a FACS Canto HTS system (BD Biosciences). Mean fluorescence intensity values in the R-PE channel were collected and fitted for maximal and half maximal binding concentration using a nonlinear least squares regression method and the SOLVER function in Microsoft Excel. The half maximal binding for the anti-CCR5 mAb fit to a value of EC50 = 7.8 nM and 6.4 nM in two independent assay runs.
Preparation of gel microdroplets
GMDs were prepared using a bulk emulsion method as describedCitation40 with minor modifications. P. pastoris cells were grown in YPD media at 30°C overnight. 1.5 × 108 P. pastoris cells were harvested by centrifugation and washed twice with PBS. Low melting temperature agarose (0.15 g) was fully dissolved in 10 mL BMDY media by heating with stirring at 70°C, and then cooled to 42°C before adding A431 and yeast cells under sterile conditions. The co-encapsulation efficiency of yeast and A431 cells was optimized empirically, yielding 5–10% co-encapsulation rates at cell concentrations of 2.5×107 P. pastoris and 6×106 A431 cells per mL in the molten agarose. The co-encapsulation was carried out by mixing 1.25×108 P. pastoris cells and 3×107 A431 cells in 5 mL of filtered agarose solution. The agarose mixture was then added dropwise by pipet into a vigorously stirred solution of 11.82 mL of light mineral oil (Fisher: O121-4) containing 0.18 mL of Span80. The solution was emulsified for 6 minutes with vigorously stirring, and then cooled to 4°C with gentle stirring. The emulsions were broken by washing three times with mineral oil and four times with PBS. After washing, GMDs were pelleted by centrifugation, resuspended in PBS, and filtered first through a 70 µm cell strainer (Fisher: 08-771-2) and then a 40 µm cell strainer (Fisher: 08-771-1). Finally, GMDs that passed the 70 µm cell strainer but were retained by the 40 µm cell strained were recovered in PBS, yielding microdroplets with diameters between 40 µm and 70 µm. GMDs were subsequently suspended in BMDY media and cultured at 30°C for 12 hours, for co-encapsulating A431 cells and yeast expressing anti-EGFR antibodies of different affinities, or 18 hours, for co-encapsulating yeast expressing anti-EGFR mAb and cell lines with different EGFR expression levels. While the viability of P. pastoris expression hosts in the GMDs was not explicitly quantified in these studies, we note that, following induction and viewing via microscopy, most Pichia-containing GMDs in the current work contained dividing yeast cells (). Additionally, in prior studies Saccharomyces cerevisiae expression hosts maintained high viability during similar encapsulation experiments, as measured by live-dead cell staining.Citation40,41 Thus, it does not appear that GMD encapsulation causes excessive loss of viability for yeast expression hosts.
Flow cytometric sorting of gel microdroplets
Following induction, GMDs were stained with 5 µg/mL goat anti-human IgG-PE on ice for 30 minutes. Stained GMDs were washed twice with PBSF and then sorted on a Sony iCyt Synergy SY3200 (excitation 488 nm; emission 585/40 nm) equipped with a 130 micron acrylic nozzle. Backscatter and forward scatter signals were used to exclude free cells and droplet debris. Sorting gates were set to capture the highest 0.05% (high stringency) or 0.5% (low stringency) of GMDs based on PE signal. GMDs were analyzed at >1000 events/second. Flow data was collected using Winlist 3D 7.0, exported as FSC files and analyzed using FlowJo v10. P. pastoris cells from sorted droplets were recovered by outgrowth in YPD media containing 100 μg/mL zeocin.
Enrichment of the “spike-in” library
Sorted P. pastoris cells were grown to saturation in YPD media containing 100 μg/mL Zeocin and spread on YPD-agar plates. After 48 hours incubation at 30°C, 24 clones were picked and inoculated into 1 mL of growth medium in deep-well 96-well plates. P. pastoris were first grown at 30°C in YPD media for 24 hours and then induced at 30°C in BMDY media for 24 hours. The specificities of the secreted antibodies were determined by incubating the supernatant of the P. pastoris cultures with fixed A431 cells. After 30 minutes incubation with supernatant, A431 cells were washed twice with PBS, stained with goat anti-human IgG-PE, and analyzed on a MACSQuant (Miltenyi Biotec) (excitation 488 nm; emission 585/40 nm).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Piotr Bobrowicz for extensive technical discussions and material assistance. We also thank Nels Nielson for performing preliminary flow cytometry analysis and Mr. Nielson and Dr. Eric Krauland for thoughtful discussions. We thank Andrew Crowley for assistance optimizing GMD encapsulation conditions. We thank Sarat Pudi, Tao Wang, and Stéphanie Durand for help constructing the GAP-mAb vector. We thank Courtney Blickarz for the transfer of the A431 cell line and protocols for cell culturing and immunoassays. We appreciate the guidance of Dr. Sarah Dostal and Dr. Thomas Scanlon regarding preparation of GMDs. In addition, we are grateful for Dr. Yingda Xu for measurements of antibody binding affinity. This work was supported in part by NCI grant number 1 U54 CA151662–01, in part by NIGMS grant number P20-GM113132, and in part by NIAID grant numbers 1R01AI123372, 1R21AI094391, and 1R01AI102691. Finally, we gratefully acknowledge the financial support of the Thayer School of Engineering.
References
- Reichert JM. Antibodies to watch in 2015. mAbs. 2015;7:1-8. doi:10.4161/19420862.2015.988944. PMID:25484055.
- Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. mAbs. 2015;7:9-14. doi:10.4161/19420862.2015.989042. PMID:25529996.
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315-7. doi:10.1126/science.4001944. PMID:4001944.
- Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature. 1989;341:544-6. doi:10.1038/341544a0. PMID:2677748.
- Huse WD, Sastry L, Iverson SA, Kang AS, Alting-Mees M, Burton DR, Benkovic SJ, Lerner RA. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science. 1989;246:1275-81. doi:10.1126/science.2531466. PMID:2531466.
- Clackson T, Hoogenboom HR, Griffiths AD, Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352:624-8. doi:10.1038/352624a0. PMID:1907718.
- Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, Rogers GK. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Nat Acad Sci U S A. 2004;101:9193-8. doi:10.1073/pnas.0400187101. PMID:15197275.
- Mazor Y, Van Blarcom T, Iverson BL, Georgiou G. E-clonal antibodies: selection of full-length IgG antibodies using bacterial periplasmic display. Nat Protoc. 2008;3:1766-77. doi:10.1038/nprot.2008.176. PMID:18948976.
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553-7. doi:10.1038/nbt0697-553. PMID:9181578.
- Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A. 2000;97:10701-5. doi:10.1073/pnas.170297297. PMID:10984501.
- Chen B, Lim S, Kannan A, Alford SC, Sunden F, Herschlag D, Dimov IK, Baer TM, Cochran JR. High-throughput analysis and protein engineering using microcapillary arrays. Nat Chem Biol. 2016;12:76-81. doi:10.1038/nchembio.1978. PMID:26641932.
- Doerner A, Rhiel L, Zielonka S, Kolmar H. Therapeutic antibody engineering by high efficiency cell screening. FEBS lett. 2014;588:278-87. doi:10.1016/j.febslet.2013.11.025. PMID:24291259.
- Burden RE, Caswell J, Fay F, Scott CJ. Recent advances in the application of antibodies as therapeutics. Future Med Chem. 2012;4:73-86. doi:10.4155/fmc.11.165. PMID:22168165.
- Menzel C, Schirrmann T, Konthur Z, Jostock T, Dubel S. Human antibody RNase fusion protein targeting CD30+ lymphomas. Blood. 2008;111:3830-7. doi:10.1182/blood-2007-04-082768. PMID:18230757.
- Carter T, Sterling-Levis K, Ow K, Doughty L, Hattarki M, Shapira D, Hewish D, Kortt AA, Russell PJ. Biodistributions of intact monoclonal antibodies and fragments of BLCA-38, a new prostate cancer directed antibody. Cancer Immunol, Immunother: CII. 2004;53:533-42. doi:10.1007/s00262-003-0460-1. PMID:14722669.
- Di Fede G, Bronte G, Rizzo S, Rolfo Cervetto C, Cocorullo G, Gulotta G, Bazan V, Russo A. Monoclonal antibodies and antibody fragments: state of the art and future perspectives in the treatment of non-haematological tumors. Expert Opin Biol Ther. 2011;11:1433-45. doi:10.1517/14712598.2011.594436. PMID:21663530.
- Bender E, Woof JM, Atkin JD, Barker MD, Bebbington CR, Burton DR. Recombinant human antibodies: linkage of an Fab fragment from a combinatorial library to an Fc fragment for expression in mammalian cell culture. Hum Antibodies Hybridomas. 1993;4:74-9. PMID:8518367.
- Liang M, Dubel S, Li D, Queitsch I, Li W, Bautz EK. Baculovirus expression cassette vectors for rapid production of complete human IgG from phage display selected antibody fragments. J Immunol Methods. 2001;247:119-30. doi:10.1016/S0022-1759(00)00322-7. PMID:11150543.
- Nelson CD, Palermo LM, Hafenstein SL, Parrish CR. Different mechanisms of antibody-mediated neutralization of parvoviruses revealed using the Fab fragments of monoclonal antibodies. Virology. 2007;361:283-93. doi:10.1016/j.virol.2006.11.032. PMID:17217977.
- Mazor Y, Van Blarcom T, Mabry R, Iverson BL, Georgiou G. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli. Nat Biotechnol. 2007;25:563-5. doi:10.1038/nbt1296. PMID:17435747.
- Zhou C, Jacobsen FW, Cai L, Chen Q, Shen WD. Development of a novel mammalian cell surface antibody display platform. mAbs. 2010;2:508-18. doi:10.4161/mabs.2.5.12970. PMID:20716968.
- Rakestraw JA, Aird D, Aha PM, Baynes BM, Lipovsek D. Secretion-and-capture cell-surface display for selection of target-binding proteins. Protein Eng, Des Sel: PEDS. 2011;24:525-30. doi:10.1093/protein/gzr008.
- Shaheen HH, Prinz B, Chen MT, Pavoor T, Lin S, Houston-Cummings NR, Moore R, Stadheim TA, Zha D. A dual-mode surface display system for the maturation and production of monoclonal antibodies in glyco-engineered Pichia pastoris. PloS One. 2013;8:e70190. doi:10.1371/journal.pone.0070190. PMID:23875020.
- Rhiel L, Krah S, Gunther R, Becker S, Kolmar H, Hock B. REAL-Select: full-length antibody display and library screening by surface capture on yeast cells. PloS one. 2014;9:e114887. doi:10.1371/journal.pone.0114887. PMID:25501029.
- Wilkinson TC, Gardener MJ, Williams WA. Discovery of functional antibodies targeting ion channels. J Biomol Screen. 2015;20:454-67. doi:10.1177/1087057114560698. PMID:25473082.
- Sun H, Li M. Antibody therapeutics targeting ion channels: are we there yet? Acta pharmacol Sin. 2013;34:199-204. doi:10.1038/aps.2012.202. PMID:23381110.
- Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. mAbs. 2010;2:594-606. doi:10.4161/mabs.2.6.13420. PMID:20864805.
- Mujic-Delic A, de Wit RH, Verkaar F, Smit MJ. GPCR-targeting nanobodies: attractive research tools, diagnostics, and therapeutics. Trends Pharmacol Sci. 2014;35:247-55. doi:10.1016/j.tips.2014.03.003. PMID:24690241.
- Hoogenboom HR, Lutgerink JT, Pelsers MMAL, Rousch MJMM, Coote J, van Neer N, De Bruïne A, Van Nieuwenhoven FA, Glatz JF, Arends JW. Selection-dominant and nonaccessible epitopes on cell-surface receptors revealed by cell-panning with a large phage antibody library. Eur J Biochem. 1999;260:774-84. doi:10.1046/j.1432-1327.1999.00214.x. PMID:10103007.
- Siva AC, Kirkland RE, Lin B, Maruyama T, McWhirter J, Yantiri-Wernimont F, Bowdish KS, Xin H. Selection of anti-cancer antibodies from combinatorial libraries by whole-cell panning and stringent subtraction with human blood cells. J immunol Methods. 2008;330:109-19. doi:10.1016/j.jim.2007.11.008. PMID:18096183.
- Wang XX, Cho YK, Shusta EV. Mining a yeast library for brain endothelial cell-binding antibodies. Nat Methods. 2007;4:143-5. doi:10.1038/nmeth993. PMID:17206151.
- Panagiotou V, Love KR, Jiang B, Nett J, Stadheim T, Love JC. Generation and screening of Pichia pastoris strains with enhanced protein production by use of microengraving. Appl Environ Microbiol. 2011;77:3154-6. doi:10.1128/AEM.00104-11. PMID:21378037.
- Love KR, Politano TJ, Panagiotou V, Jiang B, Stadheim TA, Love JC. Systematic single-cell analysis of Pichia pastoris reveals secretory capacity limits productivity. PloS One. 2012;7:e37915. doi:10.1371/journal.pone.0037915. PMID:22685548.
- Love KR, Bagh S, Choi J, Love JC. Microtools for single-cell analysis in biopharmaceutical development and manufacturing. Trends Biotechnol. 2013;31:280-6. doi:10.1016/j.tibtech.2013.03.001. PMID:23582471.
- El Debs B, Utharala R, Balyasnikova IV, Griffiths AD, Merten CA. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc Natl Acad Sci U S A. 2012;109:11570-5. doi:10.1073/pnas.1204514109. PMID:22753519.
- Akbari S, Pirbodaghi T. A droplet-based heterogeneous immunoassay for screening single cells secreting antigen-specific antibodies. Lab on a chip. 2014;14:3275-80. doi:10.1039/C4LC00082J. PMID:24989431.
- Weaver JC, McGrath P, Adams S. Gel microdrop technology for rapid isolation of rare and high producer cells. Nat Med. 1997;3:583-5. doi:10.1038/nm0597-583. PMID:9142132.
- Powell KT, Weaver JC. Gel microdroplets and flow cytometry: rapid determination of antibody secretion by individual cells within a cell population. Bio/technology. 1990;8:333-7.
- Tumarkin E, Tzadu L, Csaszar E, Seo M, Zhang H, Lee A, Peerani R, Purpura K, Zandstra PW, Kumacheva E. High-throughput combinatorial cell co-culture using microfluidics. Integr Bio: quantitative biosciences from nano to macro. 2011;3:653-62. doi:10.1039/c1ib00002k. PMID:21526262.
- Scanlon TC, Dostal SM, Griswold KE. A high-throughput screen for antibiotic drug discovery. Biotechnol Bioeng. 2014;111:232-43. doi:10.1002/bit.25019. PMID:23955804.
- Dostal SM, Fang Y, Guerrette JC, Scanlon TC, Griswold KE. Genetically enhanced lysozyme evades a pathogen derived inhibitory protein. ACS Chem Biol. 2015;10:1110-7. doi:10.1021/cb500976y. PMID:25607237.
- Konitzer JD, Pramanick S, Pan Q, Augustin R, Bandholtz S, Harriman W, Izquierdo S. Generation of a highly diverse panel of antagonistic chicken monoclonal antibodies against the GIP receptor. mAbs. 2017;9:536-49. doi:10.1080/19420862.2016.1276683. PMID:28055305.
- Mettler Izquierdo S, Varela S, Park M, Collarini EJ, Lu D, Pramanick S, Rucker J, Lopalco L, Etches R, Harriman W. High-efficiency antibody discovery achieved with multiplexed microscopy. Microscopy (Oxford, England). 2016;65:341-52. PMID:27107009.
- Derer S, Bauer P, Lohse S, Scheel AH, Berger S, Kellner C, Peipp M, Valerius T. Impact of epidermal growth factor receptor (EGFR) cell surface expression levels on effector mechanisms of EGFR antibodies. J Immunol. 2012;189:5230-9. doi:10.4049/jimmunol.1202037.
- Rakestraw JA, Baskaran AR, Wittrup KD. A flow cytometric assay for screening improved heterologous protein secretion in yeast. Biotechnol prog. 2006;22:1200-8. doi:10.1021/bp0600233. PMID:16889399.
- Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol/Hematol. 2001;38:17-23. doi:10.1016/S1040-8428(00)00134-7. PMID:11255078.
- Zhang F, Wang S, Yin L, Yang Y, Guan Y, Wang W, Xu H, Tao N. Quantification of epidermal growth factor receptor expression level and binding kinetics on cell surfaces by surface plasmon resonance imaging. Anal Chem. 2015;87:9960-5. doi:10.1021/acs.analchem.5b02572. PMID:26368334.
- Haigler H, Ash JF, Singer SJ, Cohen S. Visualization by fluorescence of the binding and internalization of epidermal growth factor in human carcinoma cells A-431. Proc Natl Acad Sci U S A. 1978;75:3317-21. doi:10.1073/pnas.75.7.3317. PMID:356052.
- Kearns JD, Bukhalid R, Sevecka M, Tan G, Gerami-Moayed N, Werner SL, Kohli N, Burenkova O, Sloss CM, King AM, et al. Enhanced Targeting of the EGFR Network with MM-151, an Oligoclonal Anti-EGFR Antibody Therapeutic. Mol Cancer Ther. 2015;14:1625-36. doi:10.1158/1535-7163.MCT-14-0772. PMID:25911688.
- Boland WK, Bebb G. Nimotuzumab: a novel anti-EGFR monoclonal antibody that retains anti-EGFR activity while minimizing skin toxicity. Expert Opin Biol Ther. 2009;9:1199-206. doi:10.1517/14712590903110709. PMID:19624281.
- Garrido G, Tikhomirov IA, Rabasa A, Yang E, Gracia E, Iznaga N, Fernández LE, Crombet T, Kerbel RS, Pérez R. Bivalent binding by intermediate affinity of nimotuzumab: a contribution to explain antibody clinical profile. Cancer Biol Ther. 2011;11:373-82. doi:10.4161/cbt.11.4.14097.
- Koskela EV, de Ruijter JC, Frey AD. Following nature's roadmap: folding factors from plasma cells led to improvements in antibody secretion in S. cerevisiae. Biotechnol j. 2017;12(8). doi:10.1002/biot.201600631.
- Madsen CK, Vismans G, Brinch-Pedersen H. The PARS sequence increase the efficiency of stable Pichia pastoris transformation. J Microbiol Methods. 2016;129:1-7. doi:10.1016/j.mimet.2016.07.015. PMID:27444547.
- Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi BK, et al. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science. 2006;313:1441-3. doi:10.1126/science.1130256. PMID:16960007.
- Estep P, Reid F, Nauman C, Liu Y, Sun T, Sun J, Xu Y. High throughput solution-based measurement of antibody-antigen affinity and epitope binning. mAbs. 2013;5:270-8. doi:10.4161/mabs.23049. PMID:23575269.