
ABSTRACT
The commercial success of bispecific antibodies generally has been hindered by the complexities associated with generating appropriate molecules for both research scale and large scale manufacturing purposes. Bispecific IgG (BsIgG) based on two antibodies that use an identical common light chain can be combined with a minimal set of Fc mutations to drive heavy chain heterodimerization in order to address these challenges. However, the facile generation of common light chain antibodies with properties similar to traditional monoclonal antibodies has not been demonstrated and they have only been used sparingly. Here, we describe the design of a synthetic human antibody library based on common light chains to generate antibodies with biochemical and biophysical properties that are indistinguishable to traditional therapeutic monoclonal antibodies. We used this library to generate diverse panels of well-behaved, high affinity antibodies toward a variety of epitopes across multiple antigens, including mouse 4-1BB, a therapeutically important T cell costimulatory receptor. Over 200 BsIgG toward 4-1BB were generated using an automated purification method we developed that enables milligram-scale production of BsIgG. This approach allowed us to identify antibodies with a wide range of agonistic activity that are being used to further investigate the therapeutic potential of antibodies targeting one or more epitopes of 4-1BB.
Introduction
The ability of bispecific antibodies to bind two different antigens or epitopes enables a wide range of clinical applications that cannot be addressed through traditional monospecific monoclonal antibodies. These applications include redirecting effector cells to engage tumor cells,Citation1 circumventing escape mechanisms,Citation2 targeting heterogeneous tumors,Citation3 actively crossing the blood-brain barrier,Citation4 and enhancing tissue specificity.Citation5 Even though they were first described in the literature over 50 ago,Citation6 many bispecific antibodies that have entered the clinic, and the only two (catumaxomab, blinatumomab) that have been approved for therapy in the US or Europe, redirect T cells to engage tumor cells.Citation7 Discovering antibodies for this therapeutic mechanism of action has generally been performed by pairing a given CD3-specific antibody with panels of tumor-associated-antigen-specific antibodies. The resulting bispecific antibodies are then screened in T cell-mediated tumor killing assays to select ones that cause efficient synapse formation, which is dependent on the proximity of the epitope to the membrane.Citation8 However, antibodies that modulate receptor signaling are susceptible to geometric constraints in an unpredictable manner, so their properties can only be identified empirically through antibody generation and functional screening.Citation9-11 Therefore, it may be naïve to assume this sequential approach to bispecific antibody discovery, as used for T-cell redirecting antibodies, will be successful for other mechanisms of action or result in a drug with the most optimal properties.
In order to discover bispecific antibodies with superior therapeutic properties, a greater degree of antibody generation and screening may be requiredCitation12 compared to traditional monoclonal antibody discovery because of additional degrees of freedom. This process has been hindered by the complexities associated with generating appropriate molecules for both discovery and manufacturing purposes. Although over 60 different bispecific antibody formats that use various approaches to circumvent production issues have been described in the literature,Citation13 many of these formats do not have the same level of “developability”, a term used to describe the ability to successfully manufacture a well-behaved drug.Citation13 The bispecific IgG (BsIgG) format that most closely resembles monoclonal antibodies is considered by manyCitation14-19 to be the preferred choice, since these are more likely to maintain the desirable properties associated with monoclonal antibodies without the need for individual customization for each antibody pair selected.Citation7
BsIgG are challenging to produce in a single host cell because the random pairing of the two heavy and two light chains results in the expression of 10 different IgG species with only one being the BsIgG of interest. This reduces the overall yields of the BsIgG, and its relatively low concentration amongst the byproducts results in the need for elaborate purification techniques that still result in some amount of contamination with other IgG species, which poses potential clinical and regulatory risks. While alternative strategies exist to overcome this issue, they can be resource intensiveCitation14,16 or do not completely eliminate mispairing while introducing a significant mutational load, which may lead to immunogenicity.Citation5,17,20,21
An alternative approach for single cell manufacturing of BsIgG is to use antibodies that are based on the same light chain in combination with a minimal set of Fc mutations to drive heavy chain heterodimerization.Citation22 The resulting BsIgG can readily be purified from the two undesired byproducts.Citation1623 While this “common light chain” approach was introduced two decades agoCitation22,24 and has more recently been explored due to the renewed interest in bispecific antibodies,Citation12,22-28 it has yet to be shown to be a reliable approach for the generation of diverse, high-affinity panels of antibodies to a variety of antigens. For example, pioneering work in this field illustrated that it was possible to generate antibodies toward multiple antigens using a library with a restricted light chain repertoire,Citation22 but a decade later the highest affinity common light chain antibody reported without individual optimization is only 35 nM.Citation26 More recent work described common light chain antibodies with low nanomolar and even picomolar affinities, but this was limited to just two antibodies to each of two antigens.Citation25 Nevertheless, we expected the generation of diverse panels of high-affinity antibodies targeting a broad array of antigens would be feasible by applying the appropriate library design and synthesis techniques, since cartilaginous fish and camelids naturally generate diverse panels of high affinity single variable domain antibodies.Citation29 Further, a recent report demonstrating the ability to generate large panels of high affinity antibodies based on common heavy chain antibodies further enforced this potential.Citation18 Manufacturing of BsIgG, however, typically requires a custom purification protocol for each BsIgG,Citation16,23 which makes the generation of smaller quantities of numerous high-purity bispecific antibodies required for biophysical characterization and functional screening time consuming and labor intensive. The development of methods to expedite this process is long overdue.
We describe here a synthetic human antibody library based on a set of common light chains that was used to generate diverse panels of high affinity antibodies toward multiple antigens. To facilitate screening of BsIgG based on various combinations of antibodies, we developed a methodology including automated purification that enables the milligram-scale production of dozens of high-purity BsIgG per week. This method was utilized to generate hundreds of antibodies toward mouse 4-1BB, which were tested for agonistic activity in vitro. This approach allowed us to identify antibodies with a wide range of agonistic activity that are being used to further investigate the therapeutic potential of antibodies targeting 4-1BB.
Results
Library design
A total of four heavy chain and three light chain human germline frameworks were selected as the backbone for the synthetic library. VH1-69, VH3-15, VH3-23, VH5-51, Vκ1-39, Vκ3-20 and Vλ1-47 were chosen based on their frequency in the memory compartment of the immune repertoire obtained from 10 healthy donors, natural pairing frequencies,Citation30-32 canonical complementarity-determining region (CDR) structural diversityCitation33 and stability.Citation34,35 These criteria were chosen to allow the generation of antibodies targeting a broad array of antigens with desirable biophysical properties while minimizing the risk of immunogenicity. The undiversified common light chain sequences were based on germline V and J segments lacking junctional diversity. This resulted in CDR-L3 lengths of 9 for VK1-39 and VK3-20 and 11 for VL1-47, which, not surprisingly, also correlate with the most frequently found lengths in nature for each germline (Fig. S1A). The resulting light chain sequences for all three germlines were found in all 10 donors, suggesting a low probability of immunogenicity.
The diversity introduced into the heavy chain was designed to further support the above-mentioned design criteria. Positions to diversify were limited to the CDRs and selected based on structural proximity to the antigen and abundance of natural amino acid variability.Citation36,37 This was accomplished by analyzing representative crystal structures for each heavy chain germline found in the Protein Databank Bank (PDB) in conjunction with the immune repertoire data from over 228 donors. This analysis resulted in the generation of germline-specific designs for CDR-H1 and CDR-H2 (). The amino acid diversity to include was based on the natural amino acid distribution of non-redundant naïve B cell sequences from the immune repertoire of these donors to avoid biasing the results based on clonal expansion.Citation37 For each position selected for diversification, all amino acids found at a frequency above 1% were included with the exception of cysteine, which was excluded to avoid potential protein expression issues. The germline amino acid was used for all other positions.
Figure 1. Human synthetic antibody library design. (A) Amino acid usage in the library design for VH3-23 CDR-H3 length 12. Kabat numbering and germline reference amino acid are shown for each position. (B and C) Library post-translational modification (PTM) motif reduction. (B) Theoretical frequency of potentially detrimental PTM motifs in the library design for each germline before and after removal from CDR-H1 and CDR-H2. (B) Results for individual PTM motifs for CDR-H3 length 12. (C) Theoretical frequency of all PTM motifs before (x-axis) and after (y-axis) removal from CDR-H1 and CDR-H2 across all CDR-H3 lengths. Each symbol represents a different CDR-H3 length for the indicated germline and the arrow indicates how the frequencies increase with increasing CDR-H3 length.
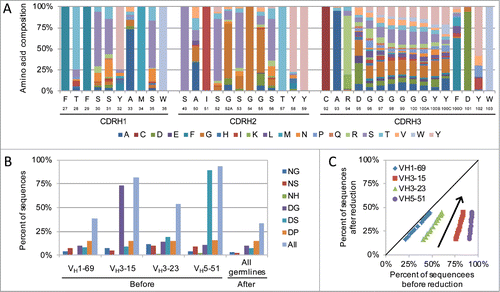
For CDR-H3, the design consisted of a cassette that spanned lengths of 6 to 17 amino acids (Kabat positions 95–102), which accounted for 81% of the unique sequences found in the immune repertoire analyzed (Fig. S1B). The same cassette was used for each germline, since for a particular CDR-H3 length, there is a high degree of amino acid usage similarity across germlines (Pearson's r = 0.93± 0.01 across lengths of 6–17 amino acids, Fig. S1D). Synthetic libraries typically use a CDR-H3 length distribution profile that mimics the bell-shaped distribution found in natureCitation30,37 (Fig. S2A). However, combining this approach with the natural amino acid distribution found at each position results in upwards of 100,000 copies for the shorter CDR-H3 sequences, which decrease the overall library size, while longer CDR-H3 lengths containing the most diversity are under-sampled (Fig. S2B).Citation37 To increase the number of unique CDR-H3s present in the library, this redundancy and under-sampling was minimized by using a sigmoidal-shaped length distribution profile (Fig. S2A), and the amino acid frequency profiles were smoothed by applying a damping coefficient to decrease the frequency of the more frequent clones while maintaining the hierarchal amino acid usage in each position (Fig. S2C). This reduced the redundancy of CDR-H3 sequences by up to 372-fold (Fig. S2B) and increased the overall diversity of the library.
Library synthesis
The library was constructed using the Slonomics® gene synthesis technology, which relies on a set of universal building blocks called anchors.Citation37,38 In total, there are 4096 unique anchors that encode all combinations of DNA codons in adjacent positions (642 = 4096). Pools of anchor molecules are assembled in a series of ligation and digestion reactions to synthesize small stretches of DNA one codon at a time, which are then combined to generate longer stretches of DNA. Anchors used in library synthesis were selected using an algorithm designed to incorporate the desired amino acids at the desired frequencies while minimizing the presence of restriction sites that are used in Slonomics®-based DNA synthesis and engineered into the frameworks of each germline for cloning. The library design also takes into consideration codon usage to maximize protein expression in the desired hosts, in this case E. coli and mammalian expression systems.
Library synthesis accuracy was improved by determining the incorporation efficiency of each anchor through the synthesis of 128 libraries each containing 32 different anchors at equal molar ratios to account for the 4096 unique anchors. These libraries were subjected to Roche 454 sequencing and the relative incorporations of each anchor were used to generate correction factors to compensate for their incorporation efficiency (Fig. S3A). These correction factors were used to adjust the frequency of each anchor within each pool of anchors used to assembly the library and increased the accuracy of amino acid incorporation at each position (Χ2 = 35.9 vs 41.0, Fig. S3B). The positional amino acid accuracy increase was compounded when looking at the combinatorial amino acid frequency across all positions (Χ2 = 5.0 vs 13.8, Fig. S3C), which is a more critical assessment of library synthesis accuracy since this is where the library diversity grows exponentially.
All combinatorial library designs result in the presence of common troublesome post-translational modifications including deamidation (NS, NG NH), isomerization (DS, DG) and protease cleavage (DP).Citation30 The frequencies of these motifs vary across the different germline libraries and emerge with higher frequencies when they are present in the germline CDR sequences. For example, VH5-51 CDR-H2 has a DS motif in its germline sequence and the library design would result in this motif appearing in 88% of the CDR-H2 sequences, while VH1-69 does not have any of these motifs in its germline CDR sequences and the library design would result in each of these motifs appearing in less than 8% of the CDR-H1 and CDR-H2 sequences combined (Fig. S4). To address this issue, anchor molecules encoding these post-translational modifications were removed prior to the synthesis of CDR-H1 and CDR-H2 for each germline. This strategy significantly reduced the overall frequency of antibodies containing these motifs, while minimizing the loss of antibodies that may require these motifs in CDR-H3 for antigen specificity ( and ).
The heavy chain CDRs were assembled with undiversified framework regions that were codon optimized for both E. coli and mammalian expressionCitation39,40 to maximize phage display levels for antibody isolation and future expression as full-length IgGs for functional testing, respectively. Roche 454 sequencing was used to assess the quality of the libraries prior to transformation into E. coli. In general, the amino acid frequencies following library synthesis correlated very well with the design, amino acid liabilities were reduced, and the desired amino acid diversity was generated (data not shown). The libraries were individually cloned based on germline and CDR-H3 length into a phagemid containing each of the three common light chains codon optimized for expression in E. coli. A total of 106 transformations were used for each heavy chain and light chain pairing, resulting in a total library of 9 × 1011 transformants (Table S1).
Antibody generation
The libraries were used to generate antibodies toward a panel of antigens, including human EGFR, mouse 4-1BB and mouse ROR2 (). This was accomplished by subjecting the phage displayed libraries to 4 rounds of panning using antigen that was site-specifically biotinylated through an Avi-tag at the C-terminus in order to minimize potential disruption of epitopes through random conjugation or alteration of protein folding following adsorption onto 96-well plates.Citation41 The biotinylated antigen was captured on streptavidin or neutravidin-coated 96-well plates to enrich for antigen-specific antibodies. A 5th and final round of panning was performed in solution with antigen concentrations as low as 5 nM to preferentially enrich for higher affinity clones.
Table 1. Library panning and antibody screening summary.
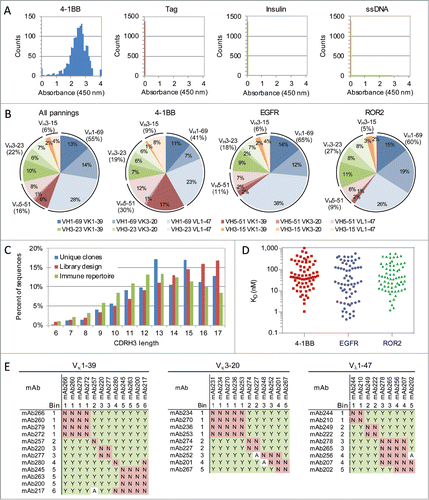
Following the final round of panning, up to 3072 clones were randomly chosen for each antigen panned (). Each clone was sequenced and its binding to four antigens was performed using an automated phage ELISA (). In addition to the antigen used for panning, antigen specificity was determined by measuring binding to an irrelevant protein containing the same tags used for purification and biotinylation (i.e., a “tag control”) as well as insulin and single-stranded DNA (ssDNA) to provide a preliminary polyreactivity assessment.Citation42 While at least 94% of the unique antibodies from each panning bound the panned antigen, some of them also bound insulin or ssDNA resulting in 85%, 92% and 94% of all clones being assigned as antigen-specific for 4-1BB, EGFR and ROR2, respectively (). This library was also synthesized with the light chain CDRs diversified using a method analogous to that described above for heavy chain CDR diversification (data not shown). The libraries had nearly identical diversities of 9 × 1011 transformants for the common light chain library and 7 × 1011 transformants for the diversified light chain library, which enabled a fair comparison between the resulting antibodies obtained from both libraries. Interestingly, the percentage of antigen-specific clones for each antigen from this common light chain library was higher than those from the diversified light chain library (85% vs 62% for 4-1BB, 92% vs 33% for EGFR, 94% vs 21% for ROR2). Overall, at least 42% of all clones screened were unique and antigen specific, leading to at least 555 unique antigen-specific antibodies for each antigen for further characterization.
Figure 2. Antibody binding and sequence analysis. (A) Antibody binding specificity ELISA. Results from 1536 antibodies isolated after the 4th and 5th round of panning toward 4-1BB. (B) Germline usage of unique antigen-specific antibodies isolated following library panning toward three different antigens. A total of 887, 601 and 1244 unique antibodies toward 4-1BB, EGFR and ROR2 were identified and included in the analysis (). (C) CDR-H3 length distribution profile of the unique antigen-specific antibodies across all three panning antigens (blue), the library design (red) and the immune repertoire from 218 human donors (green). (D) Affinities of unique and antigen-specific scFvs toward all three antigens. Antibodies were expressed as full-length IgGs and their monovalent affinities determined at 37 ˚C using SPR. (E) Epitope binning for 32 anti-ROR2 antibodies. The antibodies are grouped based on their common light chain. For each antibody pair it is indicated if they sandwich (Y), do not sandwich (N) or if the results were ambiguous (A).
Antibody characterization
Antigen-specific antibodies were identified from all heavy and light chain combinations, as well as all CDR-H3 lengths included in the library ( and ). The cumulative length distribution profile from antibodies toward all three antigens significantly correlated with the library design (R2 = 0.76, Pearson's one-sided p < 0.001), resulting in an average length of 13.5 compared to 12.8 in the natural repertoire (). While the individual and combinatorial usage of each heavy and light chain germline was highly variable across the different panning antigens, VH1-69 and Vλ1-47 were the most used heavy and light chains across all pannings (55% and 48%, respectively) and their pairing consistently provided the most hits (28% overall).
All unique antibodies with the desired binding specificity were selected and subjected to a phage competition ELISA to assess their relative affinities based on percent inhibition ().Citation43 In order to increase assay throughput, phage competition ELISA was not normalized for phage concentrations and only one antigen concentration was used. The results between technical replicates are highly correlated (Pearson's one-sided p < 0.0005) and the estimated affinities reasonably predict the actual affinities (Pearson's one-sided p < 0.0005, Fig. S6). Up to 93 clones with the highest relative affinities and diverse sequences (Fig. S7) were expressed as full-length IgGs in mammalian cells and their affinities more accurately determined using a surface plasmon resonance (SPR) biosensor assay. Nearly 90% of clones (232 of 258) were successfully expressed and able to bind their respective antigens (). Their affinities at 37°C ranged from <1 nM to >1 µM, with over half having affinities less than 100 nM (137 of 258, ). These affinities are nearly indistinguishable from antibodies generated toward the same antigens from a library containing the same heavy chain diversity, but also containing light chain diversity (Fig. S8)
A subset of 32 antibodies targeting each of these antigens based on a variety of heavy and light chain pairings was selected for epitope binning to assess their relative epitopic diversityCitation44 (, Fig. S9). A total of 12–22 bins and 182–269 sandwiching pairs of antibodies were identified for each antigen (Table S2). This is similar to results seen for antibodies based on diversified light chains,Citation45 suggesting the use of undiversified light chains has a minimal effect on antigen epitope coverage, which is likely attributed to CDRH3 playing a dominant role in antigen binding.Citation46 To take advantage of single cell manufacturing of BsIgG using common light chain antibodies, the antibodies need to be derived from the same common light chain. Reanalyzing the epitope binning results based on their individual light chains reduces the epitope bins and sandwiching pairs per light chain to 1–9 and 0–70, respectively. However, the maximum number of bins and sandwiching pairs is dependent upon the number of unique antibodies tested. When this is taken into consideration, the fraction of possible bins identified is only reduced from 0.53 to 0.44 and the fraction of possible sandwiching pairs actually increases from 0.45 to 0.46.
A representative antibody for each of the three light chains was randomly selected for basic biophysical characterization to ensure they behaved similarly to other monoclonal antibodies. The molecular weights and the amount of higher molecular weight species (HMWS) were determined using size-exclusion chromatography with multi-angle light scattering (SEC-MALS). All antibodies were determined to have an expected molecular weight of approximately 150 kDa and contained less than 0.6–1.2% HMWS (Fig. S10A). Their stability was assessed by differential scanning calorimetry (DSC) to determine the melting temperature (Tm, Fig. S10B). The Tm of the first transition ranged from 69–71°C for each of the antibodies, which are similar to other antibodies.Citation16
Higher affinity antibody generation
While the number of common light chain antibodies generated with low to sub-nanomolar affinities is substantially greater than previously reported, we were interested in determining if even higher affinity antibodies could be readily generated. Affinity optimization of individual antibodies is a routine, but time consuming practice. Alternative methods of affinity maturation have been employed to address this bottleneck, such as simultaneous affinity maturation of antibody pools.Citation47,48 A common approach, referred to as light chain shuffling, involves taking the heavy chain of one or more antibodies and combining them with a library of light chains with the goal of identifying a more optimal pairing.Citation47,49,50 This approach is obviously not feasible with common light chain antibodies since the goal is to maintain an identical light chain with multiple antibodies.
However, the common light chain library design limits the majority of CDR-H3 sequences to pair with a single CDR-H1 and CDR-H2 combination. To identify a more optimal pairing, the CDR-H3 sequences obtained following the 4th round of panning against 4-1BB were recombined with the original library of CDR-H1 and CDR-H248 in a process called H3-shuffling. Germline-specific primers were used to recover the CDR-H3 sequences while maintaining the VH and VL germline pairings of each clone. Library generation was readily accomplished due to the modular nature of the original library design, which incorporated unique germline-specific restriction sites in framework 3 of the heavy chains and a generic restriction site in framework 4 of the heavy chain while minimizing their occurrences in the diversified CDRs (). This maintains the CDR-H3 and VH germline pairing and prevents the introduction of non-human sequences in framework 3, which could arise through a PCR-based assembly. Instead of returning to the polyvalent scFv phagemid used to maximize the chance of antibody recovery from the original library, a primarily monovalent Fab display phagemid was used to increase the probability of identifying high affinity clones.Citation51 The resulting shuffled library contained 3 × 1010 transformants and was subjected to 1 round of plate panning, then 3 rounds of solution panning as described above.
Figure 3. Higher affinity antibody generation. (A) Germline-specific primers were used to recover the CDR-H3 sequences while maintaining the VH and VL germline pairings of each clone. The resulting fragments were cloned into a Fab display phagemid containing the original germline pairings for each CDR-H3 sequence. (B) Germline usage of unique antigen-specific antibodies isolated following library panning toward 4-1BB using the original library or CDR-H3 shuffled library. A total of 748 and 135 unique CDR-H3 sequences were identified following panning the original library and CDR-H3 shuffled library, respectively, and included in the analysis. (C) Affinities of unique and antigen-specific scFvs toward 4-1BB from the original library (red) and CDR-H3 shuffled library (purple). Antibodies were expressed as full-length IgGs and their monovalent affinities determined at 37 ˚C using a biosensor. (D) Epitope binning for 32 CDR-H3 shuffled anti-4-1BB antibodies. The antibodies are grouped based on their common light chain. For each antibody pair, it is indicated if they sandwich (Y), do not sandwich (N) or if the results were ambiguous (A).
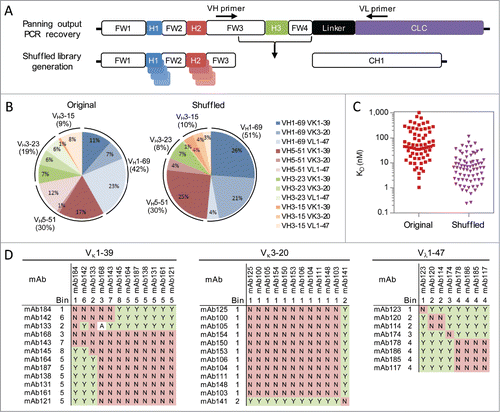
Following the 4th and final round of panning, 1536 clones were sequenced and their binding toward 4-1BB, a tag control, insulin, and ssDNA were determined using the same automated phage ELISA described above. The total number of unique antigen-specific antibodies based on the VH sequence increased from 750 (49% total) for the original library to 1180 (77% total) for the shuffled library (). However, only a small fraction of these shuffled library antibodies (135, 11%) had unique CDR-H3 sequences, while the majority (748, 99.7%) from the original library were unique. This was expected since the original library was designed to minimize CDR-H3 sequence redundancy while the shuffled library was designed to promote this redundancy in a search for more optimal pairings with CDR-H1 and CDR-H2. For example, 309 antibodies were identified from the shuffled library that used the same CDR-H3 sequence, but differed in CDR-H1 and CDR-H2. The heavy and light chain germline usage of these antibodies was also rather different pre- and post-shuffling ().
A diverse set of 760 antigen-specific antibodies including at least one antibody from each unique CDR-H3 were subjected to a phage competition ELISA. Significantly more antibodies (94%) had percent inhibitions >50% from the shuffled library compared to the original library (6%, Fig. S11A). Antibodies sharing the same CDR-H3 sequence but differing in their CDR-H1 and CDR-H2 sequences can exhibit a nearly two order of magnitude range in relative affinities as estimated using their percent inhibitions (Fig. S11B). A set of 91 antibodies with diverse CDR-H3 sequences and higher percent inhibitions were expressed as full-length IgGs in mammalian cells and their affinities more accurately determined using an SPR biosensor assay (Fig. S11C). Nearly all the antibodies (96%) were successfully expressed and able to bind 4-1BB (). Their affinities ranged from 0.2 nM to 117 nM with an average affinity of 13 nM compared to a range of 1 nM to 1 μM and an average affinity of 128 nM for the antibodies from the original library ().
A total of 16 unique epitope bins were identified across a panel of 32 antibodies including 292 sandwiching pairs (Table S2, Fig. S12). Based on individual light chains, these numbers were again reduced to 2–8 epitope bins and 11–23 sandwiching partners, but the fraction of possible bins and sandwiching pairs was only reduced from 0.50 to 0.44 and from 0.59 to 0.42, respectively. Surprisingly, the number of epitope bins and sandwiching pairs identified from the shuffled library was actually higher than from the anti-4-1BB antibodies obtained from the original library. This indicates that the shuffling process provides a straightforward method to significantly increase antibody affinity without negatively affecting the epitopic diversity.
Bispecific antibody generation
Identifying the two antibodies that result in a BsIgG with optimal function requires empirically testing numerous antibody combinations. While large quantities of highly pure individual BsIgG can readily be obtained by combining Protein A and ion exchange chromatography (IEC),Citation14-16,23 many BsIgG require the development of custom purification conditions because the isoelectric points (pI) of the two antibodies are too similar, leading to poor separation by IEC. This hinders the generation of large, diverse panels of high purity BsIgG for further testing and characterization. To circumvent this problem, we developed universal purification conditions that can be used for all antibodies regardless of their isoelectric points. Specifically, we modified our existing BsIgG technologyCitation16 by incorporating a polyhistidine tag on the C-terminus of the heavy chain with a higher pI (hIgG2 RRRR) to accentuate the pI difference from the heavy chain with a lower pI (hIgG2 EEE, ).
Figure 4. Universal method for BsIgG expression and purification. (A) Schematic of the method. Both heavy chains (higher pI in blue, lower pI in red) and their shared common light chain (gray) are co-expressed from a single cell. The heavy chains contain complementary mutations in their constant regions to facilitate preferentially heterodimer formation.Citation16 A polyhistidine tag was incorporated on the C-terminus of the heavy chain with the lower pI. The clarified cell culture supernatant was subjected to an automated four-step tandem purification process on an FPLC to purify the BsIgG from the monospecific contaminants. (B) Ion exchange chromatography (IEC) elution profiles of individually expressed mAb A hIgG2 EEE (red) and mAb B hIgG2 RRRR (blue), and co-expression of mAb A and mAb B (green). The linear gradient of percent buffer B (black) is also shown. The number of histidines in the polyhistidine tag on mAb B hIgG2 RRRR is indicated.
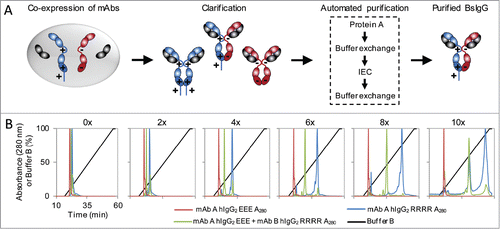
Two model antibodies that utilize the same common light chain, but bind to different antigens were cloned into either the hIgG2 RRRR construct with up to a 10x polyhistidine tag or the hIgG2 EEE construct, then expressed individually or co-expressed in mammalian cells. The clarified cell culture supernatants were purified via fast protein liquid chromatography (FPLC) using Protein A and IEC. The IEC fractions expected to be the BsIgG were collected and dialyzed in phosphate-buffered saline, then confirmed to be the BsIgG by antigen binding using bio-layer interferometry (BLI). As expected, the separation of the BsIgG from the monospecific contaminants increased proportionally to the length of the polyhistidine tag, leading to robust separation of the BsIgG from the monospecific contaminants (). While the 10x polyhistidine tag resulted in the most robust separation, the inclusion of the polyhistidine tag raised concerns that it may increase non-specific binding. This concern was addressed by incubating each BsIgG at up to 100 nM with cells that do not express either target antigen, and then detecting bound IgG with anti-human IgG Alexa Fluor 647 by flow cytometry. Only the BsIgG with the 10x polyhistidine tag showed evidence of non-specific binding (Fig. S13).
Based on these results, an ion exchange protocol was developed for the 8x polyhistidine-tag BsIgG that could be used for all BsIgG regardless of their variable domain isoelectric points. This enabled the development of a fully automated FPLC purification process that produces high purity BsIgG in the final formulation buffer from a 50 mL culture in 2.5 hours (). This process was used to generate 285 BsIgGs toward 4-1BB with an average yield of 2.0 ± 1.1 mg. In situations where lower quantities of antibody are required and higher throughput is desired, the process throughput can be increased using high performance liquid chromatography (HPLC). Interestingly, these BsIgGs were already 79 ± 13% pure after Protein A purification.
Antibody-induced 4-1BB signaling
4-1BB is a co-stimulatory receptor expressed on the surface of activated immune cells, including T cells.Citation52 Interaction with its ligand, 4-1BBL, activates the NFκB signaling cascade, resulting in increased T cell cytotoxicity and survival, and thus enhanced tumor clearance.Citation52 WeCitation53 and othersCitation54 have shown that agonistic antibodies toward 4-1BB can be used to mimic this interaction, and agonistic anti-4-1BB antibodies have been shown to have a clinical benefit in multiple cancer indications.Citation55 However, these antibodies are specific for human 4-1BB, which makes it challenging to dissect their mechanism of action using fully immunocompetent mouse models. To address this issue, the common light chain antibodies described above were generated toward mouse 4-1BB. These surrogate antibodies can be used as tools to increase our understanding of the underlying biology of this therapeutic approach and potentially improve upon it.Citation56
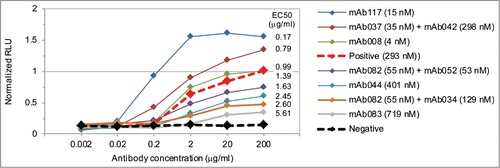
Common light chain antibodies with a range of affinities and epitopes toward mouse 4-1BB were expressed as either traditional IgGs or BsIgG, and then their ability to induce 4-1BB signaling was determined using an in vitro cell based assay (Fig. S14). Since agonistic antibodies toward 4-1BB and other receptors frequently depend upon Fc gamma receptor (FcγR) engagement to induce signaling,Citation9,53 the antibodies were immobilized to mimic FcγR-mediated crosslinking, then incubated with HEK293T cells transduced with mouse 4-1BB and an NFκB luciferase reporter. The results from this type of cell line correlate well with those from stimulated primary T cells and allow direct comparison of antibodies across multiple assays by avoiding donor T cell variability.Citation53 Both monoclonal and bispecific antibodies with a wide range of activity with regards to both EC50 and maximum signaling intensity were identified (). Surprisingly, there was no apparent correlation between EC50 and antibody affinity or epitope, and the most potent molecule was a monoclonal antibody (mAb117). None of the antibodies induced 4-1BB signaling when co-incubated in solution with the HEK293T reporter cell line (data not shown) as expected from similar experiments conducted with antibodies targeting human 4-1BB.
Figure 5. Antibody induced 4-1BB. Both monospecific and bispecific antibodies with a range of EC50 and maximum signaling levels were generated. The positive control is the commercially available anti-mouse 4-1BB antibody MAB9371 (red dashed) and the negative control is an antibody toward an irrelevant antigen (black dashed). The sample name includes the antibody affinity. All antibodies were generated with human IgG2dA D265A Fcs with or without the bispecific EEE or RRRR mutations. The relative luminescence units (RLU) for each antibody was normalized to the positive control included on the same assay plate. High throughput screening limited the number of replicates to an N of 1 or 2 for all antibodies with the exception of MAB9371 which had an average standard deviation of 0.06 normalized RLU over 10 replicates at each of 6 concentrations (Fig. S13).
Discussion
We developed a synthetic human antibody library based on undiversified common light chains to generate antibodies with biophysical and biochemical properties indistinguishable from traditional monoclonal antibodies, which is critical for successful therapeutic development. We demonstrated how this library can be used to generate large panels of high affinity antibodies that bind diverse epitopes on a variety of antigens. In contrast to previous work,Citation22,24-26 at least 10 antibodies with single-digit nanomolar affinities were isolated directly from the libraries toward each antigen and at least one antibody based on each of the three common light chains was identified. Further, the binding kinetics were determined at 37°C so the affinity improvement compared to these previous reports is likely even higher since they used lower temperatures, which typically results in lower KD values.Citation57
Our common light chain antibodies have similar affinities and epitope coverage compared to antibodies with diversified light chains, and they can also be affinity matured in pools using a parallel library-based approach analogous to that employed for traditional antibodies.Citation48 This approach resulted in antibodies with 10-fold higher average affinities compared to antibodies isolated directly from the library, which is similar to what others have accomplished when affinity maturing common light chain antibodies on an individual basis, but it drastically expedites the process since hundreds of antibodies are affinity matured in parallel.Citation26 Further, these antibodies maintained their specificity and breadth of epitope coverage, which is critical for obtaining antibodies with the desired function. This was readily accomplished since the library contained germline-specific restrictions sites in the framework regions that were used to recombine the antigen-specific antibody CDR-H3 sequences obtained from the initial panning with up to nearly a billion CDR-H1 and CDR-H2 sequence combinations to find a more optimal pairing. The incorporation of this step further justifies minimizing CDR-H3 redundancy in the library design. In essence, this process mimics the natural antibody diversification process that occurs in B cells where the heavy and light chain pairing is established then the antibody is affinity matured through rounds of somatic hypermutation.
Generating sufficient quantities of BsIgGs has been a major limitation to their discovery. Here, we demonstrated that single cell expression of common light chain BsIgGs with complementary Fcs that facilitated preferential heterodimerization are 79% pure following protein A purification alone, which is dramatically higher than the 25% expected for BsIgGs requiring two different light chains. In instances where the functional screen is tolerant of this level of purity, thousands of BsIgGs could be generated using 96-well plate expression and purification approaches for initial screening. However, this level of purity is neither sufficient for all functional assays nor acceptable for accurate functional assessment and additional characterization. To address this issue, we developed a simple approach coupled with an automated continuous purification process based on existing bispecific antibody manufacturing techniquesCitation16 to enable the purification of highly pure common light chain BsIgG at the milligram scale in 2.5 hours without the need for per-antibody customization. The key to developing this process was the incorporation of a small polyhistidine tag on the C-terminus of one of the heavy chains, which results in robust and reproducible separation of the contaminating monospecific antibodies from the desired BsIgG during the IEC step. While our approach was based on accentuating the pI difference that exists between the two heavy chains that preferentially heterodimerize using our BsIgG technology,Citation16 it could be used with any Fc heterodimerization technology, such as the commonly used knobs-into-holes approach.Citation58
It has become apparent that the therapeutic mechanisms of actions of many cancer immunotherapies are complex and will require the use of immuno-competent mouse models to more comprehensively understand the underlying biology.Citation59,60 Many therapeutic antibodies, however, do not cross-react with their mouse counterparts due to low sequence homology. This includes antibodies toward 4-1BB, which has been shown to be a therapeutically important T cell costimulatory receptor.Citation53,54 While the most advanced antibodies showed early evidence of clinical activity, they have different safety profiles, which could be the result of antibody intrinsic differences, such as their affinities, epitopes or Fc isotypes, or extrinsic differences, such as the patient populations treated.Citation61 Surrogate antibodies that recognize mouse 4-1BB are required to investigate these questions in syngeneic mouse models. To help with this future work, we used the methods described above to generate traditional monoclonal antibodies toward mouse 4-1BB, as well as dozens of highly pure bispecific antibodies per week and hundreds overall with minimal human involvement. This enabled us to find antibodies that induce mouse 4-1BB signaling with a range of activity. While more potent antibodies were identified compared to existing antibodies, none of the antibodies were able to induce FcγR-independent activation,Citation9 including BsIgGs derived from antibodies with non-overlapping epitopes. These antibodies will serve as valuable tools to enable further dissection of antibody-mediated modulation of 4-1BB in immunocompetent mouse models in an attempt to find more optimal therapeutic antibodies.
Another important aspect of the common light chain antibodies generated here is the origin of their synthetic antibody sequences. Not only are the heavy chains sequences designed to look indistinguishable from antibodies derived from humans,Citation37 but the common light chain sequences used are based on the germline sequences of light chains that are frequently used by humans,Citation31 and the exact sequence was found in nearly all individuals sequenced. They are thus potentially less immunogenic than traditional monoclonal antibodies that contain non-germline light chain sequences. Further, they have biophysical properties typical of therapeutic IgGs which makes them suitable for both therapeutic monoclonal and bispecific antibody development.
Overall, we developed a process to generate high affinity common light chain antibodies with broad epitope coverage by mimicking the natural antibody evolutionary process. BsIgG based on common light chain antibodies can be easily discovered and generated, resulting in antibodies with biochemical and biophysical properties that are indistinguishable from traditionally generated human antibodies. This approach allows more antibodies to be evaluated preclinically, and will hopefully lead to BsIgGs with more optimal therapeutic properties.
Materials and methods
Antibody discovery
Antigens containing a C-terminal 8x polyhistidine tag and AviTag™ (Avidity LLC) were expressed using the Expi293 system (Thermo Fisher Scientific), and then purified on an AKTA Avant (GE Healthcare) using a HisTrap excel (GE Healthcare) column followed by a Superdex 200 Increase GL (GE Healthcare) column according to the manufacturer's protocols. Site-specific biotinylation through the AviTag™ (Avidity, Cat. No. BirA-500) was performed according to the manufacturer's protocols. The VH libraries were synthesized using the Slonomics® technology (MorphoSys) in a manner similar to that previously described.Citation37 The phage displayed library was prepared and biopanning performed with site-specifically biotinylated antigen using an established protocol as a guideline.Citation37 Automated phage ELISAs were based on previously described methods.Citation62 For higher affinity antibody generation, the CDR-H3 sequences were recovered from phages from the 4th round of biopanning by PCR using VH FW3 and VL FW1 germline-specific primers, subcloned into a phagemid containing the cognate VL and CDR-H1 and CDR-H2 library; the phage-displayed library was then prepared, biopanning performed and antibody screening performed using phage ELISAs in a manner similar to what is described above. Full details are described in Supplemental materials.
Antibody expression and purification
The variable heavy chain domain from the antibody fragments were recovered by PCR and subcloned into a mammalian expression vector for full-length human IgG expression, then expressed using the Expi293 system. Purification was performed using an AKTA Avant (GE Healthcare) equipped for multistep tandem purification in a manner similar to that previously described.Citation63 Traditional IgGs were purified from conditioned medium with a HiTrap™ MabSelect SuRe (GE Healthcare) column followed by a HiPrep™ Desalting Column (GE Healthcare) according to the manufacturer's protocols. A280-based watch conditions were implemented during the HiTrap MabSelect Sure purification elution phase to direct the eluate to the HiPrep™ Desalting Column. For bispecific IgG purification, the eluate from the HiTrap™ MabSelect SuRe column was directed to the HiPrep™ Desalting Column followed by purification with MonoS GL (GE Healthcare) HiPrep™ and an additional HiPrep™ Desalting Column purification step. A280-based watch conditions were implemented during the initial HiPrep™ Desalting Column purification to direct the eluate to the MonoS GL column. Unless otherwise indicated, the MonoS GL purification was performed with a multistep protocol consisting of a step gradient to 25% Buffer B for 5 column volumes, a linear gradient of 25–75% Buffer B over 40 column volumes and a step gradient to 100% Buffer B for 5 column volumes with A280-based watch conditions implemented during the linear gradient phase to direct the eluate to the HiPrep™ Desalting Column. Full details are described in in Supplemental materials.
Antibody characterization
The molecular weights were determined using multi-angle light scattering and the thermal stability determined using differential scanning calorimetry as previously described.Citation16 Antibody binding kinetics and affinities were determined by SPR using a Biacore 4000 biosensor (GE Lifesciences) similar to a manner previously described.Citation64 Antibody epitope binning was performed using a previously described sandwiching techniqueCitation44 with the exception of modifications for antibody capture and regeneration and a combination of SPR and BLI was used. An automated mouse 4-1bb signaling assay was based on a previously described method.Citation53 Full details are described in in Supplemental materials.
Abbreviations
| BLI | = | Bio layer interferometry |
| BsIgG | = | bispecific IgG |
| CDR | = | complementarity determining region |
| CDR-H | = | heavy chain CDR |
| CDR-L | = | light chain CDR |
| DSC | = | differential scanning calorimetry |
| EGFR | = | epidermal growth factor receptor |
| FcγR | = | Fc gamma receptor |
| FPLC | = | fast protein liquid chromatography |
| FW | = | framework |
| HMWS | = | higher molecular weight species |
| HPLC | = | high-performance liquid chromatography |
| IEC | = | ion exchange chromatography |
| pI | = | isoelectric point |
| ROR2 | = | receptor tyrosine kinase-like orphan receptor 2 |
| scFv | = | single-chain variable fragment |
| SEC-MALS | = | size exclusion chromatography with multi-angle light scattering |
| ssDNA | = | single-stranded DNA |
| SPR | = | surface plasmon resonance |
| VH | = | heavy chain variable domain |
| VL | = | light chain variable domain |
| 4-1BB | = | tumor necrosis factor receptor superfamily member 9 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1406570_Suppl_Mat.zip
Download Zip (1.6 MB)Acknowledgments
We would like to thank Dr. Ralf Strohner and Dr. Stefan Arnold from Morphosys for training us on the Slonomics® technology. We are grateful to Dilduz Telman and Lora Zhao of Pfizer Inc. for their technical assistance with 454 pyrosequencing and Yik Andy Yeung of Pfizer Inc. for help developing the phage panning protocols. We are appreciative of Timothy Fisher of Pfizer Inc. for sharing the reporter cell line we used to functionally characterize some of the 4-1BB antibodies. We would also like to thank Hoang Tran from GE Healthcare for helping set-up the tandem purification using the Avant FPLC (GE). And finally, we would like to thank Barbra Sasu, Shahram Salek-Ardakani and Yik Andy Yeung of Pfizer Inc. for their careful review of the manuscript.
References
- Nagorsen D, Bargou R, Ruttinger D, Kufer P, Baeuerle PA, Zugmaier G. Immunotherapy of lymphoma and leukemia with T-cell engaging BiTE antibody blinatumomab. Leukemia & lymphoma. 2009;50:886–91. doi:10.1080/10428190902943077.
- McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, Zhang B, Luus L, Overland R, Nguyen S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Molecular cancer therapeutics. 2012;11:582–93. doi:10.1158/1535-7163.MCT-11-0820.
- Dong J, Sereno A, Aivazian D, Langley E, Miller BR, Snyder WB, Chan E, Cantele M, Morena R, Joseph IB, et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. mAbs. 2011;3:273–88. doi:10.4161/mabs.3.3.15188.
- Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3:84ra44. doi:10.1126/scitranslmed.3002230.
- Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs. 2015;7:377–89. doi:10.1080/19420862.2015.1007816.
- Nisonoff A, Rivers MM. Recombination of a mixture of univalent antibody fragments of different specificity. Archives of biochemistry and biophysics. 1961;93:460–2. doi:10.1016/0003-9861(61)90296-X.
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Molecular immunology. 2015;67:95–106. doi:10.1016/j.molimm.2015.01.003.
- Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, Clark V, Hristopoulos M, Cook R, Slaga D, et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer cell. 2017;31:383–95. doi:10.1016/j.ccell.2017.02.001.
- White AL, Chan HT, French RR, Willoughby J, Mockridge CI, Roghanian A, Penfold CA, Booth SG, Dodhy A, Polak ME, et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer cell. 2015;27:138–48. doi:10.1016/j.ccell.2014.11.001.
- Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Pluckthun A. Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2. Structure. 2013;21:1979–91. doi:10.1016/j.str.2013.08.020.
- Moraga I, Wernig G, Wilmes S, Gryshkova V, Richter CP, Hong WJ, Sinha R, Guo F, Fabionar H, Wehrman TS, et al. Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands. Cell. 2015;160:1196–208. doi:10.1016/j.cell.2015.02.011.
- Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, Yoshihashi K, Okuyama-Nishida Y, Saito H, Tsunoda H, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nature medicine. 2012;18:1570–4. doi:10.1038/nm.2942.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017:9:182–212.
- Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5145–50. doi:10.1073/pnas.1220145110.
- Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nature biotechnology. 2013;31:753–8. doi:10.1038/nbt.2621.
- Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. Journal of molecular biology. 2012;420:204–19. doi:10.1016/j.jmb.2012.04.020.
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nature biotechnology. 2014;32:191–8. doi:10.1038/nbt.2797.
- Fischer N, Elson G, Magistrelli G, Dheilly E, Fouque N, Laurendon A, Gueneau F, Ravn U, Depoisier JF, Moine V, et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nature communications. 2015;6:6113. doi:10.1038/ncomms7113.
- Labrijn AF, Meesters JI, Priem P, de Jong RN, van den Bremer ET, van Kampen MD, et al. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nature protocols. 2014;9:2450–63. doi:10.1038/nprot.2014.169.
- Dillon M, Yin Y, Zhou J, McCarty L, Ellerman D, Slaga D, et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. mAbs. 2017;9:213–30.
- Liu Z, Leng EC, Gunasekaran K, Pentony M, Shen M, Howard M, Stoops J, Manchulenko K, Razinkov V, Liu H, et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. The Journal of biological chemistry. 2015;290:7535–62. doi:10.1074/jbc.M114.620260.
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nature biotechnology. 1998;16:677–81. doi:10.1038/nbt0798-677.
- Sharkey B, Pudi S, Wallace Moyer I, Zhong L, Prinz B, Baruah H, et al. Purification of common light chain IgG-like bispecific antibodies using highly linear pH gradients. mAbs. 2017;9:257–68.
- Ward ES. VH shuffling can be used to convert an Fv fragment of anti-hen egg lysozyme specificity to one that recognizes a T cell receptor V alpha. Molecular immunology. 1995;32:147–56. doi:10.1016/0161-5890(94)00119-L.
- Krah S, Schroter C, Eller C, Rhiel L, Rasche N, Beck J, et al. Generation of human bispecific common light chain antibodies by combining animal immunization and yeast display. Protein Eng Des Sel. 2017;30:291–301. doi:10.1093/protein/gzw077.
- Jackman J, Chen Y, Huang A, Moffat B, Scheer JM, Leong SR, et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. The Journal of biological chemistry. 2010;285:20850–9. doi:10.1074/jbc.M110.113910.
- Nissim A, Hoogenboom HR, Tomlinson IM, Flynn G, Midgley C, Lane D, et al. Antibody fragments from a ‘single pot’ phage display library as immunochemical reagents. The EMBO journal. 1994;13:692–8.
- Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Scientific reports. 2015;5:17943. doi:10.1038/srep17943.
- Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nature biotechnology. 2005;23:1126–36. doi:10.1038/nbt1142.
- Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs. 2013;5:445–70. doi:10.4161/mabs.24218.
- Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20216–21. doi:10.1073/pnas.0909775106.
- DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, et al. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nature medicine. 2015;21:86–91. doi:10.1038/nm.3743.
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. Journal of molecular biology. 1987;196:901–17. doi:10.1016/0022-2836(87)90412-8.
- Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. Journal of molecular biology. 2005;347:773–89. doi:10.1016/j.jmb.2005.01.053.
- Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. Journal of molecular biology. 2003;325:531–53. doi:10.1016/S0022-2836(02)01237-8.
- Raghunathan G, Smart J, Williams J, Almagro JC. Antigen-binding site anatomy and somatic mutations in antibodies that recognize different types of antigens. Journal of molecular recognition: JMR. 2012;25:103–13. doi:10.1002/jmr.2158.
- Zhai W, Glanville J, Fuhrmann M, Mei L, Ni I, Sundar PD, et al. Synthetic antibodies designed on natural sequence landscapes. Journal of molecular biology. 2011;412:55–71. doi:10.1016/j.jmb.2011.07.018.
- Van den Brulle J, Fischer M, Langmann T, Horn G, Waldmann T, Arnold S, et al. A novel solid phase technology for high-throughput gene synthesis. BioTechniques. 2008;45:340–3. doi:10.2144/000112953.
- Fath S, Bauer AP, Liss M, Spriestersbach A, Maertens B, Hahn P, et al. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PloS one. 2011;6:e17596. doi:10.1371/journal.pone.0017596.
- Maertens B, Spriestersbach A, von Groll U, Roth U, Kubicek J, Gerrits M, et al. Gene optimization mechanisms: a multi-gene study reveals a high success rate of full-length human proteins expressed in Escherichia coli. Protein Sci. 2010;19:1312–26. doi:10.1002/pro.408.
- Miersch S, Sidhu SS. Synthetic antibodies: concepts, potential and practical considerations. Methods. 2012;57:486–98. doi:10.1016/j.ymeth.2012.06.012.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, et al. Biophysical properties of the clinical-stage antibody landscape. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:944–9. doi:10.1073/pnas.1616408114.
- Lee CV, Sidhu SS, Fuh G. Bivalent antibody phage display mimics natural immunoglobulin. Journal of immunological methods. 2004;284:119–32. doi:10.1016/j.jim.2003.11.001.
- Fagerstam LG, Frostell A, Karlsson R, Kullman M, Larsson A, Malmqvist M, et al. Detection of antigen-antibody interactions by surface plasmon resonance. Application to epitope mapping. Journal of molecular recognition: JMR. 1990;3:208–14. doi:10.1002/jmr.300030507.
- Abdiche YN, Harriman R, Deng X, Yeung YA, Miles A, Morishige W, et al. Assessing kinetic and epitopic diversity across orthogonal monoclonal antibody generation platforms. mAbs. 2016;8:264–77. doi:10.1080/19420862.2015.1118596.
- Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi:10.1016/S1074-7613(00)00006-6.
- Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu TY, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel. 2013;26:663–70. doi:10.1093/protein/gzt047.
- Prassler J, Steidl S, Urlinger S. In vitro affinity maturation of HuCAL antibodies: complementarity determining region exchange and RapMAT technology. Immunotherapy. 2009;1:571–83.
- Clackson T, Hoogenboom HR, Griffiths AD, Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352:624–8. doi:10.1038/352624a0.
- Kang AS, Jones TM, Burton DR. Antibody redesign by chain shuffling from random combinatorial immunoglobulin libraries. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:11120–3. doi:10.1073/pnas.88.24.11120.
- Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods in enzymology. 2000;328:333–63. doi:10.1016/S0076-6879(00)28406-1.
- Cheuk AT, Mufti GJ, Guinn BA. Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer gene therapy. 2004;11:215–26. doi:10.1038/sj.cgt.7700670.
- Fisher TS, Kamperschroer C, Oliphant T, Love VA, Lira PD, Doyonnas R, et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer immunology, immunotherapy: CII. 2012;61:1721–33. doi:10.1007/s00262-012-1237-1.
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nature medicine. 1997;3:682–5. doi:10.1038/nm0697-682.
- Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:5349–57. doi:10.1158/1078-0432.CCR-17-1243.
- Wakefield I, Stephens S, Foulkes R, Nesbitt A, Bourne T. The use of surrogate antibodies to evaluate the developmental and reproductive toxicity potential of an anti-TNFalpha PEGylated Fab' monoclonal antibody. Toxicological sciences: an official journal of the Society of Toxicology. 2011;122:170–6. doi:10.1093/toxsci/kfr083.
- Zeder-Lutz G, Zuber E, Witz J, Van Regenmortel MH. Thermodynamic analysis of antigen-antibody binding using biosensor measurements at different temperatures. Anal Biochem. 1997;246:123–32. doi:10.1006/abio.1996.9999.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–21. doi:10.1093/protein/9.7.617.
- Labrijn AF, Meesters JI, Bunce M, Armstrong AA, Somani S, Nesspor TC, et al. Efficient Generation of Bispecific Murine Antibodies for Pre-Clinical Investigations in Syngeneic Rodent Models. Scientific reports. 2017;7:2476. doi:10.1038/s41598-017-02823-9.
- Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E2646–54. doi:10.1073/pnas.1604268113.
- Perez-Ruiz E, Etxeberria I, Rodriguez-Ruiz ME, Melero I. Anti-CD137 and PD-1/PD-L1 Antibodies En Route toward Clinical Synergy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:5326–8. doi:10.1158/1078-0432.CCR-17-1799.
- Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. Journal of molecular biology. 2004;338:299–310. doi:10.1016/j.jmb.2004.02.050.
- Yoo D, Provchy J, Park C, Schulz C, Walker K. Automated high-throughput protein purification using an AKTApurifier and a CETAC autosampler. Journal of chromatography A. 2014;1344:23–30. doi:10.1016/j.chroma.2014.04.014.
- Safsten P, Klakamp SL, Drake AW, Karlsson R, Myszka DG. Screening antibody-antigen interactions in parallel using Biacore A100. Anal Biochem. 2006;353:181–90. doi:10.1016/j.ab.2006.01.041.