
ABSTRACT
During the development of monoclonal antibodies (mAbs) and other therapeutic proteins, immunogenicity, in particular the induction of anti-drug antibodies (ADAs), is an important concern, and thus immunogenicity assessment is a requirement for their approval. Establishment of appropriate methods for detecting and characterizing ADAs is necessary for immunogenicity assessment, but the lack of commonly available reference standards makes it difficult to compare and evaluate the methods. It is also difficult to compare the data with those obtained by other methods or facilities without reference standards. Here, we developed a panel of ADAs against anti-CD20 rituximab (Rituxan®, MabThera®); the panel consisted of eight clones of recombinant human-rat chimeric mAbs that target rituximab. The anti-rituximab mAbs showed different binding properties (specificity, epitope and affinity), and different neutralization potencies for CD20 binding, complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity. The molecular size of the immune complex consisting of rituximab and the anti-rituximab mAb differed among the clones, and was well correlated with their level of Fcγ-receptor activation. These results suggest that the ADAs chosen for the newly developed panel are suitable surrogates for human ADAs, which exhibit different potential to affect the efficacy and safety of rituximab. Next, we used this panel to compare several ADA-detecting assays and revealed that the assays had different abilities to detect the ADAs with different binding characteristics. We conclude that our panel of ADAs against rituximab will be useful for the future development and characterization of assays for immunogenicity assessment.
Introduction
Monoclonal antibody (mAb) therapeutics have shown great success as treatments for various diseases, including tumors and inflammatory diseases.Citation1-3 Because of their higher target specificity, mAb treatments are generally considered to pose a lower risk of adverse reactions than chemical drugs. However, administration of mAbs and other therapeutic proteins may cause immunogenicity, and in particular the induction of anti-drug antibodies (ADAs),Citation4-6 which can adversely affect the efficacy, pharmacokinetics and/or safety profiles of drugs. A prominent example was the development of human anti-mouse antibodies in patients who received murine mAbs, which was the major obstacle to continued use of this therapy.Citation7 To resolve this problem, chimeric and humanized mAbs, in which the rodent-derived sequences are reduced by recombinant DNA technologies, have been developed. In addition, transgenic-mouse and phage-display technologies have enabled the generation of fully human mAbs. These advances in mAbs-production technologies have generally succeed in reducing immunogenicity, and contributed to the clinical use of therapeutic mAbs with lower risk of unwanted immune responses.Citation8,Citation9 However, inductions of ADAs are still reported in patients administered chimeric, humanized and human mAbs.Citation10,Citation11 Thus, based on the existing data, even when human mAbs, in which the rodent-derived sequences have been completely eliminated, are used in therapy, the risk of inductions of ADAs, including human anti-human antibodies (HAHAs) is not completely eliminated.
ADAs directed against therapeutic mAbs can affect efficacy, pharmacokinetics and/or safety profiles.Citation4,Citation12,Citation13 In the case of infliximab (human-mouse chimeric mAb targeting tumor necrosis factor (TNF)), it has been reported that the presence of ADAs is associated with lower serum drug concentrations, reduced clinical responses and increased risk of adverse events, including infusion reactions.Citation14-16 A correlation between the serum concentration of rituximab (a human-mouse chimeric anti-CD20 mAb) and human anti-chimeric antibody,Citation17 and the relationship of HAHA to the biodistribution of sibrotuzumab (a humanized anti-FAP mAb)Citation18 have also been reported. An anti-idiotypic IgE against basiliximab (a human-mouse chimeric anti-interleukin-2 receptor mAb) has been reported to trigger anaphylactic shock.Citation19
Because of these risks of ADAs, immunogenicity assessment is a regulatory requirement for approval of therapeutic mAbs.Citation20, Citation21 The strategy for immunogenicity assessment consists of several steps, including screening assay, confirmatory assay and characterization. In the screening and confirmation steps, ADAs with the potential to bind to the tested drugs are detected using various assay platforms,Citation22,Citation23 including radioimmunoassay (RIA), enzyme-linked immunosorbent assay (ELISA), electrochemiluminescence (ECL) immunoassay, surface plasmon resonance (SPR) assay and bio-layer interferometry (BLI) assay. Each of these binding assays has different sensitivity for the detection of ADAs in human clinical samples, including serum or plasma. In addition, the ability to detect ADAs that show weak binding affinities or fast dissociation rates varies depending on the type of assay format (e.g., the presence or absence of a washing step). Cell-based functional assays are the preferred approach for characterizing whether a detected ADA is a neutralizing antibody (NAb), and they are also useful for estimating the impact of the presence of an ADA on clinical efficacy. However, the sensitivity of cell-based assays to detect ADAs is often inferior to that of binding assays, and thus the neutralizing activity of an ADA with low concentration may not be detected.Citation24 Accordingly, these assays have different performance characteristics for detecting or characterizing ADAs. When establishing the methods for immunogenicity assessment, it is thus important to choose and validate an assay that is suitable for the purpose. Nonetheless, there is currently no commonly available reference standard of ADAs against mAb therapeutics, despite the potential importance of such a standard for evaluating and comparing the assays.
In this study, we generated recombinant human-rat chimeric anti-rituximab mAbs and developed a panel of ADAs against rituximab. The panel consists of anti-rituximab mAbs with various binding characteristics and neutralizing activities. Using this ADA panel, we then showed that the molecular size of an immune-complex consisting of rituximab and anti-rituximab mAb is well correlated with the activation of Fcγ receptor (FcγR), which may lead to the immune response in vivo. In addition, we experimentally demonstrated the differential ability of the ADA assays to detect ADAs with different binding characteristics.
Results
Generation of anti-rituximab mAbs
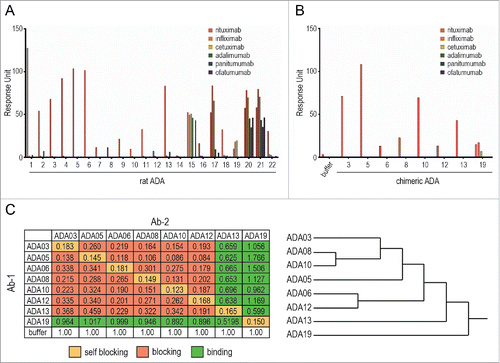
To create ADAs against rituximab, rats were immunized with an F(ab')2 fragment of rituximab, and hybridomas were generated by using the iliac lymph node method. The hybridomas were screened for their activity to bind rituximab, resulting in the establishment of 22 clones expressing rat anti-rituximab mAbs (rat ADAs). To estimate the binding specificity of the rat ADAs, we performed an SPR-binding assay in which a human-mouse chimeric therapeutic mAb (rituximab, infliximab or cetuximab) or fully human therapeutic mAb (adalimumab, panitumumab or ofatumumab) was captured on the sensor chip, and the binding of rat ADAs was analyzed (). Most of the established clones (except for ADA15, 17, 19, 20 and 21) showed specific binding to rituximab, indicating these clones recognize the complementarity-determining region (CDR) of rituximab and that they are anti-idiotypic antibodies. ADA19 bound to human-mouse chimeric mAbs, but not to fully human mAbs, suggesting that ADA19 recognizes the murine-derived sequences in human-mouse chimeric mAbs. ADA15, 17, 20 and 21, which bound to both human-mouse chimeric and fully human mAbs, are thought to recognize the constant region of human IgG. We next prepared the human-rat chimeric ADAs against rituximab (chimeric ADAs) by grafting the variable regions of rat ADA to human IgG1. The binding specificity of each chimeric ADA was evaluated by SPR analysis, and we confirmed that, as well as original rat clones, ADA03, 05, 06, 08, 10, 12 and 13 bind specifically to rituximab, and ADA19 binds to chimeric mAbs with a murine variable region (). We used this set of human-rat chimeric ADAs in all experiments hereafter.
Figure 1. Binding characteristics of anti-rituximab mAbs. (A, B) The binding specificity of anti-rituximab mAbs was analyzed by an SPR-binding assay. The binding of each clone to rituximab, infliximab, cetuximab (human-mouse chimeric IgG1), adalimumab (human IgG1), panitumumab (human IgG2) or ofatumumab (human IgG1) was represented as a response unit. (C) Epitope binning assay. The binding response of Ab-2 (a competing Ab) to Ab-1 (the saturating Ab)-bound rituximab is shown in the matrix. Self-blocking, blocking (<0.5) and binding (≥0.5) are indicated in yellow, red and green, respectively. The dendrogram generated by cluster analysis indicates the difference in the binding epitopes of each clone.
Epitope analysis of anti-rituximab mAbs
We next performed an epitope binning assay to characterize the binding epitope of anti-rituximab mAbs by using the BLI method. The epitope binning assay consists of two association steps: 1) binding of the saturating mAb (Ab-1) to a rituximab-immobilized sensor chip, and 2) binding of a competing Ab (Ab-2) to rituximab in the presence of Ab-1. The binding of Ab-2 in the presence of Ab-1, which was normalized to the binding in the absence of Ab-1, is shown in with the dendrogram generated by cluster analysis. When Ab-1 and Ab-2 were the same mAb, self-blocking was observed as expected. ADA03, 05, 06, 08, 10, and 12 prevented each other from binding to rituximab, suggesting that they share the same binding epitope. In contrast, ADA13 blocked the binding of ADA03, 05, 06, 08, 10 and 12 to rituximab, but was bound to rituximab in the presence of these ADAs, indicating that ADA13 had a different binding epitope than the other clones. ADA19, which recognizes murine-derived sequences in chimeric mAbs, did not block the binding of other rituximab-specific mAbs, and showed binding to rituximab even in the presence of other rituximab-specific mAbs.
Binding affinities of anti-rituximab mAbs
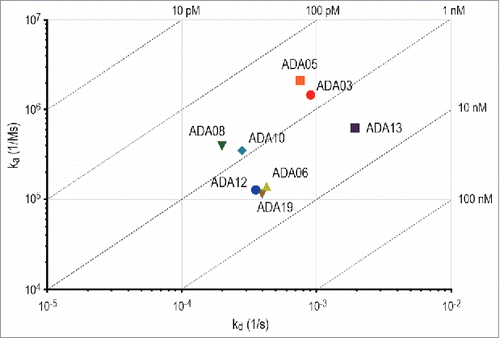
To assess the rituximab-binding affinity of anti-rituximab mAbs, the binding between rituximab and anti-rituximab mAbs was analyzed by the BLI method. The binding sensorgrams and analyzed kinetic parameters are shown in Supplemental Figure 1 and , respectively. All of the clones bound to rituximab in a dose-dependent manner, and their dissociation constants (KD) varied from 100 pM to 10 nM. The on-off rate map, in which the kinetic parameters of each clone are visualized, indicated that the kinetics parameters (ka and kd) differed among the clones (). In particular, ADA13 showed a higher kd value than the other clones.
Table 1. Summary of binding kinetics parameters of anti-rituximab mAbs.
Figure 2. On-off rate map indicating the binding kinetics parameters of anti-rituximab mAbs analyzed by BLI assay. The association rate constant (ka) is plotted against the dissociation rate constant (kd). The diagonal lines indicate the equilibrium dissociation constant (KD).
Neutralizing activity of anti-rituximab mAbs
When considering the effects of ADAs on clinical efficacy, it is important to evaluate whether an ADA is a NAb. Rituximab binds to CD20 expressed on the cell surface of B-cell lymphoma and exerts anti-tumor activity by killing the target cells via complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC).Citation25,Citation26 We first estimated the neutralizing activity of anti-rituximab mAbs against the CD20-binding activity of rituximab. Binding of rituximab to CD20-expressing Raji cells was dose-dependently inhibited by all clones of the anti-rituximab mAbs, indicating that they neutralize the CD20-binding activity of rituximab (). ADA13 showed lower neutralization potency than the other clones, whereas all the clones except ADA13 exhibited similar potency. We next assessed the neutralization potency against the CDC activity of rituximab by using Raji cells as target cells. As in the binding assay, all clones inhibited the CDC activity of rituximab, and the potency of ADA13 appeared to be lower than that of the other clones (). For the measurement of ADCC activity, we used Raji cells and Jurkat/FcγRIIIa/NFAT-Luc reporter cells as target and effector cells, respectively. Human FcγRIIIa is one of the activating FcγRs expressed on various human immune cells, and plays a pivotal role in the natural killer cell-mediated ADCC activity of mAbs.Citation27 By using Jurkat/FcγRIIIa/NFAT-Luc reporter cells, activation of FcγRIIIa by the antigen-mAb complex can be measured as luciferase activity, and this assay is useful as a surrogate of the ADCC assay.Citation28,Citation29 As shown in , the activation of FcγRIIIa by rituximab bound to CD20 expressed on Raji cells was inhibited by anti-rituximab mAbs. The strength of the neutralization potency was in the order of ADA05 > ADA03, ADA08, ADA12 > ADA06 > ADA19 > ADA10 > ADA13. The IC50 values of each clone in the binding, CDC and ADCC assays are summarized in . These results suggest that the anti-rituximab mAbs in our ADA panel have different abilities to neutralize the pharmacological activities of rituximab.
Figure 3. Neutralization activity of anti-rituximab mAbs against CD20 binding (A), CDC activity (B) and ADCC activity (C) of rituximab. The percentage of activity is plotted against the concentration of ADA (n = 3, bars indicate SEM). An anti-TNF mAb, infliximab, was used as a negative control in the binding assay.
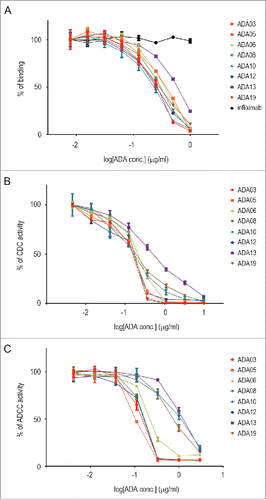
Table 2. Summary of neutralization activities of anti-rituximab mAbs.
Immune-complex formation of anti-rituximab mAbs
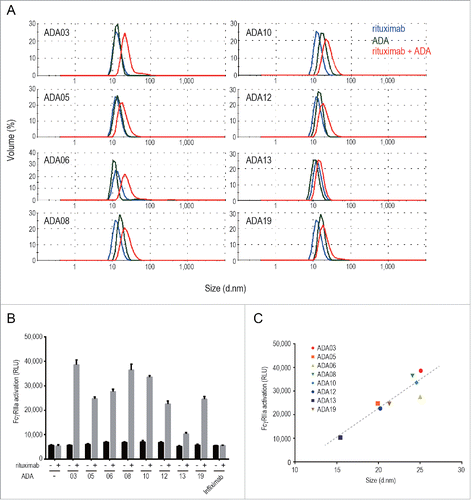
The formation of immune complexes between ADAs and therapeutic proteins is a major cause of the immune-mediated adverse reactions attributed to ADAs.Citation12 Drug/ADA immune complexes can activate a complement cascade and FcγR-mediated immune-cell signaling, resulting in immune-cell activation and inflammatory cytokine release. In order to evaluate the formation of immune complexes between rituximab and anti-rituximab mAbs, we first analyzed the molecular size of anti-rituximab mAbs in the presence or absence of rituximab by the dynamic light scattering (DLS) method. The results showed that the size of molecules in the mixture of rituximab and anti-rituximab mAbs was larger than that of rituximab or anti-rituximab mAbs alone (), indicating the formation of rituximab/ADA immune complexes. The molecular size of the rituximab/ADA immune complexes varied among the clones. To examine whether rituximab/ADA immune complexes can activate immune cells, we next evaluated the activation of FcγR by the immune complexes by using Jurkat/FcγRIIa/NFAT-Luc cells. Human FcγRIIa is expressed on various immune cells including monocytes, macrophages, neutrophils and dendritic cells, and is involved in the activation of these cells by antigen-bound IgGs. Anti-rituximab mAbs did not activate FcγRIIa in the absence of rituximab, but did activate FcγRIIa in the presence of rituximab (), suggesting that immune complexes consisting of rituximab and anti-rituximab mAbs have the potential to activate immune cells via FcγRIIa. Interestingly, immune complexes of larger molecular size exhibited stronger FcγRIIa activation, and a positive correlation (R square = 0.848) was observed between the molecular size of the immune complex and FcγRIIa activation (). These results suggest that the molecular size of the rituximab/ADA immune complex could be one of the important characteristics of ADA that is correlated with immune-mediated adverse reactions caused by unwanted activation of immune cells.
Figure 4. Molecular size and FcγR activation property of the rituximab/ADA immune complex, (A) DLS analysis of the rituximab/ADA immune complex. The size distribution of rituximab (blue), ADA (green) and the rituximab/ADA complex at a molar ratio of 1:1 (red) is shown. (B) FcγRIIa activation by ADAs in the presence or absence of rituximab was measured by using Jurkat/FcγRIIa/NFAT-Luc reporter cells. Infliximab was used as a negative control. Data represent the mean + SEM (n = 3). (C) Correlation between the FcγRIIa activation and the size of the immune complex. The FcγRIIa activation (in relative light units: RLU) is plotted against the size of the immune complex (diameter). The dashed line is a regression line (R square is 0.848).
Comparison of the methods for detecting ADAs against rituximab
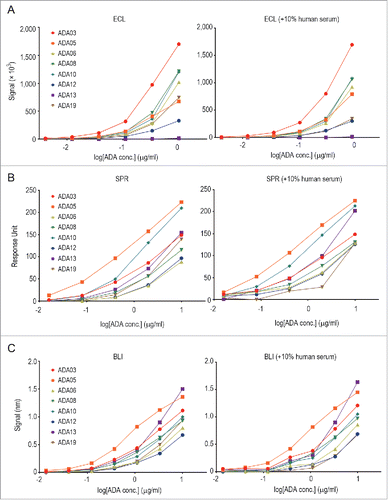
As described above, we developed an ADA panel consisting of anti-rituximab mAbs with different binding-epitope, binding-affinity and neutralization-activity characteristics, and we applied this panel to a comparison of different methods for detecting ADAs: ECL immunoassay, SPR assay and BLI assay. The assays were performed in the presence or absence of human serum. The details of these assays are described in the Materials and Methods. As shown in , the detectability of each clone of the anti-rituximab mAbs differed among the assays. In particular, there was a remarkable difference in the detection of ADA13, which showed a higher kd value and seemed to dissociate more rapidly than the other clones. The ECL immunoassay could not detect ADA13 at all (), whereas the SPR and BLI assay could detect it in a dose-dependent manner ( and ). In addition, ADA05 showed the strongest response in the SPR and BLI assay, and a weaker response in the ECL immunoassay. In this way, the SPR and BLI methods, but not the ECL immunoassay, showed similar tendencies in terms of the detection of ADAs with different binding characteristics. Addition of 10% human serum in the samples did not influence the detectability of almost clones, but the response signal of ADA19 was apparently decreased in the presence of 10% human serum in all three methods.
Figure 5. Comparison of the methods for detecting anti-rituximab mAbs. Each serially diluted clone of anti-rituximab mAbs was measured by ECL immunoassay (A), SPR assay (B) and BLI assay (C) in the presence or absence of 10% human serum as described in the Materials and Methods.
Discussion
Development of a method for detecting and characterizing ADAs is a critical step in the immunogenicity assessment of biopharmaceuticals, including therapeutic mAbs. Because the human ADAs induced in patients are polyclonal antibodies with different binding epitopes and binding affinities, it is important when developing ADA-detection methods to ensure that the methods can detect ADAs with various characteristics in a range of clinically relevant concentrations. A polyclonal antibody prepared from drug-immunized animals is generally used as a surrogate positive control in assay development, even though animal-derived polyclonal antibodies generally exhibit different characteristics in binding affinity or binding epitopes compared to human ADAs. Human antibodies that show similar characteristics to the ADAs induced in patients are desirable surrogates, but they are not commonly available. Recently, the first World Health Organization erythropoietin antibody reference panel was established.Citation30,Citation31 This panel consists of nine fully human mAbs against erythropoietin with various characteristics, and is useful for evaluating assay performance as well as for assay validation and standardization.Citation32
In this study, we developed a panel of anti-rituximab mAbs that consists of 8 clones with various characteristics in binding epitope, binding affinity and neutralization activity. This is the first report on the development of an ADA panel for rituximab. When human-murine chimeric mAbs, including rituximab, are administrated to human, both CDRs and framework regions in murine-derived variable regions can be immunogenic determinants,Citation33 therefore we chose seven anti-idiotypic clones that specifically bind to rituximab (ADA03, 05, 06, 08, 10, 12 and 13) and a clone that broadly binds to human-murine chimeric mAbs (ADA19). The epitope binning assay indicated that the anti-idiotypic clones could be further classified into two groups in terms of the binding epitope (ADA13 and the others). The binding affinities of anti-rituximab mAbs to rituximab (KD) varied widely, ranging from 100 pM to 10 nM. When using the ADAs in this panel as positive control reagents for the development or characterization of various assays, it is important that the recombinant ADAs show characteristics similar to those of human ADAs developed in patients. Animal-derived antibodies with adjuvant-based immunization are generally considered to show higher binding affinity than human ADAs. Although the analysis of detailed characteristics of human ADAs is technically difficult due to their limited availability, it has been reported that patient-derived anti-adalimumab mAbs show high binding affinity (KD between 0.6 and 233 pM) to adalimumab,Citation34 suggesting that the binding affinities of our anti-rituximab mAbs are not very different from those of patient-derived ADAs.
Among our anti-rituximab mAbs, ADA13 shows different characteristics from the other clones in terms of its biological activity. ADA13 binds to rituximab with an affinity (KD) similar to ADA06, 12 and 19, but dissociates from rituximab more rapidly because of its higher off-rate (kd). The neutralizing potency of ADA13 in CD20 binding and CDC and ADCC activity is significantly lower than that of the other clones. Thus, ADA13 seems to hold promise as a surrogate for an ADA with lower potency that is not sufficiently affinity maturated. All of the anti-idiotypic clones except for ADA13 showed similar neutralization activity in the CD20-binding assay, but there was a difference in the potency to neutralize CDC and ADCC activity. In the CDC assay, ADA06 and 10 showed somewhat weaker potency than ADA03, 05, 08 and 12. In the ADCC assay, the difference between the clones was more remarkable: the order of the neutralizing potency against ADCC activity was ADA05 > ADA03, ADA08, ADA12 > ADA06 > ADA10. These results indicate that the anti-idiotypic clones in our panel exhibit various levels of activity for neutralizing the pharmacological activity of rituximab, and also suggest that the cell-based assays have differential ability to measure the neutralization activity.
The drug/ADA immune complex can induce the activation of various immune cells, and this activation correlates with the impact of ADAs on the drug pharmacokinetics and/or safety profile. As shown in , the anti-rituximab mAbs in our developed ADA panel formed immune complexes with rituximab, and the molecular sizes of the rituximab/ADA immune complexes differed among the clones. The size of the rituximab/ADA03 immune complex, which was the largest complex formed by the clones, was approximately 25 nm in diameter. Based on the estimated molecular weight calculated by the DLS software, the immune complex seems to be a tetrameric complex consisting of two rituximab and two ADA03 molecules. These results are consistent with a previous report in which therapeutic mAbs and ADA were shown to form relatively smaller immune complexes (e.g., dimeric or tetrameric complexes) due to steric hindrance.Citation12,Citation34,Citation35 It has been reported that small immune complexes can persist in circulation, while large immune complexes can be cleared efficiently by immune cells via FcγRs,Citation12,Citation36,Citation37 although the relationship between the molecular size of the immune complex and the FcγRs-activation profile is not fully understood. We recently reported that the molecular size of immune complexes formed by TNF and anti-TNF mAbs was correlated with the ability of the complexes to activate FcγRs.Citation38 In this study, we revealed that the rituximab/ADA immune complexes directly activated FcγRIIa, and that their ability to activate FcγRIIa was correlated with their molecular size. This is the first report to experimentally demonstrate the direct activation of FcγRs by mAb/ADA immune complexes. Although the contribution of FcγRIIa-activation measured by our reporter assay on the activation of immune cells in vivo is not clear, our results suggest that small mAb/ADA immune complexes can activate FcγR-expressing immune cells, which may lead to faster clearance of mAbs by phagocytosis or induce adverse reactions mediated by inflammatory cytokines. Interestingly, the order of the ability of anti-idiotypic clones to activate FcγRIIa by the rituximab/ADA immune complex was different from the order of the neutralization activity of the clones. For example, ADA10 exhibited weaker neutralization activity against ADCC than most of the other anti-idiotypic clones, but the rituximab/ADA10 immune complex strongly activated FcγRIIa. These results suggest that the impact of ADAs on the drug efficacy may not always be correlated with the impact on drug safety: ADAs with weak neutralization activity have the potential to induce adverse reactions via FcγR-mediated immune-cell activation.
Our ADA panel was also shown to be useful for comparing or evaluating the methods for detecting ADAs against rituximab. When comparing the detectability of three different assays (ECL immunoassay, SPR assay and BLI assay) for the ADAs against rituximab, only the ECL immunoassay failed to detect ADA13. The ECL immunoassay in bridging format is one of the most popular methods for detecting ADAs because of its high sensitivity and throughput. On the other hand, the washing steps required in the ECL immunoassay often make it difficult to detect ADAs with a low binding affinity or high dissociation rate. As shown in , ADA13 binds rituximab with a similar binding affinity (KD) to ADA06, 12 and 19, but dissociates more rapidly than the others. Thus, the undetectability of the ECL immunoassay for ADA13 can be explained by the higher kd value of ADA13: the binding of ADA13 to rituximab may not be maintained in the washing steps in the ECL immunoassay. Both the SPR assay and BLI assay are label-free assays that do not require washing steps, and they showed a similar ability to detect various clones of anti-rituximab mAbs in our panel. We also examined the effect of human serum in the samples on the detectability of ADAs against rituximab. In all three methods, the dose-dependent responses of most clones (except for ADA19) were not affected by the addition of 10% human serum, suggesting that these clones were useful for the characterization of ADA assays performed in the presence of human serum. The response signal of ADA19 was apparently decreased in the samples containing human serum regardless of the type of ADA assays. As revealed by the binding analysis, ADA19 broadly binds to human-murine chimeric mAbs and its binding specificity seems to be lower than other clones (). Thus, it is considered that the binding of ADA19 to immunoglobulins or other proteins in human serum interfered with its detection.
In conclusion, we generated human-rat chimeric anti-rituximab mAbs with various biological characteristics and developed a panel of ADAs against rituximab. By using this panel, we revealed the relationship between the size of the mAb/ADA immune complex and FcγR-mediated immune-cell activation, and also experimentally demonstrated the difference in the detectability of ADAs between the ECL immunoassay, SPR assay and BLI assay. Our panel consists of human-rat chimeric mAbs with human IgG1 subclass, whereas ADAs with various immunoglobulin subclasses, including IgG4 and IgM, can be induced in patients, and the detection of ADAs with IgG4 and IgM subclass is often more complicated than the detection of ADAs with IgG1. Thus, substitution of the immunoglobulin subclass of anti-rituximab mAbs to IgG4 and IgM will strengthen the usefulness of our ADA panel for the assessment of assays detecting ADAs.
We are not aware of any study in which the detailed characteristics of patient-derived ADAs against rituximab were analyzed, and we cannot assess whether our recombinant ADAs exhibit similar characteristics to ADAs induced in patients. In addition, no matter how similar recombinant ADAs are to clinical ADAs in their biological functions, they are not the same as the clinical samples, which consist of polyclonal antibodies with various characteristics. Although there are limitations in the use of recombinant ADAs generated by animal immunization, we believe that the panel of control ADAs with diverse repertoire of different binding properties and neutralization activities is a promising tool that may facilitate decision-making on assay selection by the comparison and evaluation of ADA assays. Our approach can be applied to other mAb therapeutics, and will assist in the development and characterization of various assays for immunogenicity assessment.
Materials and methods
Generation of rat anti-rituximab mAbs
Immunization of rats and generation of hybridomas were performed by ITM (Nagano, Japan). Briefly, rituximab F(ab')2 prepared by pepsin digestion of MabThera® was crosslinked by glutaraldehyde and used as an immunogen. WKY rats were immunized with Freund's Complete Adjuvant-conjugated immunogen. After two weeks, the iliac lymph nodes were collected from the immunized rats, followed by separation of lymphocytes and fusion to myeloma. The resultant hybridomas were screened by ELISA, and the hybridomas secreting rat anti-rituximab mAbs were established. The hybridomas were cultured in GIT medium (WAKO) and rat anti-rituximab mAbs were purified from the culture medium by using a HiTrap Protein G HP column (GE Healthcare).
Generation of human-rat chimeric anti-rituximab mAbs
For sequence analysis of the variable regions of rat anti-rituximab mAbs, total RNA was purified from rat hybridomas by using an RNeasy Plus Micro Kit (QIAGEN). cDNA synthesis and 5′-RACE PCR were performed by using a SMARTer RACE 5′/3′ Kit (Clontech) according to the manufacturer's instructions. DNA sequencing was performed by TAKARA BIO (Shiga, Japan). DNA fragments encoding the variable region of the heavy chain and light chain were synthesized by GenScript Japan (Tokyo, Japan), and subcloned into pFUSE-CHIg-hG1 and pFUSE2-CLIg-hk vector (Invivogen), respectively, resulting in the expression vectors of human-rat chimeric anti-rituximab mAbs. The expression and purification of recombinant mAbs were performed as described previously.Citation28, Citation29 Briefly, FreeStyle CHO-S cells (Invitrogen) were transfected with mAb expression vectors by using FreeStyle MAX reagent (Invitrogen) and cultured in FreeStyle Expression Medium (Invitrogen) (37°C, 8% CO2). After 7 days of culture, the culture supernatants were collected by centrifugation, and the recombinant mAbs were purified using HiTrap Protein G HP columns.
Biotinylation of mAbs therapeutics
Rituximab (MabThera®), infliximab (Remicade®), cetuximab (Erbitux®), adalimumab (Humira®), panitumumab (Vectibix®) and ofatumumab (Arzerra®) were purchased from reagent distributors, and biotinylated using an EZ-Link Sulfo-NHS-LC-Biotinylation Kit (Pierce) according to the manufacturer's instructions.
SPR analysis
A Biacore T200 SPR biosensor (GE Healthcare) and Biotin CAPture Kit (GE Healthcare) were used for SPR analysis. The capturing of biotinylated ligands and regeneration of the sensor chip were performed according to the manufacturer's instructions. All measurements were performed at 25°C and HBS-EP+ (GE Healthcare) was used as a running buffer. To evaluate the binding specificity of anti-rituximab mAbs, biotinylated mAb therapeutics (rituximab, infliximab, cetuximab, adalimumab, panitumumab and ofatumumab) were captured on the Sensor Chip CAP, then anti-rituximab mAbs diluted to 1 ug/ml were injected for 90 sec. In the ADA detection assay, anti-rituximab mAbs were serially diluted in running buffer or running buffer containing 10% human serum (Sigma-Aldrich, H4522), and were injected on the sensor chip loaded with biotinylated rituximab for 600 sec. Binding levels (RU) were calculated by using Biacore T200 Software (GE Healthcare).
BLI analysis
BLI analysis was performed by using an Octet RED384 system (Paul ForteBio) and Dip and Read Streptavidin (SA) Biosensors (Paul ForteBio). All experiments were performed at 30°C and 1000 rpm in HBS-EP+ buffer. The epitope binning assay was performed using an in-tandem format. Biotinylated rituximab diluted to 1 µg/ml was loaded on the SA biosensors for 60 sec, and 20 ug/ml anti-rituximab mAbs (saturating Abs: Ab-1) were applied to the biosensors for 600 sec, resulting in the saturated binding to rituximab. Then, 20 µg/ml anti-rituximab mAbs (competing Abs: Ab-2) were applied for 300 sec, and their binding to rituximab was calculated by using Data Analysis HT 9.0 software (Paul Fortebio). The binding levels of Ab-2 in the presence of Ab-1 were normalized to the binding levels in the absence of Ab-1. Cluster analysis was performed by using JMP software (SAS Institute Japan). For the binding kinetics analysis, biotinylated rituximab diluted to 1 µg/ml was loaded onto the SA biosensors for 60 sec, and serially diluted anti-rituximab mAbs were applied to the biosensors for 120 sec followed by a 130-sec dissociation phase. The kinetics parameters (ka, kd and KD) were calculated from the sensorgrams using a 1:1 binding model by Data Analysis 9.0 software (Paul Fortebio). In the ADA detection assay, anti-rituximab mAbs were serially diluted in HBS-EP+ buffer or HBS-EP+ buffer containing 10% human serum, and were applied to the biosensors loaded with biotinylated rituximab for 180 sec, and the binding levels (nm) were calculated using Data Analysis 9.0 software.
Cell-based CD20-binding assay
The human Burkitt lymphoma cell line Raji (JCRB9012) was obtained from the JCRB Cell Bank (NIBIO, Japan) and cultured in RPMI1640 medium (Invitrogen) supplemented with 10% fetal bovine serum. An ECL cell-based assay was performed to evaluate the neutralization activity against CD20-binding. Raji cells suspended in phosphate-buffered saline (PBS; 3 × 104 cells/well) were seeded into a Multi-Array 96-well High Bind Plate (Meso Scale Discovery) and incubated at 37°C for 1 hour. After removing the supernatant, the plates were blocked by adding 3% MSD Blocker A (Meso Scale Discovery) for 1 hour. Mixtures of 0.5 µg/ml biotinylated rituximab, 0.5 µg/ml SULFO-TAG Streptavidin (Meso Scale Discovery) and serially diluted anti-rituximab mAbs were prepared in 1% MSD Blocker A and incubated at room temperature for 1 hour. The pre-incubated mixtures (25 µl/well) were added to each well of the Raji-coated plate and incubated at room temperature for 1 hour. After washing with PBS, MSD Read Buffer T (Meso Scale Discovery) (150 µl/well) was added to each well, and the ECL signal was measured by using MESO QuickPlex SQ120 (Meso Scale Discovery). The percentage of binding activity was calculated by normalizing the signals of anti-rituximab mAbs-treated samples to that of the negative control and plotted against the concentration of anti-rituximab mAbs. The IC50 value of each mAb was calculated by 4-parameter logistic regression (PRISM 6; Graphpad Software).
CDC assay
Mixtures of 50 µg/ml rituximab and the serially diluted anti-rituximab mAbs were prepared in PBS and incubated at 37°C for 30 min. Raji cells were suspended in Opti-MEM (Invitrogen) supplemented with 16% human serum (SIGMA) and seeded into a 96-well plate (1 × 104 cells/80 µl/well). 20 µl of the pre-incubated mixtures were added to each well and incubated at 37°C for 2 hours. Cytotoxicity was measured by using a CytoTox-Glo Cytotoxicity Assay (Promega) according to manufacturer's instructions. The percentage of CDC activity was calculated by normalizing the cytotoxicity of each sample to that of the negative control and plotted against the concentration of anti-rituximab mAbs. The IC50 value of each mAb was calculated by 4-parameter logistic regression.
ADCC assay (FcγRIIIa activation assay)
An FcγRIIIa activation assay was performed as a surrogate for the ADCC assay.Citation28,Citation39 Raji and Jurkat/FcγRIIIa/NFAT-Luc cells were used as target and effector cells, respectively. Mixtures of 1 µg/ml rituximab and serially diluted anti-rituximab mAbs were prepared in PBS and incubated at 37°C for 30 min. Raji and Jurkat/FcγRIIIa/NFAT-Luc cells suspended in Opti-MEM were seeded into a 96-well plate (90 µl/well, the effector:target ratio was 10:1), and then the pre-incubated mixtures were added to each well (10 µl/well), followed by incubation at 37°C for 5 hours. Luciferase activity was measured by using a ONE-Glo Luciferase Assay Reagent (Promega) and Ensight multimode plate reader (PerkinElmer). The luciferase activity was normalized to the negative control, and the percentage of activity was plotted against the concentration of anti-rituximab mAbs. The IC50 value of each mAb was calculated by 4-parameter logistic regression.
Measurement of the size and FcγR activation ability of immune complexes
Mixtures of 100 µg/ml rituximab and 100 µg/ml of anti-rituximab mAbs were prepared in PBS and incubated at 37°C for 30 min. The size distribution of pre-incubated mixtures was measured by DLS analysis at 25°C using a ZetaSizer Nano (Malvern). For measuring FcγR activation by immune complexes, pre-incubated mixtures were added to Jurkat/FcγRIIa/NFAT-Luc cells at a final concentration of 1 µg/ml. After 5 hours of incubation at 37°C, luciferase activity was measured by using ONE-Glo Luciferase Assay Reagent and an Ensight multimode plate reader.
ECL bridging assays for ADA detection
Rituximab was labeled with ruthenium using MSD GOLD SULFO-TAG NHS-Ester Conjugation Packs (Meso Scale Discovery) according to the manufacturer's instructions. Mixtures of 0.5 µg/ml biotinylated rituximab, 0.5 µg/ml ruthenium-labeled rituximab and serially diluted anti-rituximab mAbs were prepared in assay diluent (1% Blocker A in PBS) in the presence or absence of 10% human serum, and incubated at room temperature for 2 hours with shaking. An MSD GOLD 96-well Streptavidin QUICKPLEX Plate (Meso Scale Discovery) was blocked with 3% Blocker A in PBS for 2 hours. After three washings with 0.05% Tween/PBS, 50 µl of the pre-incubated mixtures were added to each well and incubated at room temperature for 1 hour with shaking. The plate was washed with 0.05% Tween/PBS and 150 µl of (2x) MSD Read Buffer T was added to each well, followed by the detection of ECL signals using MESO QuickPlex SQ120.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
supp_data_1424610.tif
Download TIFF Image (1.6 MB)Acknowledgments
This research was partially supported by a grant for Research on Regulatory Science of Pharmaceuticals and Medical Devices from the Japan Agency for Medical Research and development (AMED). We thank Ms. Chizuru Miyama for technical assistance.
Additional information
Funding
References
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16. doi:10.1038/nri2761.
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs. 2012;4:413–5. doi:10.4161/mabs.19931.
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–87. doi:10.1038/nrc3236.
- Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14:296–302. doi:10.1208/s12248-012-9340-y.
- De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28:482–90. doi:10.1016/j.it.2007.07.011.
- Rosenberg AS, Sauna ZE. Immunogenicity assessment during the development of protein therapeutics. J Pharm Pharmacol. 2017. doi:10.1111/jphp.12810.
- Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, Krusemeier M, Kay NE. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin Diagn Lab Immunol. 1994;1:365–72.
- Ducancel F, Muller BH. Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs. 2012;4:445–57. doi:10.4161/mabs.20776.
- Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi:10.1016/j.ymeth.2005.01.001.
- Getts DR, Getts MT, McCarthy DP, Chastain EM, Miller SD. Have we overestimated the benefit of human(ized) antibodies? MAbs. 2010;2:682–94. doi:10.4161/mabs.2.6.13601.
- Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–65. doi:10.4161/mabs.2.3.11641.
- Krishna M, Nadler SG. Immunogenicity to Biotherapeutics – The Role of Anti-drug Immune Complexes. Front Immunol. 2016;7:21. doi:10.3389/fimmu.2016.00021.
- Wang YM, Wang J, Hon YY, Zhou L, Fang L, Ahn HY. Evaluating and Reporting the Immunogenicity Impacts for Biological Products–a Clinical Pharmacology Perspective. AAPS J. 2016;18:395–403. doi:10.1208/s12248-015-9857-y.
- Baert F, Noman M, Vermeire S, Van Assche G, G DH, Carbonez A, Rutgeerts P. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N Engl J Med. 2003;348:601–8. doi:10.1056/NEJMoa020888.
- Krintel SB, Grunert VP, Hetland ML, Johansen JS, Rothfuss M, Palermo G, Essioux L, Klause U. The frequency of anti-infliximab antibodies in patients with rheumatoid arthritis treated in routine care and the associations with adverse drug reactions and treatment failure. Rheumatology (Oxford). 2013;52:1245–53. doi:10.1093/rheumatology/ket017.
- Moots RJ, Xavier RM, Mok CC, Rahman MU, Tsai WC, Al-Maini MH, Pavelka K, Mahgoub E, Kotak S, Korth-Bradley J, et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS One. 2017;12:e0175207. doi:10.1371/journal.pone.0175207.
- Maeda T, Yamada Y, Tawara M, Yamasaki R, Yakata Y, Tsutsumi C, Onimaru Y, Kamihira S, Tomonaga M. Successful treatment with a chimeric anti-CD20 monoclonal antibody (IDEC-C2B8, rituximab) for a patient with relapsed mantle cell lymphoma who developed a human anti-chimeric antibody. Int J Hematol. 2001;74:70–5. doi:10.1007/BF02982552.
- Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639–47.
- Baudouin V, Crusiaux A, Haddad E, Schandene L, Goldman M, Loirat C, Abramowicz D. Anaphylactic shock caused by immunoglobulin E sensitization after retreatment with the chimeric anti-interleukin-2 receptor monoclonal antibody basiliximab. Transplantation. 2003;76:459–63. doi:10.1097/01.TP.0000073809.65502.8F.
- U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH). Assay Development and Validation for Immunogenicity Testing of Therapeutic Protein Products, Guidance for Industry, DRAFT GUIDANCE. 2016.
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on Immunogenicity assessment of therapeutic proteins EMEA/CHMP/BMWP/14327/2006 Rev 1. 2017.
- Lu Y, Khawli LA, Purushothama S, Theil FP, Partridge MA. Recent Advances in Assessing Immunogenicity of Therapeutic Proteins: Impact on Biotherapeutic Development. J Immunol Res. 2016;2016:8141269. doi:10.1155/2016/8141269.
- Partridge MA, Purushothama S, Elango C, Lu Y. Emerging Technologies and Generic Assays for the Detection of Anti-Drug Antibodies. J Immunol Res. 2016;2016:6262383. doi:10.1155/2016/6262383.
- Tatarewicz SM, Mytych DT, Manning MS, Swanson SJ, Moxness MS, Chirmule N. Strategic characterization of anti-drug antibody responses for the assessment of clinical relevance and impact. Bioanalysis. 2014;6:1509–23. doi:10.4155/bio.14.114.
- Boross P, Leusen JH. Mechanisms of action of CD20 antibodies. Am J Cancer Res. 2012;2:676–90.
- Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366:2008–16. doi:10.1056/NEJMct1114348.
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi:10.1038/nri2206.
- Tada M, Ishii-Watabe A, Suzuki T, Kawasaki N. Development of a cell-based assay measuring the activation of FcgammaRIIa for the characterization of therapeutic monoclonal antibodies. PLoS One. 2014;9:e95787. doi:10.1371/journal.pone.0095787.
- Takakura M, Tada M, Ishii-Watabe A. Development of cell-based assay for predictively evaluating the FcgammaR-mediated human immune cell activation by therapeutic monoclonal antibodies. Biochem Biophys Res Commun. 2017;485:189–94. doi:10.1016/j.bbrc.2017.02.050.
- Mytych DT, Barger TE, King C, Grauer S, Haldankar R, Hsu E, Wu MM, Shiwalkar M, Sanchez S, Kuck A, et al. Development and characterization of a human antibody reference panel against erythropoietin suitable for the standardization of ESA immunogenicity testing. J Immunol Methods. 2012;382:129–41. doi:10.1016/j.jim.2012.05.013.
- Wadhwa M, Mytych DT, Bird C, Barger T, Dougall T, Han H, Rigsby P, Kromminga A, Thorpe R, Participants of the Study. Establishment of the first WHO Erythropoietin antibody reference panel: Report of an international collaborative study. J Immunol Methods. 2016;435:32–42. doi:10.1016/j.jim.2016.05.005.
- Shibata H, Nishimura K, Miyama C, Tada M, Suzuki T, Saito Y, Ishii-Watabe A. Comparison of different immunoassay methods to detect human anti-drug antibody using the WHO erythropoietin antibody reference panel for analytes. J Immunol Methods. 2017;452:73–7. doi:10.1016/j.jim.2017.09.009.
- van Schie KA, Wolbink GJ, Rispens T. Cross-reactive and pre-existing antibodies to therapeutic antibodies–Effects on treatment and immunogenicity. MAbs. 2015;7:662–71. doi:10.1080/19420862.2015.1048411.
- van Schouwenburg PA, Kruithof S, Votsmeier C, van Schie K, Hart MH, de Jong RN, van Buren EE, van Ham M, Aarden L, Wolbink G, et al. Functional analysis of the anti-adalimumab response using patient-derived monoclonal antibodies. J Biol Chem. 2014;289:34482–8. doi:10.1074/jbc.M114.615500.
- van Schouwenburg PA, van de Stadt LA, de Jong RN, van Buren EE, Kruithof S, de Groot E, Hart M, van Ham SM, Rispens T, Aarden L, et al. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann Rheum Dis. 2013;72:104–9. doi:10.1136/annrheumdis-2012-201445.
- Rojas JR, Taylor RP, Cunningham MR, Rutkoski TJ, Vennarini J, Jang H, Graham MA, Geboes K, Rousselle SD, Wagner CL. Formation, distribution, and elimination of infliximab and anti-infliximab immune complexes in cynomolgus monkeys. J Pharmacol Exp Ther. 2005;313:578–85. doi:10.1124/jpet.104.079277.
- Kim MS, Lee SH, Song MY, Yoo TH, Lee BK, Kim YS. Comparative analyses of complex formation and binding sites between human tumor necrosis factor-alpha and its three antagonists elucidate their different neutralizing mechanisms. J Mol Biol. 2007;374:1374–88. doi:10.1016/j.jmb.2007.10.034.
- Krayukhina E, Noda M, Ishii K, Maruno T, Wakabayashi H, Tada M, Suzuki T, Ishii-Watabe A, Kato M, Uchiyama S. Analytical ultracentrifugation with fluorescence detection system reveals differences in complex formation between recombinant human TNF and different biological TNF antagonists in various environments. MAbs. 2017;9:664–79. doi:10.1080/19420862.2017.1297909.
- Tada M, Tatematsu K, Ishii-Watabe A, Harazono A, Takakura D, Hashii N, Sezutsu H, Kawasaki N, et al. Characterization of anti-CD20 monoclonal antibody produced by transgenic silkworms (Bombyx mori). MAbs. 2015;7:1138–50. doi:10.1080/19420862.2015.1078054.