
ABSTRACT
Immunoglobulin G–like bispecific antibodies with asymmetric architecture are among the most widely used bispecific antibody formats for diagnostic and therapeutic applications. The primary technical challenge for this format is how to achieve correctly paired assembly of four unique polypeptide chains. Advances in protein engineering and process development are being used to overcome these challenges and are driving a corresponding demand for sensitive analytical tools to monitor and control mispaired species. Here, we report a systematic approach for analysis and characterization of mispairing in asymmetric bispecific antibodies. This approach consists of three orthogonal components, the first of which is a liquid chromatography (LC)-mass spectrometry (MS)–based method to measure the mass of intact antibodies. This method is used for fast analysis of mispairing and requires minimal method development, which makes it an ideal choice for early-stage development. The second component is a hydrophobic interaction chromatography (HIC)–based mispairing method that is suitable for lot release testing. The HIC method is robust and quality control friendly, and offers great linearity, precision, and accuracy. The third component is a two-dimensional LC-MS method for on-line chromatographic peak identification, which not only expedites this task but also reduces the risk of undesirable modifications during conventional fraction collection. These three methods dovetail to form the foundation of a complementary toolbox for analysis and characterization of mispairing in asymmetric bispecific antibodies and provide guidance and support for process development throughout the drug development life cycle.
Introduction
Bispecific antibodies have become widely used formats in recent years for diagnostic and therapeutic applications.Citation1-Citation3 As of 2017, ~ 60 bispecific antibodies were in clinical studies,Citation4 and two have received approval from the US Food and Drug Administration: blinatumomab (Blincyto®; Amgen/Micromet) and emicizumab-kxwh (Hemlibra® Chugai/Genentech). The more than 100 bispecific antibody formats that have been reported in the literatureCitation5,Citation6 can be divided into three categories. The first group comprises bispecific antibodies possess two antigen binding sites connected by a linker, but no Fc region, as exemplified by the bispecific T-cell engagerCitation7-Citation10 format employed for Blincyto. The second group consists of immunoglobulin (IgG)-like bispecific antibodies with asymmetric architecture, in which the two binding arms of the antibody adapt different structures to target different receptorsCitation11 (e.g., Hemlibra). The third class of molecules are appended IgGs with symmetric architecture in which the second binding site is fused to either the IgG heavy or light chain in a symmetrical fashion.Citation12-Citation15
The primary technical challenge for IgG-like bispecific antibodies with asymmetric architecture is how to improve the fidelity of polypeptide chain pairing. One manufacturing process used in past generates a hybrid-hybridoma (quadroma) by fusing two antibody-producing cell lines, which allows the combination of the heavy and light chains of two different antibodies.Citation11,Citation16 This process was used for the production of catumaxomab (Removab®), a bispecific antibody therapeutic approved in the European Union in 2009 that was subsequently withdrawn from the market. The main drawback of the quadroma approach is the high level of product-related variants and impurities expressed with the target product. In addition to the desired form, nine undesirable forms are also generated by this process. Chromatographic purification is required to remove these undesirable species, resulting in a low overall yield.Citation16
Various strategies have been introduced in recent years to improve the efficiency of correct pairing. The first efforts focused on improvement of the heterodimerization between two different heavy chains by using protein engineering techniques, which rely on either steric or electrostatic steering effects. One prominent example is knobs-into-holes (KIH) technology, in which a steric effect is used to promote heavy-chain heterodimerization.Citation17–Citation20 In one study, KIH technology combined with additional sequence mutation to allow additional disulfide bond formation resulted in >95% heterodimer formation.Citation20 Similarly, electrostatic interactions have been used to promote heterodimerization between two heavy chains.Citation21,Citation22 By mutating charged groups in the Fc region or introducing new charged groups in the hinge region, one heavy chain is enriched with positively charged functionality and vice versa. Homodimerization is thus minimized due to electrostatic repulsion, and the desired heterodimerization is maximized due to electrostatic attraction. Near-perfect heterodimerization can be achieved by utilizing this electrostatic steering effect.Citation21 Subsequent efforts to improve fidelity have focused on pairing between cognate heavy and light chains. Technologies introduced to improve the correct heavy chain–light chain pairing include the use of a common light chainCitation20 (which tends to compromise specificity of each binding arm and limit the diversity of bispecific antibodies); CrossMab,Citation23 in which the light chain of one antigen-binding fragment (Fab) arm is exchanged for the anti-human IgG (Fd) region of the corresponding heavy chain; and DuetMab,Citation24,Citation25 in which the correct heavy chain–light chain pairing is achieved by replacing the native disulfide bond in one of the Fab arms with an engineered disulfide bond.
The percentage of correctly paired bispecific antibodies is significantly improved by utilizing these protein engineering technologies. The low levels of mispaired species are typically removed by chromatography purifications.Citation26 To simplify the purification process and make it more conducive to platform development, the two unique light chains can be engineered to possess sequence homology from different subfamilies (kappa and lambda). Affinity matrices such as LambdaFabSelect (LFS) and KappaSelect can then be used to selectively capture asymmetric bispecific antibodies that contain at least one light chain within the target subfamily.Citation26,Citation27
Advances in protein engineering and process development have driven a corresponding demand for sensitive analytical tools to monitor and control mispairing. Here, we report a systematic approach for characterization and analysis of mispairing in asymmetric bispecific antibodies. This approach consists of three orthogonal components: 1) a fast LC-MS method to assess mispaired species during early process development, such as clone selection and purification; 2) a robust and quality control (QC)–friendly method for lot release testing of mispaired species; and 3) a two-dimensional (2D) liquid chromatography (LC)–mass spectrometry (MS) method for chromatographic peak identification. This three-step approach provides a complementary toolbox for analysis and characterization of mispairing in asymmetric bispecific antibodies that can be used to support process development.
Results
Fast LC-MS based method to assess mispaired species during early process development
The development of a product-specific chromatography- or electrophoresis-based method to monitor mispaired species is often a resource- and time-consuming task. However, because biopharmaceutical analytical and process development cycles often run in parallel, broadly applicable techniques requiring little development are needed. MS-based measurement of the mass of intact antibodies (referred to herein as “intact mass measurement”), coupled with reversed-phase LC separation, can serve as a platform for the development of an analytical method for early-stage development. As shown in , many mispaired bispecific antibody forms are substantially different from the target form, and can therefore be expected to be well separated by reversed-phase LC. Even when LC selectivity is not achieved, intact-mass measurement can be used to distinguish mispaired species from the correctly paired product because their masses usually differ by hundreds to thousands of daltons (). Due to the nature of the mass spectrometer, this LC-MS method is used mainly for qualitative purposes and its quantitative results are not suitable for use in a Good Manufacturing Practice (GMP) QC environment. Nevertheless, during the early development cycle, this LC-MS–based method provides critical guidance for process development. To illustrate this, we describe the development of a bispecific antibody purification process as an example to demonstrate how this LC-MS–based method can be used to guide process development.
Scheme I. Correctly paired bispecific antibody and potential mispaired species.
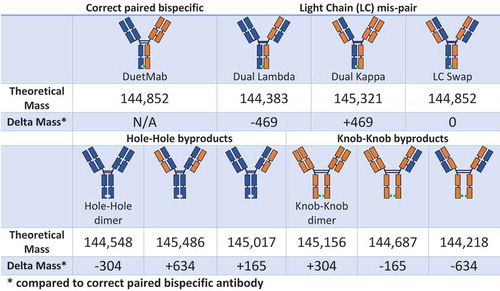
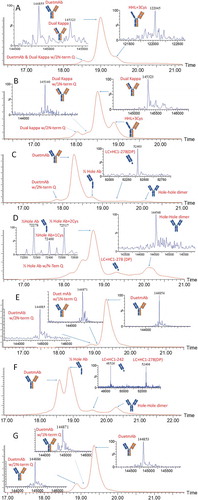
Bis-A (DuetMabCitation24,Citation25) is an asymmetric bispecific antibody with a KIH Fc design to promote correct heavy-chain heterodimerization. Bis-A contains two light chains, a lambda light chain and a kappa light chain. shows the initial purification process design for Bis-A. The harvest cell culture fluid (HCCF) was first captured with protein A, yielding the protein A pool sample. LC-MS analysis detected three chromatographic peaks for the protein A pool (). Intact mass measurement of the main peak indicated coelution of two species: the correctly paired bispecific antibody product and a mispaired species with dual kappa light chains. This is a good example of the method’s capability to elucidate mispaired species even when they coelute with the desired product. Intact-mass measurement of the pre-peak () indicated that it was the N-terminal Q form, rather than the pyro-Q form, of the main peak (including both correctly paired and dual kappa species). The post-peak was determined to be a product fragment missing the lambda light chain (heavy-heavy-light [HHL]). Since neither major impurity species contained the lambda light chain, LFS resin was chosen as the main affinity capture step to selectively remove these impurities.
Figure 1. LC-MS analysis of Bis-A samples obtained from purification processes as shown in . (A) Protein A pool; (B) LFS flow-through/protein A; (C) LFS elute; (D) kappa flow-through; (E) kappa elute; (F) polishing step washing; (G) polishing step elute. Detailed mass data for each sample are provided in Supplemental Table S1.
Scheme II. Downstream process used to purify bispecific antibodies.
and C show the results of LC-MS analysis for the LFS flow-through and elute samples, respectively. Because the LFS flow-through samples contained many process- and host-related impurities, they were further processed by a secondary protein A purification step to remove these host impurities. As expected, the LFS flow-through–protein A sample contained dual kappa light-chain mispaired species and an HHL fragment (a missing lambda light chain) (). shows the major species that were detected for the LFS elute sample. As expected, the major impurities in HCCF (a dual kappa and HHL fragment) were not present in the LFS elute sample. In addition to the desired bispecific antibody and its N-terminal Q form, hole-hole dimer, hole half-antibody, and hole half-antibody fragments were also detected in the LFS elute samples. A hole-hole dimer was also detected in the HCCF–protein A sample, but its intensity was low due to signal suppression by other species. Hole half-antibody and its fragments were not detected in the HCCF–protein A sample, possibly because these species have incomplete Fc regions and hence no affinity for protein A. Because hole-hole dimer, hole half-antibody, and hole half-antibody fragments did not contain a kappa light chain, KappaSelect resin (GE Healthcare, Chicago, IL) was used for the next purification step to remove these impurities.
and E show results of the LC-MS analysis for the kappa flow-through and kappa elute samples, respectively. Hole-hole dimer, hole half-antibody, and hole half-antibody fragments were enriched in the kappa flow-through sample. The final product from the kappa elute pool contained the correctly paired bispecific antibody product and its N-terminal Q species. The finding that there were no other mispaired species or impurities detected in this final product demonstrates the success of the initial purification process scheme.
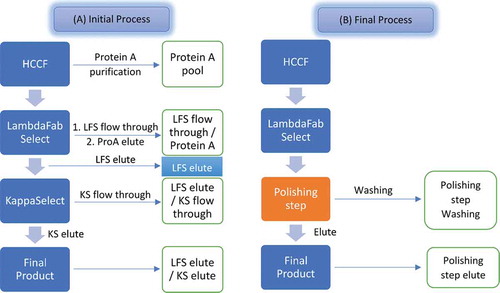
One drawback of the initial purification process was its reliance on two expensive affinity resins. As a consequence, we sought a less costly purification step that could replace the second affinity step, which utilized KappaSelect. In the final process, a polishing step of mixed-mode chromatography replaced the secondary affinity step used in the initial process (). To improve the performance of the polishing step, a washing operation was included. shows the LC-MS analysis results for the sample obtained from the washing operation. All impurities in the LFS elute sample, such as hole-hole dimer, hole half-antibody, and hole half-antibody fragments, were enriched in the wash fraction, demonstrating the effectiveness of this step. Although ~ 2–3% of the desired product was also detected in this wash fraction, a low amount of loss of the desired bispecific antibody was deemed acceptable, considering the benefits of removing the secondary affinity step. shows the LC-MS analysis results obtained for the product when the final process was used. Only the desired bispecific antibody and its N-terminal Q species were detected, demonstrating the success of the final purification process scheme.
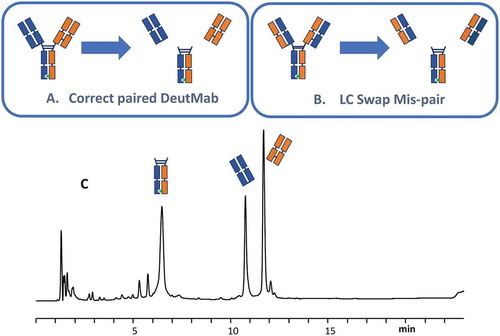
One limitation of the MS-based method for measuring the mass of intact antibodies is that it cannot detect bispecific antibodies with swapped light chains because the swapping of the light chains introduces no mass change from the desired product (). To address this limitation, an LC-MS subunit analysis experiment was performed (). The final bispecific antibody product was incubated with the enzyme GingisKHAN, which specifically cuts between the K and T residues in the sequence (…KSCDK/THTCPPCP…) above the hinge region. After digestion, two different Fabs and one Fc were generated. The two Fabs generated from the species in which the light chain was swapped were different from the two Fabs from the correctly paired product. shows the results of LC-MS analysis of the final process product after GingisKHAN digestion. The two Fabs detected by LC-MS matched the Fab generated from the correctly paired bispecific antibody, which confirmed the correct structure for the final products.
Figure 2. LC-MS subunit analysis. (A) Subunits generated from correctly paired DuetMab; (B) subunits generated from LC swap mispair species; (C) LC-MS analysis results.
HIC based method for lot release testing of mispaired species
A convenient method is required for routine release testing to monitor the level of mispaired species in each manufacturing lot. The LC-MS method just discussed is not designed for precise quantitative measurements and therefore is not appropriate in a QC environment. Hence, a more robust and QC-friendly chromatography-based method is needed to support lot release. Charge-basedCitation28 or mixed-modeCitation29 chromatography methods have been reported to separate and quantify mispaired species. The results of our studies suggest that the hydrophobicity difference between mispaired and correctly paired bispecific antibodies is significant and can be exploited to develop a hydrophobic interaction chromatography (HIC)–based mispairing assay. Here we describe the use of a different DuetMab,Citation24,Citation25 Bis-B, as an example to illustrate the development of the HIC method.
The first step in the development of the HIC method was to select the appropriate stationary phase. Different columns with ether, butyl, phenyl, and alkyl amide stationary phases were screened. A MAbPac HIC-10 column showed the best performance (mispaired species were very well separated from correctly paired bispecific antibodies) and was selected for further optimization. Since the separation of proteins in HIC is mainly based on the salting-out principle,Citation30 mobile-phase pH, salt type, and concentration are key parameters for HIC performance.Citation31,, demonstrates the performance of the HIC method with the use of different salts. Sodium chloride provided very poor resolution and was eliminated from further investigation. No resolution between the main peak and the pre-peak was observed with the use of ammonium sulfate, which was also ruled out. The best performance was obtained with sodium sulfate, which was selected for further development of the HIC method. shows the performance of the HIC−10 column when mobile phases with different pH levels were used. The performance of the HIC−10 column was not optimal at pH ≤5.5; a complete loss of resolution between the main peak and the pre-peak occurred at pH 5.0 and a partial loss occurred at pH 5.5 (., Superior resolution was obtained at pH 6.0, 6.5, and 7.0, demonstrating the robustness of this method in a wide pH range. The midpoint, pH 6.5, was selected as the operating pH. shows the HIC performance at different initial salt concentrations. Similar resolution was obtained with 1.0 M and 2.0 M salt. To reduce both the column back pressure and wear on the instrument, a low salt concentration of 1.0 M was selected for the optimized method.
Figure 3. HIC method development for Bis-B. (A) Salt screening; (B) pH screening; (C) salt concentration screening. Seven peaks were separated in the final method, which are labeled in panel C. The HIC profile for final purified materials is also shown in panel C.
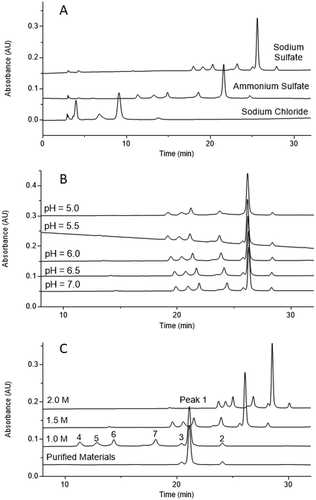
The robustness of the optimized method was tested by using multiple HIC column lots and high-performance LC instruments. The optimized method was qualified according to International Council on Harmonisation Q2 guidelines.Citation32 The results demonstrated the method’s robustness, linearity, accuracy, and precision. The method qualification results are shown in . Excellent linearity (r2 = 0.9954) was obtained, in the range of 1.1–5.1% mispaired species (). The repeatability (n = 6) and intermediate precision (n = 6; different analyst, column, instrument, and day) were 2.0% and 5.7%, respectively. A spike/recovery study was conducted to evaluate the accuracy of this method, and the recovery ranged between 80% and 90%.
Table 1. Summary of HIC method qualification results.
2D LC-MS–based analytical tools for chromatographic peak identification
After development of the chromatography-based method to monitor bispecific antibody mispairing, our next objective was to identity each chromatographic peak to form the basis of an appropriate specification for mispaired species. The traditional approach to this task involves fractionation of each chromatographic peak, followed by characterization with MS. The drawbacks of this approach include the labor intensiveness of the process and the risk of introducing undesirable modifications during the fraction collection and subsequent sample preparation steps. To expedite chromatographic peak identification and reduce the risk of undesirable modifications, we developed an online 2D LC-MS method that included two separate experiments.
In the first experiment, the peak identified by one-dimensional (1D) HIC was trapped by an online solid-phase extraction cartridge, desalted, and eluted to the mass spectrometer for intact mass measurements. In the second experiment, the peak from the 1D HIC was trapped with an online solid-phase extraction cartridge in a manner similar to that used in the first experiment. The trapped proteins were reduced in the trapping cartridge by conditioning with 2.5 mM tris[2-carboxyethyl]phosphine (TCEP) in 0.1% trifluoroacetic acid (TFA) (aqueous) as a reducing solvent. The reduced proteins were then desalted and subjected to 2D reversed-phase separation to resolve the heavy-chain and light-chain subunits. By combining the intact and subunit masses obtained from the two 2D LC-MS experiments, the mispaired species, resolved by 1D HIC, could be identified with a high degree of confidence. The peak identifications for the HIC separation () are used to illustrate this concept.
shows the results of intact mass measurement (the first experiment) for peak 1 to peak 7 from HIC (). shows the subunit analysis data obtained from the second 2D LC-MS experiment for the corresponding peaks. The observed masses were compared with the theoretical masses of proposed structures to confirm their identities.
Figure 4. Intact mass measurements for Bis-B from the 2D LC-MS experiment for (A) peak 1, (B) peak 2, (C) peak 4, (D) peak 5, (E) peak 6, and (F) peak 7 from the HIC method (, final method).
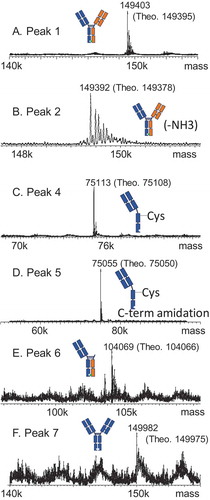
Figure 5. Subunit analysis data for Bis-B from the 2D LC-MS experiment for (A) peak 1, (B) peak 2, (C) peak 4, (D) peak 5, (E) peak 6, and (F) peak 7 from the HIC method (, final method). Detailed mass data for each peak are provided in Supplemental Table S2.
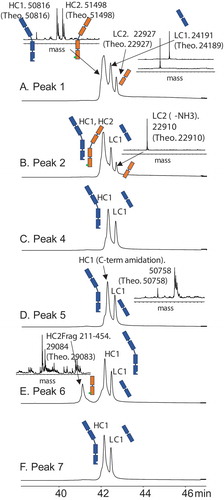
Peak 1 was the main peak in HIC. In the reducing experiment, three subunit component peaks were observed in the 2D reversed-phase chromatography results (). MS results indicated that two species were coeluted in the first peak, a hole heavy chain and a knob heavy chain. The second and third peaks were a kappa light chain and a lambda light chain, respectively. Combining all four subunit components in peak 1 generated an antibody with a mass of 149,403 Da, which was a good match with the intact mass measurement for peak 1 (149,395 Da; ). The most probable connection pattern for peak 1 was the desired correct pairing. Another possible connection that could generate the same intact mass measurement was the light-chain swap discussed previously, which was ruled out by the LC-MS subunit analysis experiment described previously. Peak 1 was therefore identified as the correctly paired bispecific antibody.
Peak 2 was the post-peak of the main peak. Like peak 1, four subunit components were detected in the reducing experiment for peak 2 (). Peak 1 and peak 2 had an identical hole heavy chain, knob heavy chain, and kappa light chain, but differed in the mass of the lambda light chain, which was 17 Da lighter in peak 2 due to the succinimide intermediate formed in its lambda light chain. Peak 2 was therefore identified as the correctly paired bispecific antibody with succinimide in the lambda light chain.
Peak 3 was the pre-peak of the main peak and was identified by traditional fraction collection followed by peptide map analysis (data not shown). Peak 3 was identified as the correctly paired antibody with deamidation in the lambda light chain, as its deamidation site was the same amino acid as the succinimide site in peak 2. The main reason why peak 3 was not identified by 2D LC-MS was the small mass difference of only 1 Da, corresponding to deamidation. The 2D LC-MS method in this study had a mass accuracy of larger than 1 Da, and therefore could not unambiguously detect a mass difference this small.
Two subunit component peaks were observed for peak 4 (), a hole heavy chain and a kappa light chain. Intact mass measurement indicated the mass of peak 4 to be 75,113 Da (), and connecting the hole heavy chain and the kappa light chain generated a mass of 74,989 Da (hole half-antibody). The difference between the hole half-antibody and the intact mass measurement was 124 Da, which was probably caused by cysteinylation (5-Da difference from the theoretical mass). The structure of peak 4 was thus identified as a cysteinylated hole half-antibody.
Similarly, two subunit component peaks were detected for peak 5 (). The kappa light chains were identical for both peak 5 and peak 4, whereas the hole heavy chain for peak 5 was 58 Da less than that of peak 4. This difference was due to the C-terminal amidation formed in the hole heavy chain in peak 5. Intact mass measurement of peak 5 was 75,055 Da, which was also 58 Da less than that of peak 4 (). The structure of peak 5 was thus identified as a cysteinylated hole half-antibody with heavy-chain C-terminal amidation.
Three subunit component peaks were observed for peak 6 in the reducing experiment (). The first subunit component peak was unique and was observed only for peak 6. The mass of this unique component peak was 29,084 Da, and its mass profile indicated that it was glycosylated (). Because only heavy chain had a glycosylation site, this unique component was possibly a heavy-chain fragment. According to the detected mass, this unique component could be identified as the knob heavy-chain Fc fragment N211-G454 (theoretical mass of 29,083 Da vs observed mass of 29,084 Da). The clip site for this knob heavy-chain Fc fragment was above the hinge region, and it still had two hinge cysteines to link to the other heavy chain before it was reduced. The other two subunit component peaks observed for peak 6 were a hole heavy chain and a kappa light chain (). Because the intact mass measurement of peak 6 was 104,069 Da () and combining all three components generated a protein subunit with a mass of 104,066 Da, the structure of peak 6 was identified as a hole half-antibody (a hole heavy chain and a kappa light chain) connected to the knob heavy-chain fragment N211-G454 through hinge disulfide bonds.
Peak 7 had two subunit component peaks detected in the reducing experiment, which were identified as a hole heavy chain and a kappa light chain (). These results were identical to those for peak 4, which was identified as a hole half-antibody. The difference between peak 7 and peak 4 was determined by intact mass measurement, which showed that peak 7 was 149,982 Da () and peak 4 was 75,113 Da (). Since the hole heavy chain and the kappa light chain were the only two components detected for peak 7, and it was approximately double the size of peak 4, the structure of peak 7 was identified as a hole-hole dimer.
Discussion
We report a systematic approach for the characterization and analysis of mispaired species in bispecific antibodies with asymmetric architecture. This approach includes three steps, the first of which is an LC-MS–based method to measure the mass of intact antibodies that can be used to support early process development, such as clone selection and purification. The advantage of this generalized method is its fast analysis and minimal requirement for method development, which makes it an ideal choice for early-stage development. With the improvement in the resolution power of mass spectrometry, mispaired species can be characterized with better accuracy and sensitivity even when mass differences are small.Citation33
The second step is a robust and QC-friendly method for lot release testing. The hydrophobicity difference between the correctly paired and mispaired bispecific antibodies was exploited with an HIC-based assay for GMP lot release. The capability to resolve different antibodies is a prerequisite for a generalized separation approach for mispaired bispecific antibodies. Hydrophobicity is one of the most differentiating characteristics among different monoclonal antibodies (mAbs). In fact, HIC is often employed in a high-throughput fashion to triage candidates during lead selection and developability studies. Fekete et al. have shown the wide retention time range of 17 commercial therapeutic mAbs, fusion proteins, and ADCs by using a generic fast HIC method.Citation34 In that study, only two cases of coelution were observed. The isoelectric point is another product-specific characteristic that may be used to resolve different proteins. However, the inherent charge variants would greatly reduce the separation window, and hence limit the general application of charge-based methods for the separation of different mAbs. During our internal method evaluation, using other projects in our pipeline, we found HIC to be applicable in many more cases than the charge-based method.
The third step is a convenient 2D LC-MS analytical tool for efficient identification of chromatographic peaks. To expedite peak identification, an online 2D LC-MS method was developed and applied to elucidate the mispaired species that were resolved by the HIC method. It is clear that the 2D LC-MS approach should be equally applicable for the peak characterization of other separation modes.
In summary, the three-step systematic approach presented here forms a complementary toolbox for analysis and characterization of mispaired species from asymmetric bispecific antibodies and provides guidance and support for process development throughout the drug development life cycle.
Materials and methods
Materials
The bispecific antibodies used for this study, Bis-A and Bis-B, were generated using Chinese hamster ovary mammalian cells at Medimmune (Gaithersburg, MD). The design and production of these bispecific antibodies have been disclosed previously.Citation24,Citation25 LC and LC-MS grade solvents were purchased from EMD (Billerica, MA). Chemical reagents, including TFA, formic acid, sodium phosphates, sodium sulfate, and TCEP, were purchased from Sigma-Aldrich (St. Louis, MO).
LC-MS method for measurement of intact-antibody mass
LC-MS measurements of the mass of intact antibodies were acquired by using an ultra–high-performance LC system and a Synapt G2 mass spectrometer (Waters, Milford, MA). A PLRP-S column (4,000 Å, 8 µm, 150 × 2.1 mm; Agilent, Santa Clara, CA) was used for reverse-phase separation. Mobile phases A and B were 0.05% TFA in water and acetonitrile, respectively. Mass spectra were collected at a mass-to-charge ratio range of 800–4,500. Molecular mass was determined by deconvolution of the mass spectra data, using the MaxEnt I software package.
LC-MS analysis of antibody subunits
Bispecific antibody subunits were generated by using GingisKHAN enzyme (Genovis) under manufacturer-recommended conditions and were analyzed by LC-MS, using a BEH C4 column (1.7 µm, 2.1 × 150 mm; Waters). Mobile phases were aqueous/acetonitrile with 0.1% TFA.
Hydrophobic interaction chromatography
The optimized HIC method was performed with a MAbPac HIC-10 column (4.6 × 250 mm, 5 µm; Thermo Fisher Scientific). Mobile phase A consisted of 50 mM sodium phosphate, 1 M sodium sulfate, pH 6.5; mobile phase B was 50 mM sodium phosphate, pH 6.5. The column temperature was maintained at 30 °C, and the flow rate was 1.0 mL/min with a gradient from 0% B to 100% B in 30 min.
2D LC-MS method for measurement of intact-antibody mass
The experiments were conducted on an ultra–high-performance LC system (Waters). The same HIC conditions described above were used for the 1D separation, and a six-port, two-position valve was used for heart cutting. The cuts were trapped in an AdvancBio Desalting-RP cartridge (2.1 × 12.5 mm; Agilent) and subjected to 2D desalting with a 300 Diphenyl RRHD column (1.8 µm, 2.1 × 100 mm; Agilent) before MS analysis with a Xevo G2-SX quadrupole time-of-flight mass spectrometer (Waters).
2D LC-MS for online-reduced subunit MS
The experiments were conducted on a 2D LC system (Agilent). The same HIC conditions as described above were used for the 1D separation, and a 1290 2D Valve G1170A (Agilent) was used for heart cutting. The cuts were trapped in an AdvancBio Desalting-RP cartridge (Agilent), and the trapped proteins were reduced in the cartridge for 10 min with 0.1% TFA–2.5 mM TCEP solution at 0.3 mL/min. The reduced protein was injected into a 300 Diphenyl RRHD column (Agilent) for the 2D separation before MS analysis with a 6530 quadrupole time-of-flight mass spectrometer (Agilent).
Abbreviations
| 1D | = | one-dimensional |
| 2D | = | two-dimensional |
| Fab | = | antigen-binding fragment |
| Fc | = | fragment crystallizable |
| GMP | = | Good Manufacturing Practice |
| HCCF | = | harvest cell culture fluid |
| HHL | = | heavy-heavy-light [chain] |
| HIC | = | hydrophobic interaction chromatography |
| IgG | = | immunoglobulin G |
| KIH | = | knob-into-hole |
| LC | = | liquid chromatography |
| LFS | = | LambdaFabSelect |
| MS | = | mass spectrometry |
| QC | = | quality control |
| TFA | = | trifluoroacetic acid |
| UPLC | = | ultra-performance liquid chromatography |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Word (85.4 KB)Acknowledgments
The authors thank Chunling Wa and Sri Hari Raju Mulagapati for their support in some of these experiments. The author also thank Deborah Shuman for help polishing this article.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Sheridan C. 2015. Amgen’s bispecific antibody puffs across finish line. Nat Biotechnol. 33:219–221. doi:10.1038/nbt0315-219.
- Dhimolea E, Reichert JM. 2012. World bispecific antibody summit, September 27–28,2011, Boston, MA. mAbs. 4:4–13. doi:10.4161/mabs.4.1.18821.
- Garber K. 2014. Bispecific antibodies rise again. Nat Rev Drug Discov. 13:799–801. doi:10.1038/nrd4478.
- Strohl WR. 2018. Current progress in innovative engineered antibodies. Protein Cell. 9:86–120. doi:10.1007/s13238-017-0457-8.
- Kontermann RE, Brinkmann U. 2015. Bispecific antibodies. Drug Discov Today. 20:838–847. doi:10.1016/j.drudis.2015.02.008.
- Brinkmann U, Kontermann RE. 2017. The making of bispecific antibodies. mAbs. 9:182–212. doi:10.1080/19420862.2016.1268307.
- Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. 2005. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 10:1237–1244. doi:10.1016/S1359-6446(05)03554-3.
- Baeuerle PA, Reinhardt C. 2009. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 69:4941–4944. doi:10.1158/0008-5472.CAN-09-0547.
- Huehls AM, Coupet TA, Sentman CL. 2015. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 93:290–296. doi:10.1038/icb.2014.93.
- Nagorsen D, Kufer P, Baeuerle PA, Bargou R. 2012. Blinatumomab: a historical perspective. Pharmacol Ther. 136:334–342. doi:10.1016/j.pharmthera.2012.07.013.
- Chelius D, Ruf P, Gruber P, Ploscher M, Liedtke R, Gansberger E, Hess J, Wasiliu M, Lindhofer H. Structural and functional characterization of the trifunctional antibody catumaxomab. mAbs. 2;2010:309–319.
- Coloma MJ, Morrison SL. 1997. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 15:159–163. doi:10.1038/nbt0297-159.
- Dimasi N, Gao C, Fleming R, Woods RM, Yao XT, Shirinian L, Kiener PA, Wu H. 2009. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J Mol Biol. 393:672–692. doi:10.1016/j.jmb.2009.08.032.
- Croasdale R, Wartha K, Schanzer JM, Kuenkele K-P, Ries C, Mayer K, Gassner C, Wagner M, Dimoudis N, Herter S, et al. 2012. Development of tetravalent IgG1 dual targeting IGF-1R-EGFR antibodies with potent tumor inhibition. Arch Biochem Biophys. 526:206–218. doi:10.1016/j.abb.2012.03.016.
- Yazaki PJ, Lee B, Channappa D, Cheung CW, Crow D, Chea J, Poku E, Li L, Andersen JT, Sandlie I, et al. A series of anti-CEA/anti-DOTA bispecific antibody formats evaluated for pre-targeting: comparison of tumor uptake and blood clearance. Protein Eng Des Sel. 2013;26:187–193. doi:10.1093/protein/gzs096.
- Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 155;1995:219–225.
- Ridgway JB, Presta LG, Carter P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Engineering. 9;1996:617–621.
- Atwell S, Ridgway JB, Wells JA, Carter P. 1997. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 270:26–35. doi:10.1006/jmbi.1997.1116.
- Shatz W, Chung S, Li B, Marshall B, Tejada M, Phung W, Sandoval W, Kelley RF, Scheer JM. 2013. Knobs-into-holes antibody production in mammalian cell lines reveals that asymmetric afucosylation is sufficient for full antibody-dependent cellular cytotoxicity. mAbs. 5:872–881. doi:10.4161/mabs.26307.
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. 1998. An efficient route to human bispecific IgG. Nat Biotechnol. 16:677–681. doi:10.1038/nbt0798-677.
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285:19637–19646. doi:10.1074/jbc.M110.117382.
- Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol. 2012;420:204–219. doi:10.1016/j.jmb.2012.04.020.
- Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, Totpal K, Wong A, Lee CV, Stawicki S, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20:472–486. doi:10.1016/j.ccr.2011.09.003.
- Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall’Acqua WF. 2015. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs. 7:461–469. doi:10.1080/19420862.2015.1022695.
- Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs. 2015;7:377–389. doi:10.1080/19420862.2015.1007816.
- Fischer N, Elson G, Magistrelli G, Dheilly E, Fouque N, Laurendon A, Gueneau F, Ravn U, Depoisier JF, Moine V, et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat Commun. 2015;6:6113. doi:10.1038/ncomms7113.
- Eifler N, Medaglia G, Anderka O, Laurin L, Hermans P. 2014. Development of a novel affinity chromatography resin for platform purification of lambda Fabs. Biotechnol Prog. 30:1311–1318. doi:10.1002/btpr.1958.
- Gramer MJ, Van Den Bremer ET, Van Kampen MD, Kundu A, Kopfmann P, Etter E, Stinehelfer D, Long J, Lannom T, Noordergraaf EH, et al. Production of stable bispecific IgG1 by controlled Fab-arm exchange: scalability from bench to large-scale manufacturing by application of standard approaches. mAbs. 2013;5:962–973. doi:10.4161/mabs.26233.
- Yang X, Zhang Y, Wang F, Wang LJ, Richardson D, Shameem M, Ambrogelly A. 2015. Analysis and purification of IgG4 bispecific antibodies by a mixed-mode chromatography. Anal Biochem. 484:173–179. doi:10.1016/j.ab.2015.06.014.
- Queiroz JA, Tomaz CT, Cabral JM. Hydrophobic interaction chromatography of proteins. J Biotechnol. 87;2001:143–159.
- Rodriguez-Aller M, Guillarme D, Beck A, Fekete S. 2016. Practical method development for the separation of monoclonal antibodies and antibody-drug-conjugate species in hydrophobic interaction chromatography, part 1: optimization of the mobile phase. J Pharm Biomed Anal. 118:393–403. doi:10.1016/j.jpba.2015.11.011.
- International Council on Harmonisation. ICH harmonised tripartite guideline: validation of analytical procedures. Q2. Switzerland: Geneva; 2005. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf.
- Yin Y, Han G, Zhou J, Dillon M, McCarty L, Gavino L, Ellerman D, Spiess C, Sandoval W, Carter PJ. 2016. Precise quantification of mixtures of bispecific IgG produced in single host cells by liquid chromatography-Orbitrap high-resolution mass spectrometry. mAbs. 8:1467–1476. doi:10.1080/19420862.2016.1232217.
- Fekete S, Veuthey J-L, Beck A, Guillarme D. 2016. Hydrophobic interaction chromatography for the characterization of monoclonal antibodies and related products. J Pharm Biomed Anal. 130:3–18. doi:10.1016/j.jpba.2016.04.004.