
ABSTRACT
For the past 10 years, the annual ‘Antibodies to watch’ articles have provided updates on key events in the late-stage development of antibody therapeutics, such as first regulatory review or approval, that occurred in the year before publication or were anticipated to occur during the year of publication. To commemorate the 10th anniversary of the article series and to celebrate the 2018 Nobel Prizes in Chemistry and in Physiology or Medicine, which were given for work that is highly relevant to antibody therapeutics research and development, we expanded the scope of the data presented to include an overview of all commercial clinical development of antibody therapeutics and approval success rates for this class of molecules. Our data indicate that: 1) antibody therapeutics are entering clinical study, and being approved, in record numbers; 2) the commercial pipeline is robust, with over 570 antibody therapeutics at various clinical phases, including 62 in late-stage clinical studies; and 3) Phase 1 to approval success rates are favorable, ranging from 17–25%, depending on the therapeutic area (cancer vs. non-cancer). In 2018, a record number (12) of antibodies (erenumab (Aimovig), fremanezumab (Ajovy), galcanezumab (Emgality), burosumab (Crysvita), lanadelumab (Takhzyro), caplacizumab (Cablivi), mogamulizumab (Poteligeo), moxetumomab pasudodox (Lumoxiti), cemiplimab (Libtayo), ibalizumab (Trogarzo), tildrakizumab (Ilumetri, Ilumya), emapalumab (Gamifant)) that treat a wide variety of diseases were granted a first approval in either the European Union (EU) or United States (US). As of November 2018, 4 antibody therapeutics (sacituzumab govitecan, ravulizumab, risankizumab, romosozumab) were being considered for their first marketing approval in the EU or US, and an additional 3 antibody therapeutics developed by Chinese companies (tislelizumab, sintilimab, camrelizumab) were in regulatory review in China. In addition, our data show that 3 product candidates (leronlimab, brolucizumab, polatuzumab vedotin) may enter regulatory review by the end of 2018, and at least 12 (eptinezumab, teprotumumab, crizanlizumab, satralizumab, tanezumab, isatuximab, spartalizumab, MOR208, oportuzumab monatox, TSR-042, enfortumab vedotin, ublituximab) may enter regulatory review in 2019. Finally, we found that approximately half (18 of 33) of the late-stage pipeline of antibody therapeutics for cancer are immune checkpoint modulators or antibody-drug conjugates. Of these, 7 (tremelimumab, spartalizumab, BCD-100, omburtamab, mirvetuximab soravtansine, trastuzumab duocarmazine, and depatuxizumab mafodotin) are being evaluated in clinical studies with primary completion dates in late 2018 and in 2019, and are thus ‘antibodies to watch’. We look forward to documenting progress made with these and other ‘antibodies to watch’ in the next installment of this article series.
Introduction
The rise in the importance of monoclonal antibody (mAb) therapeutics to medical care over the past 30 years has been extraordinary. During this time, a plethora of scientific and technological advances both large and small have facilitated the discovery and development of mAb therapeutics, over 80 of which have been granted marketing approvals. In 2018, scientists responsible for two advances critical to antibody discovery, phage display and immune checkpoint modulation, were awarded Nobel prizes. George P. Smith and Sir Gregory P. Winter were jointly awarded Nobel Prizes in Chemistry for phage display of peptides and antibodies.Citation1 George Smith first described the use of phage display in 1985,Citation2 and the proof of concept for phage-displayed peptide libraries in 1990.Citation3 Also in 1990, Sir Gregory Winter and colleagues reported the display of a folded and fully functional antibody fragment on filamentous phage. Since then, this technology has been used for research and development of antibodies by organizations located world-wide,Citation4,Citation5 yielding over 80 antibodies that entered clinical study. Of these, more than 10 have been granted marketing approvals. In 2002, adalimumab became the first mAb therapeutic derived from phage display to be granted a marketing approval. It has since become the most successful mAb on the market, and is currently prescribed for a wide variety of immune-mediated disorders (rheumatoid arthritis, juvenile rheumatoid arthritis, Crohn’s disease, psoriatic arthritis, psoriasis, axial spondyloarthritis, ulcerative colitis, uveitis, hidradenitis suppurativa and Behçet syndrome).
Compared to phage display, the discovery of immune system checkpoints in the 1990s appeared to be less directly related to the field of antibody research and development, but it is arguably now equally important. In acknowledgement of the value of this work, the 2018 Nobel Prize in Physiology or Medicine was awarded to James P. Allison and Tasuku Honjo for their discovery of cancer therapy by inhibition of negative immune regulation.Citation6 Tasuku Honjo and colleagues first described the programmed cell death 1 (PD-1) protein in 1992,Citation7 and James Allison and colleagues reported in 1996Citation8 that blocking CTLA-4’s inhibitory effects improved immune responses directed toward tumor cells. Since then, the number of proteins known to function as either stimulatory or inhibitory checkpoints of the immune system has dramatically expanded,Citation9 and numerous antibody therapeutics targeting PD-1 (cemiplimab, nivolumab, pembrolizumab), the ligand for PD-1 (PD-L1; durvalumab, avelumab, atezolizumab) or CTLA-4 (ipilimumab) have been granted marketing approvals. Because these therapeutics act by enhancing the immune system response rather than targeting a tumor antigen, they are used to treat many types of cancers, including melanoma, non-small-cell lung cancer, renal cell cancer, urothelial carcinoma, hepatocellular cancer, gastric cancer, colorectal cancer, head and neck cancer, Merkel cell carcinoma and Hodgkin’s lymphoma. Remarkably, there are ~120 antibody immune checkpoint modulators currently in clinical studies, together comprising ~20% of the total commercial clinical pipeline of antibody therapeutics. MAbs in the clinical pipeline target CTLA-4, PD-1 and PD-L1, but also other immune system checkpoints such as CD40, GITR, LAG-3, OX40, TIGIT and TIM-3.
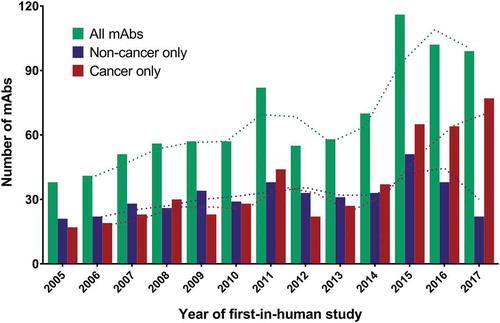
Numerous scientific discoveries, including phage display and immune checkpoint modulation, technological advances, and substantial efforts by a multitude of researchers and medical professionals enabled the increase in the number of commercially sponsored first-in-human clinical studies of antibody therapeutics from ~12 per year in the early 1990s to ~30 per year by the mid-2000s,Citation10 and then to ~70 per year in 2014 (). During 2015 to 2017, however, the number of antibody therapeutics entering a first Phase 1 study each year did not gradually increase, but rather soared upward, averaging just over 100 per year over the 3-year period. Data from this 3-year period also shows a remarkable trend toward development of antibody therapeutics for cancer at the expense of the development of such therapeutics for non-cancer indications. It remains to be seen whether these trends continue into the future.
Figure 1. Number of antibody therapeutics entering first-in-human studies per year, 2005–2017.
Green bars, all antibody therapeutics. Blue bars, antibody therapeutics for non-cancer indications only. Red bars, antibody therapeutics for cancer only. Dotted lines, 2-year moving averages. Totals include only antibody therapeutics sponsored by commercial firms; those sponsored solely by government, academic or non-profit organizations were excluded. Biosimilar antibodies and Fc fusion proteins were also excluded.
The comparatively high approval success rates for mAbs may be one reason for the growing interest in the development of these therapeutics. As reported by Hay et al.,Citation11 biologics such as mAbs have a likelihood of approval of near 1 in 4 compared to that of new molecular entities, which approach 1 in 8. Simply put, mAbs are granted marketing approvals at twice the rate of small molecule drugs.
To verify results reported in Hay et al., and to provide additional details about clinical phase transition and approval success rates for mAbs, we collected data in the public domain for over 680 antibody therapeutics that entered clinical studies sponsored by commercial firms between January 1, 2000 and December 31, 2014. The data was collected from various websites, and included sources such as company pipelines and press releases, clinical trials registries, and an open-access database of therapeutic antibodies (IMGT/mAb-DB, http://www.imgt.org/mAb-DB/). The available data was cross-checked against several commercial databases (Beacon Targeted Therapies, Therapeutic Antibody Database, BioMedTracker). Antibody therapeutics developed solely by non-commercial organizations, e.g., National Institutes of Health, and all biosimilar antibodies were excluded. Molecules with at least one binding site derived from an antibody gene were included, but Fc fusion proteins were excluded. We defined success as a first marketing approval in either the US or EU. Each molecule was assigned one therapeutic category and one of 9 possible phases of development (Phase 1, 2 or 3 clinical study; regulatory review in the US or EU; approved in the US or EU; all development terminated at Phase 1, 2 or 3, or terminated in regulatory review in the US or EU). The phase of development was determined from the data available for the most advanced phase of the molecules. For example, a molecule in Phase 3 studies was assigned to Phase 3 regardless of whether Phase 1 or Phase 2 studies were also in progress. Molecules were considered terminated if they were not listed in the relevant company pipeline or no development had recently been reported, regardless of whether the molecule was described as a licensing opportunity. Our dataset appears to be substantially larger than the mAb subset of Hay et al.; the method we used to calculate success rates was most closely aligned with their ‘lead indications’ approach.
We analyzed our data when stratified by time (entry into clinical study during 2000–2009 and 2005–2014) and by therapeutic category (non-cancer and cancer) ( and ). For all mAbs that entered clinical study during 2000–2009 (n = 357), the Phase 1 to 2, Phase 2 to 3, Phase 3 to regulatory review and regulatory review to approval transition rates were 75%, 44%, 71% and 91%, respectively, with an overall (Phase 1 to approval) success rate of 21% (). These results are nearly identical to those reported by Hay et al., for their biologics data analyzed by lead indications (75.1%, 44.0%, 71.7% and 88.0% for phase transitions in the order listed above, with an overall success rate of 20.8%). Our data for mAbs entering clinical studies during 2000–2009 indicated that mAbs developed for cancer indications had lower phase transition rates and a lower overall approval success rate compared to those developed for non-cancer indications. This trend toward lower success rates for oncology drugs was also reported by Hay et al. It should be noted that approximately one-quarter of the mAbs were in clinical studies or regulatory review at the time we calculated the success rates shown in , and thus the rates may vary somewhat when the final fates (approval or termination) for all molecules in the cohort become known.
Figure 2. Clinical phase transition and approval success rates for antibody therapeutics that entered clinical study during 2000–2009.
Green bars, all antibody therapeutics. Blue bars, antibody therapeutics for non-cancer indications only. Red bars, antibody therapeutics for cancer only. Cohorts included only antibody therapeutics sponsored by commercial firms; those sponsored solely by government, academic or non-profit organizations were excluded. Number of molecules that entered clinical study during 2000–09: all, n = 357; non-cancer only, n = 181; cancer only, n = 176. Final fates (approval or termination) are known for 76%. MAbs that had advanced to Phase 1/2 were classified as Phase 2; mAbs that had advanced to Phase 2/3 were classified as Phase 3. Two mAbs with first approvals outside the US/EU regions (italizumab (Alzumab) and Rabishield approvals in India) were classified as Phase 3. Phase transition percentages were calculated as follows: the number of antibody therapeutics that completed a given phase and transitioned to the next was divided by the arithmetic difference between the number that entered the phase and the number that remained in the phase at the time of the calculation. Phase transitions occurring between clinical studies conducted world-wide were included. Approval success were defined as a first US or EU approval; supplemental approvals were not included. Abbreviation RR, regulatory review.
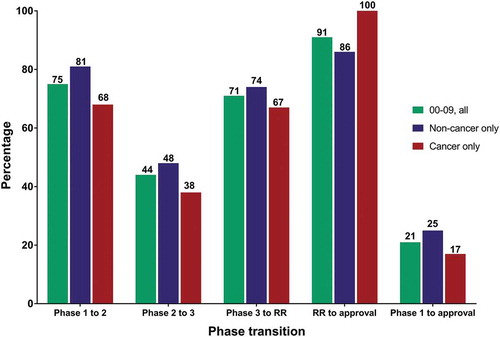
Figure 3. Clinical phase transition and approval success rates for antibody therapeutics that entered clinical study during 2005–2014.
Green bars, all antibody therapeutics. Blue bars, antibody therapeutics for non-cancer indications only. Red bars, antibody therapeutics for cancer only. Cohorts included only antibody therapeutics sponsored by commercial firms; those sponsored solely by government, academic or non-profit organizations were excluded. Number of molecules that entered clinical study during 2005–14: all, n = 569; non-cancer only, n = 295; cancer only, n = 274. Final fates (approval or termination) are known for 58%. MAbs that had advanced to Phase 1/2 were classified as Phase 2; mAbs that had advanced to Phase 2/3 were classified as Phase 3. Two mAbs with first approvals outside the US/EU regions (italizumab (Alzumab) and Rabishield approvals in India) were classified as Phase 3. Phase transition percentages were calculated as follows: the number of antibody therapeutics that completed a given phase and transitioned to the next was divided by the arithmetic difference between the number that entered the phase and the number that remained in the phase at the time of the calculation. Transitions occurring between clinical studies conducted world-wide were included. Approval success were defined as a first US or EU approval; supplemental approvals were not included. Abbreviation RR, regulatory review.
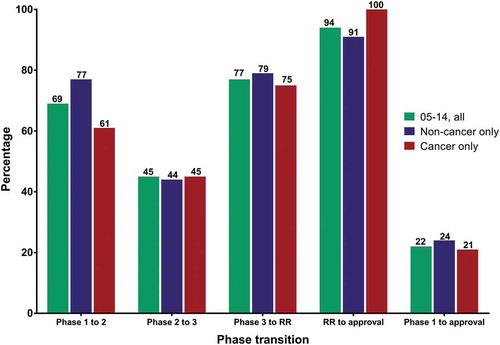
For all mAbs that entered clinical study during 2005–2014 (n = 569), the Phase 1 to 2, Phase 2 to 3, Phase 3 to regulatory review, and regulatory review to approval transition rates were 69%, 45%, 77% and 94%, respectively, with an overall success rate of 22% (). The overall approval success rates for the two periods is thus very similar, although there was some variability in phase transition rates. Our data for this cohort also showed that approval success rates were lower for mAbs developed for cancer vs non-cancer indications, but the difference (21 vs 24%, respectively) was less pronounced compared to the rates for mAbs that entered clinical study during 2000–2009 (17% vs 25%, respectively). Nearly half of the mAbs that entered clinical study during 2005–2014 were still in clinical study or regulatory review at the time of our analysis. We look forward to reporting updated success rates for this cohort when the final fates of more molecules become known in the future.
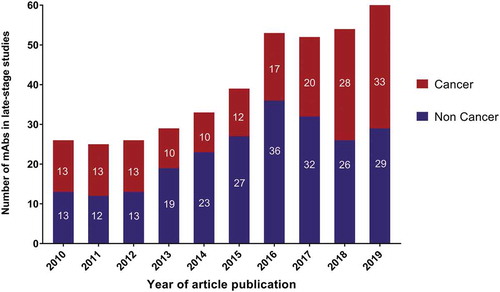
The success of antibody therapeutics has motivated hundreds of companies to engage in the development of these molecules. Collectively, companies are currently sponsoring clinical studies of more than 570 mAbs. Of these, ~90% are undergoing early-stage studies designed to assess the safety (Phase 1) or safety and preliminary efficacy (Phase 1/2 or Phase 2) of the molecules in various patient populations (). Reflecting the recent substantial increase in the number of anti-cancer antibodies entering clinical study, most (~70%) of the mAbs at Phase 1 are for cancer. The numbers of mAbs intended to treat cancer vs non-cancer indications are similar for mAbs currently at Phase 2 and late-stage clinical studies (pivotal Phase 2, Phase 2/3 or Phase 3). The ‘Antibodies to watch’ article series has documented the substantial increase in the number of mAbs in late-stage studies that has occurred over the past 10 years ().Citation12–Citation20 The 62 mAbs currently undergoing evaluation in late-stage studies represent the largest number at this stage to date, with the increase, at least in part, due to the advancement of mAbs developed by companies based in China.
Figure 4. Clinical phases for antibody therapeutics in development.
Data as of November 2018. Totals include only antibody therapeutics sponsored by commercial firms; those sponsored solely by government, academic or non-profit organizations were excluded; biosimilars and Fc fusion proteins were excluded. Phase 1/2 included with Phase 2; late-stage studies include pivotal Phase 2, Phase 2/3 and Phase 3. Tables of mAbs in late-stage studies are available at www.antibodysociety.org.
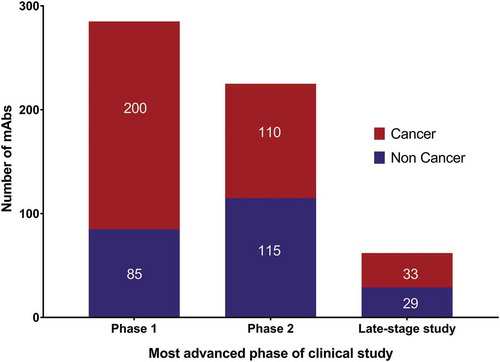
Figure 5. Antibodies to watch in 2010–2019.
Data from ‘Antibodies to watch’ articles published in mAbs. 2019 data as of November 2018. Totals include only antibody therapeutics sponsored by commercial firms; those sponsored solely by government, academic or non-profit organizations were excluded. Tables of mAbs in late-stage studies are available at www.antibodysociety.org.
Following in the tradition of the ‘Antibodies to watch’ article series, we provide here updates on recent and anticipated events relevant to antibody therapeutics clinical development. We describe the antibody therapeutics that were approved in either the US or EU during 2018, and those undergoing regulatory review in these two regions as of November 2018. In acknowledgment of the rapid progress in the development of novel antibody therapeutics in China, we also discuss several antibody immune checkpoint inhibitors undergoing regulatory review by China’s National Medical Products Administration (NMPA). Finally, we provide an overview of antibody therapeutics in late-stage clinical study that may progress to regulatory review in late 2018 or 2019, based on public disclosures by the sponsoring companies.
Antibody therapeutics approved in the US or EU in 2018
A total of 12 new antibodies were granted approvals in either the US or EU during 2018 (). Perhaps reflecting the higher approval success rate for antibodies that are treatments for non-cancer indications, the majority (75%) of the products are for such diseases, including 3 for migraine prevention and 1 for human immunodeficiency virus (HIV) infection.
Table 1. Antibody therapeutics granted first approvals in the European Union or the United States during 2018*.
Erenumab (Novartis)
On May 17, 2018, the US Food and Drug Administration (FDA) approved erenumab-aooe (Aimovig) for the preventive treatment of migraine in adults. Erenumab is a human IgG2 mAb that targets calcitonin gene-related peptide (CGRP) receptor, thereby blocking the activity of CGRP, which is involved in migraine attacks. The treatment is given by once-monthly subcutaneous (SC) injections. The approval was based on data from three clinical trials that compared erenumab-aooe to placebo. Over 2000 participants were included in the studies. In the first study (STRIVE, NCT02456740), which included 955 participants with a history of episodic migraine, patients administered erenumab-aooe experienced, on average, one to two fewer monthly migraine days than those on placebo over a 6-month period. In the second study (ARISE, NCT02483585), which included 577 patients with a history of episodic migraine, patients administered erenumab-aooe experienced, on average, one fewer migraine day per month than those on placebo over a 3-month period. In the third study (NCT02066415), which evaluated 667 patients with a history of chronic migraine, patients treated with erenumab-aooe experienced, on average, 2.5 fewer monthly migraine days than those receiving placebo over a 3-month period.Citation21 On July 26, 2018, erenumab was issued a marketing authorization in the EU for prophylaxis of migraine in adults who have at least 4 migraine days per month.
Fremanezumab (Teva Pharmaceuticals)
On September 14, 2018, the FDA approved fremanezumab-vfrm (AjovyTM) for the preventive treatment of migraine in adults. The drug may be administered as either 225 mg monthly, or 675 mg quarterly, SC doses.Citation22 Fremanezumab-vfrm is a humanized mAb that binds to CGRP, thereby blocking the binding of this ligand to its receptor. In a Phase 3 study (NCT02629861) of patients with migraines administered fremanezumab, mean migraine days per month decreased from 8.9 days to 4.9 days in the fremanezumab 225 mg dosing group, from 9.2 days to 5.3 days in the fremanezumab 675 mg dosing group, and from 9.1 days to 6.5 days in the placebo group, as assessed from baseline to 12 weeks.Citation23 A marketing authorization application for fremanezumab is undergoing review by the European Medicines Agency (EMA).
Galcanezumab (Eli Lilly & Company)
On September 27, 2018, the FDA approved galcanezumab-gnlm (Emgality®) for the preventive treatment of migraine in adults, making Emgality the third antibody therapeutic approved by FDA in 2018 for this indication. Like fremanezumab, galcanezumab is a humanized mAb that binds to CGRP. The recommended dosage of Emgality is 240 mg loading dose (administered as two consecutive injections of 120 mg each), followed by monthly doses of 120 mg. The efficacy and safety of Emgality was demonstrated in two Phase 3 clinical trials in patients with episodic migraine (EVOLVE-1 (NCT02614183), EVOLVE-2 (NCT02614196)) and one Phase 3 clinical trial in patients with chronic migraine (REGAIN (NCT02614261)). In all three studies, patients were randomized to receive once-monthly placebo, Emgality 120 mg after an initial loading dose of 240 mg, or Emgality 240 mg. In EVOLVE-1, the mean change from baseline (days) was -4.7 days (n = 210) for Emgality 120 mg compared to -2.8 days (n = 425) for placebo (p < 0.001), while in EVOLVE-2, the mean change from baseline (days) was -4.3 days (n = 226) for Emgality 120 mg compared to -2.3 days (n = 450) for placebo (p < 0.001). In the REGAIN study, the mean change from baseline (days) was -4.8 days (n = 273) for Emgality 120 mg compared to -2.7 days (n = 538) for placebo (p < 0.001).Citation24 In November 2018, the European Commission (EC) granted marketing authorization for Emgality® for the prophylaxis of migraine in adults who have at least four migraine days per month.
Burosumab (KRN23; Kyowa Hakko Kirin Co. Ltd, Ultragenyx Pharmaceutical Inc.)
On February 19, 2018, the EC issued a conditional marketing authorization in the EU for burosumab (Crysvita) as a treatment for X-linked hypophosphatemia (XLH) in children 1 year of age and older, and adolescents with growing skeletons. Burosumab is a human IgG1 mAb that binds to and inhibits the activity of fibroblast growth factor 23, thereby reducing loss of phosphate from the kidney and other metabolic abnormalities, and ameliorating bone changes that are a hallmark of the disease.Citation25 In a double-blind, placebo-controlled, Phase 3 study of symptomatic adults with XLH administered SC burosumab 1 mg/kg (n = 68) or placebo (n = 66) every 4 weeks, across midpoints of dosing intervals, 94.1% of burosumab-treated participants attained mean serum phosphate concentration above the lower limit of normal compared with 7.6% of those receiving placebo (p < 0.001).Citation26 Burosumab was granted Orphan Drug designations in the EU and US, and FDA granted the drug Breakthrough Therapy designation for pediatric XLH. Burosumab-twza was approved by the FDA on April 17, 2018.Citation27
Lanadelumab (Shire)
On August 23, 2018, the FDA approved lanadelumab-flyo (TakhzyroTM) injection for prophylaxis to prevent attacks of hereditary angioedema (HAE) in patients 12 years of age and older. This rare, genetic, and potentially life-threatening disorder can result in recurrent attacks of severe swelling in various parts of the body, including the throat. HAE affects people with low levels of, or poorly functioning, C1 esterase inhibitor proteins, which act by inhibiting plasma kallikrein and preventing spontaneous activation of the complement system. Takhzyro is a human mAb that targets plasma kallikrein, and thereby helps prevent attacks of edema. FDA’s approval of Takhzyro was based in part on data from the multicenter, randomized, double-blind, placebo-controlled, parallel-group, Phase 3 HELP study (NCT02586805), which included 125 patients with HAE. In this study, Takhzyro reduced the number of monthly HAE attacks an average of 87% (n = 27) or 73% (n = 29) vs. placebo (n = 41) when administered SC at 300 mg every two weeks or at 300 mg every four weeks, respectively (adjusted P < 0.001).Citation28 The FDA had previously granted Takhzyro Breakthrough Therapy and Orphan Drug designations. The biologics license application (BLA) for the drug was granted a priority review. Takhzyro, which was designated as an orphan medicinal product in the EU in 2015, was reviewed under EMA’s accelerated assessment program. In November 2018, the EC granted marketing authorization for Takhzyro SC injection for the routine prevention of recurrent attacks of HAE in patients aged 12 years and older.
Caplacizumab (Sanofi)
On August 31, 2018, the EC issued a marketing authorization for caplacizumab (Cablivi™), a bivalent single-domain nanobody targeting von Willebrand factor, as a treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP). During episodes of this rare, life-threatening blood clotting disorder, microclots can form, leading to low platelet counts, ischemia and organ dysfunction in aTTP patients. Caplacizumab was granted Fast Track designation in the US and Orphan Drug designations in the US and EU for the treatment of aTTP. The BLA for caplacizumab is undergoing a priority review at the FDA. A first action on the application is expected by February 6, 2019.Citation29 Caplacizumab was developed by Ablynx, a Sanofi company. The approval of caplacizumab in the EU was based in part on the Phase 3 HERCULES study (NCT02553317), a placebo-controlled, randomized study to evaluate the efficacy and safety of caplacizumab in more rapidly restoring normal platelet counts as a measure of the prevention of further microvascular thrombosis. Positive results from this study were presented at the 59th Annual Meeting of the American Society of Hematology in December 2017.Citation30 The HERCULES study recruited 145 patients with an acute episode of aTTP who were randomized 1:1 to receive either caplacizumab or placebo in addition to standard-of-care treatment, which was daily plasma exchange (PEX) and immunosuppression. Patients were administered a single intravenous (IV) bolus of 10 mg caplacizumab or placebo followed by daily SC dose of 10 mg caplacizumab or placebo until 30 days after the last daily PEX. Depending on the response, the treatment could be extended for additional 7-day periods up to a maximum of 28 days. The primary endpoint (time to platelet count response) and several secondary endpoints of HERCULES study were met. In particular, caplacizumab provided faster resolution of an aTTP episode with significantly shorter time to platelet count response, clinically relevant reduction in aTTP-related death, exacerbation of aTTP, or a major thromboembolic event, and prevention of aTTP relapses when treatment is extended until resolution of underlying disease.Citation30 A 3-year Phase 3 follow-up study (NCT02878603) of patients who completed the HERCULES study is in progress.
Mogamulizumab (Kyowa Hakko Kirin)
On August 8, 2018, the FDA approved Poteligeo® (mogamulizumab-kpkc) for IV use for the treatment of adult patients with relapsed or refractory mycosis fungoides (MF) or Sézary syndrome (SS) after at least one prior systemic therapy. The diseases are subtypes of cutaneous T-cell lymphoma (CTCL), which is a rare and difficult-to-treat type of non-Hodgkin’s lymphoma. The FDA had previously granted mogamulizumab Breakthrough Therapy and Orphan Drug designations, and the BLA for mogamulizumab received a priority review. FDA’s approval was based on an open-label, multi-center, randomized Phase 3 clinical trial (NCT01728805) of 372 patients with relapsed MF or SS who received either mogamulizumab or vorinostat. Study sites were located in the US, Europe, Japan and Australia. Median progression-free survival (PFS) was 7.6 months for patients administered mogamulizumab compared to 3.1 months for patients taking vorinostat in this clinical trial.Citation31 Poteligeo was designated as an orphan medicinal product in the EU in 2016. The EC granted a marketing authorization to Poteligeo® for the treatment of adult patients with MF or SS who have received at least one prior systemic therapy in November 2018.
Developed by Kyowa Kirin, mogamulizumab is a humanized glyco-engineered mAb that binds to CC chemokine receptor type 4 on cancer cells.Citation32 Mogamulizumab was produced using Kyowa Hakko Kirin’s proprietary POTELLIGENT® platform, which produces antibodies with low/no fucose content. Such antibodies have increased affinity to FcγRIIIa (CD16), and enhanced antibody-dependent cell-mediated cytotoxicity activity. Mogamulizumab’s first approval, in 2012, was granted by the Japanese Ministry of Health, Labour and Welfare for treatment of patients with relapsed or refractory CCR4-positive adult T-cell leukemia-lymphoma.
Moxetumomab pasudotox (Astrazeneca)
On September 13, 2018, the FDA approved moxetumomab pasudotox-tdfk (Lumoxiti) for the treatment of adult patients with relapsed or refractory hairy cell leukemia who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog. The drug is a recombinant immunotoxin targeting CD22 composed of a single-chain variable fragment (scFv) fused to a truncated form of Pseudomonas exotoxin PE38.Citation33 In a pivotal, multi-center, open-label study (NCT01829711) of moxetumomab pasudotox involving 80 patients (79% males; median age, 60.0 years), the durable complete response rate was 30%, the complete response rate was 41%, and the objective response rate (complete and partial response) was 75%.Citation34 Due to the severity and rarity of the disease, the FDA granted the application Fast Track and Priority Review designations, and moxetumomab pasudotox received US Orphan Drug designation.
Cemiplimab (Sanofi)
On September 28, 2018, the FDA approved cemiplimab-rwlc (Libtayo) for the treatment of patients with metastatic cutaneous squamous cell carcinoma (CSCC) or locally advanced CSCC who are not candidates for curative surgery or curative radiation. Cemiplimab-rwlc is the third antibody therapeutic targeting PD-1 to be granted an FDA approval, but it is the first drug to be approved in the US specifically for advanced CSCC. FDA’s approval of Libtayo was based on a combined analysis of data from an open-label, multi-center, non-randomized Phase 2 trial known as EMPOWER-CSCC-1 (Study 1540) and two advanced CSCC expansion cohorts from a multi-center, open-label, non-randomized Phase 1 trial (Study 1423). A total of 108 patients (75 with metastatic disease and 33 with locally-advanced disease) were included in the efficacy evaluation. The confirmed objective response rate for all patients treated with Libtayo was 47%.Citation35 FDA granted cemiplimab Breakthrough Therapy designation status for advanced CSCC in 2017, and the drug’s BLA was granted a priority review. A marketing authorization application for cemiplimab is undergoing EMA review.
Ibalizumab (Theratechnologies, Inc)
On March 6, 2018, the FDA approved ibalizumab-uiyk (Trogarzo) for adult patients infected with HIV who were previously treated with multiple HIV medications and whose HIV infections are resistant to currently available therapies.Citation36 Ibalizumab-uiyk, a humanized IgG4 mAb, is a CD4 domain 2-directed post-attachment HIV-1 inhibitor. The BLA was granted Breakthrough Therapy, Fast Track and Priority Review designations, and ibalizumab was granted an Orphan Drug designation by FDA. Theratechnologies Inc. and TaiMed Biologics, Inc. have an agreement to market and distribute Trogarzo in the US and Canada. The product is IV administered as a single loading dose of 2,000 mg followed by a maintenance dose of 800 mg every 2 weeks after dilution in 250 mL of 0.9% sodium chloride. The safety and efficacy of ibalizumab-uiyk were evaluated in the Phase 3 TMB-301 study (NCT02475629), which was a single arm, multicenter clinical trial conducted in 40 heavily treatment-experienced HIV-infected patients with multidrug resistant HIV-1.Citation37 At week 25, viral load <50 was achieved in 43% of patients, while 55% and 48% of patients had a ≥ 1 log10 reduction in viral load and a ≥ 2 log10 reduction in viral load, respectively. The most common adverse reactions reported in at least 5% of subjects were diarrhea, dizziness, nausea, and rash. In total, 292 patients with HIV-1 infection were exposed to ibalizumab-uiyk IV infusion during clinical studies.Citation38 A marketing authorization application for ibalizumab is undergoing EMA review.
Tildrakizumab (Sun Pharma)
On March 20, 2018, the FDA approved tildrakizumab-asmn (Ilumya) for treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. Tildrakizumab, a humanized IgG1 kappa mAb, targets IL-23p19 and blocks the interaction of IL-23 with its receptor, thereby inhibiting release of pro-inflammatory cytokines and chemokines. FDA approval was supported by results from two Phase 3 trials (reSURFACE 1 and 2) in which patients were randomized to tildrakizumab 200 mg, tildrakizumab 100 mg, or placebo (2:2:1; reSURFACE 1), or to tildrakizumab 200 mg, tildrakizumab 100 mg, placebo, or etanercept 50 mg (2:2:1:2; reSURFACE 2). In these trials, the tildrakizumab 200 mg and 100 mg doses were well tolerated and found to be efficacious compared with placebo and etanercept in the treatment of patients with moderate-to-severe chronic plaque psoriasis. The results of both studies were published in The Lancet in July 2017.Citation39 In a pooled analysis of three randomized controlled trials that included over 2000 patients, tildrakizumab therapy for up to 64 weeks was well tolerated, with low rates of serious treatment-emergent adverse events (AEs), discontinuations due to AEs, and AEs of clinical interest.Citation40 An EU-wide marketing authorization for tildrakizumab (Ilumetri) was issued to Almirall S.A. on September 17, 2018. In the EU, Ilumetri is indicated for the treatment of adults with moderate to severe plaque psoriasis who are candidates for systemic therapy.
Emapalumab (Novimmune/Swedish Orphan Biovitrum AB)
On November 20, 2018, the FDA approved emapalumab-lzsg (Gamifant) for the treatment of pediatric (newborn and above) and adult patients with primary hemophagocytic lymphohistiocytosis (HLH) who have refractory, recurrent or progressive disease or intolerance with conventional HLH therapy.Citation41 Developed by Novimmune SA, emapalumab is a human IgG1 antibody that targets interferon γ. Emapalumab received a variety of designations intended to facilitate the development of drugs for rare, serious or life-threatening diseases, including Breakthrough Therapy, Rare Pediatric Disease, and Orphan Drug designations in the US, and Priority Medicines and Orphan Drug designations in the EU. The FDA’s approval was based in part on a clinical study of 27 pediatric patients with suspected or confirmed primary HLH with either refractory, recurrent or progressive disease during conventional HLH therapy or who were intolerant of conventional HLH therapy. Results from this study showed that 63% of patients experienced a response and 70% were able to proceed to stem cell transplant. A marketing application for emapalumab is undergoing evaluation by EMA.
Antibody therapeutics undergoing regulatory review in the US or EU
As of November 2018, a total of 4 antibody therapeutics (sacituzumab govitecan, ravulizumab, risankizumab, romosozumab) are being considered for their first marketing approvals in the US or EU (). Reflecting the diversity of mAbs in clinical development, the product candidates are for diseases in a variety of therapeutic areas, such as cancer, cardiovascular/hemostasis, immune-mediated disorders, and bone/skeletal diseases.
Table 2. Investigational antibody therapeutics in regulatory review in the European Union or the United States*.
Sacituzumab govitecan (Immunomedics)
This antibody-drug conjugate (ADC) is composed of a humanized anti-Trop-2 antibody conjugated to SN38 (active metabolite of the topoisomerase I inhibitor irinotecan). A BLA for approval of sacituzumab govitecan as third-line therapy for metastatic triple-negative breast cancer was granted Priority Review by the FDA, and the action date is January 18, 2019.Citation42 Immunomedics is planning clinical studies of the ADC in other cancers. Patients are being recruited for a pivotal Phase 2 study of sacituzumab govitecan as a treatment for metastatic urothelial cancer after failure of platinum-based regimen or anti-PD-1/PD-L1 based immunotherapy (NCT03547973) and a Phase 2 study of the ADC in patients with metastatic castration-resistant prostate cancer who have progressed on second generation androgen receptor-directed therapy (NCT03725761).
Ravulizumab (Alexion Pharmaceuticals)
Also known by the drug code ALXN1210, ravulizumab is a humanized mAb targeting complement component 5 (C5). A BLA for approval of ravulizumab for treatment of patients with paroxysmal nocturnal hemoglobinuria (PNH) is undergoing review at FDA. The BLA’s action date is February 18, 2019, following an expedited eight-month review, instead of the standard 12-month review, which was allowed by Alexion’s use of a rare disease priority review voucher.Citation43 Ravulizumab is also undergoing regulatory review in the EU. The mAb was granted Orphan Drug designation in both the US and EU for the treatment of patients with PNH. In a Phase 3 study, ravulizumab demonstrated non-inferiority to Soliris® (anti-C5 eculizumab) in complement inhibitor treatment-naïve patients with PNH.Citation44 Also developed by Alexion Pharmaceuticals, Soliris® was first approved for PNH in 2007.
Risankizumab (Abbvie)
Like the recently approved products Tremfya (guselkumab) and Ilumya (tildrakizumab), risankizumab targets IL-23p19 and it was evaluated as a treatment for plaque psoriasis. The BLA for risankizumab is supported by data from four Phase 3 studies, ultIMMA-1, ultIMMa-2, IMMhance and IMMvent, that evaluated the safety and efficacy of risankizumab in more than 2,000 patients with moderate-to-severe chronic plaque psoriasis. In these four studies, all co-primary and ranked secondary outcome measures were met and no new safety signals were observed.Citation45 Results of the UltIMMa-1 (NCT02684370) and UltIMMa-2 (NCT02684357) studies were reported in The Lancet.Citation46 Assuming the BLA receives a standard review, FDA may make a decision in April 2019. Risankizumab is also undergoing regulatory review in the EU.
Romosozumab (Amgen/UCB Pharma)
This humanized IgG2 mAb, which targets sclerostin, has been evaluated as a treatment for osteoporosis in women and men. A BLA for romosozumab as a treatment of osteoporosis in postmenopausal women at high risk for fracture was submitted to the FDA in July 2016, but additional safety and efficacy data was requested in the FDA’s complete response letter, as announced by collaborators Amgen and UCB in July 2017.Citation47 In July 2018, Amgen and UCB announced that the BLA had been resubmitted,Citation48 thus starting a second review cycle. In addition to data from early-stage clinical studies, the original BLA included data from the Phase 3 placebo-controlled FRAME study, including 7,180 postmenopausal women with osteoporosis. The resubmitted BLA includes results from the more recent Phase 3 ARCH study, an alendronate-active comparator trial including 4,093 postmenopausal women with osteoporosis who experienced a fracture, and the Phase 3 BRIDGE study, which included 245 men with osteoporosis. Romosozumab is also undergoing regulatory review in the EU.
Antibody therapeutics undergoing regulatory review in China
A decade ago, when the ‘Antibodies to watch’ article series was started, the innovative antibodies in late-stage clinical studies were developed primarily by companies based in the US or EU. Since then, the Chinese biopharmaceutical industry has made substantial strides in the clinical development of antibody therapeutics, with the aim of gaining marketing approvals not only in China, but also globally. Chinese companies are engaging with FDA, and developing their product candidates according to the standards of the FDA and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (www.ich.org).Citation49 In particular, Chinese companies have progressed multiple antibodies targeting PD-1 into late-stage studies and regulatory review in China, including tislelizumab, sintilimab and camrelizumab.
Tislelizumab (BGB-A317; Beigene Ltd.)
This humanized IgG4 mAb binds to PD-1, but does not bind human FcγRI or mediate crosslinking between PD-1 and FcγRI. Interactions with FcγRI were reported to have a negative effect on anti-cancer activity of antibodies targeting PD-1.Citation50 In August 2018, BeiGene announced that China’s NMPA accepted a new drug application (NDA) for tislelizumab for the treatment of relapsed or refractory Hodgkin’s lymphoma.Citation51 The NDA, which was granted a priority review, included results from a pivotal Phase 2 study (NCT03209973) of 70 Chinese patients showing an overall response rate of 85.7%, including 61.4% complete response, with a minimum of 24 weeks of follow-up and a median follow-up time of 7.85 months at the data cutoff. Tislelizumab is also being evaluated in Phase 3 studies of patients with non-small cell lung cancer, hepatocellular carcinoma and esophageal squamous cell carcinoma.
Sintilimab (IBI308; Innovent Biologics)
Innovent Biologics and Eli Lilly and Company are jointly developing sintilimab, a human IgG4 antibody targeting PD-1, in China. In April 2018, the NDA for sintilimab for relapsed and refractory classical Hodgkin’s lymphoma was submitted in China and granted priority review status. In the Phase 2 registrational study ORIENT-1 (NCT03114683) that assessed the efficacy and safety profile of sintilimab, the overall response rate was 79.2%, with disease control rate of 97.9%, among the 96 patients with a minimum 24-week follow-up.Citation52 Sintilimab is also being evaluated as first-line treatment for patients with non-squamous non-small cell lung cancer (NCT03607539 Phase 3 ORIENT-11 study) and as first-line treatment for patients with previously untreated squamous non-small cell lung cancer (NCT03629925 Phase 3 ORIENT-12 study).
Camrelizumab (SHR-1210; Jiangsu Hengrui Medicine Co., Ltd.)
An NDA for camrelizumab, a humanized IgG4 antibody targeting PD-1, for relapsed/refractory classic Hodgkin’s lymphoma submitted by Hengrui to China’s NMPA was accepted in April 2018, and is currently under review.Citation53 Hengrui has partnered with Incyte Corporation to further develop camrelizumab. In two Phase 1/2 studies (NCT02961101, NCT03250962) of camrelizumab with or without decitabine, the addition of decitabine increased the complete response rate of patients with relapsed/refractory classic Hodgkin’s lymphoma, and significantly reversed the resistance of anti-PD-1 therapy.Citation54 The safety profile of the combination therapy was deemed acceptable. Camrelizumab is undergoing evaluation as a treatment for other types of cancer, and results have been reports for early-phase clinical studies of camrelizumab in patients with advanced non-squamous non-small cell lung cancer,Citation55 advanced hepatocellular carcinoma, gastric or esophagogastric junction cancer,Citation56 nasopharyngeal carcinoma,Citation57 advanced esophageal carcinoma,Citation58 and advanced solid tumors.Citation59
Antibodies to watch in 2019: non-cancer indications
As of November 2018, 29 novel antibody therapeutics were in late-stage clinical studies for non-cancer indications (). No single therapeutic area predominated, although, at ~40% of the total, antibodies for immune-mediated disorders were the largest group. Of the 29 product candidates, two (leronlimab, brolucizumab) may enter regulatory review by the end of 2018, and at least five (eptinezumab, teprotumumab, crizanlizumab, satralizumab, tanezumab) may enter regulatory review in 2019.
Table 3. Investigational monoclonal antibodies in late-stage clinical studies for non-cancer indications.
Leronlimab (PRO140; CytoDyn)
The chemokine receptor CCR5 is the principal HIV co-receptor, but it has potential as a drug target for other diseases, such as cancer and immune-mediated disorders.Citation60 Leronlimab, a humanized IgG4 mAb that blocks CCR5, was granted FDA’s Fast Track drug designation for the treatment of HIV infection. This designation allows a ‘rolling’ BLA submission, i.e., the BLA can be submitted in sections, which may be in progress, as CytoDyn has indicated that they made progress toward filing a BLA for leronlimab as a combination therapy for HIV and are confident that two-thirds of the BLA submission (i.e., clinical and non-clinical sections) will be completed by the end of 2018 and the last section, Chemistry, Manufacturing and Controls, by the first quarter of 2019.Citation61 Leronlimab is undergoing evaluation in late-stage clinical studies that include treatment-experienced HIV-infected patients with CCR5-tropic virus who demonstrate evidence of HIV-1 replication despite ongoing antiretroviral therapy (ART) with documented genotypic or phenotypic resistance to ART drugs within three drug classes (or within two or more drug classes with limited treatment options). The primary efficacy endpoint of a pivotal 25-week study (NCT02483078), 0.5log reduction in viral load after one week of therapy with leronlimab in combination with the patient’s failing drug regimen, was met. An open-label extension of this study is continuing to enroll patients. The successful results of this trial may serve as the basis for the BLA submission. CytoDyn is also developing leronlimab as a treatment for graft-vs.-host disease and triple-negative breast cancer.
Brolucizumab (RTH258; Novartis)
This ~26 kDa humanized scFv targets all isoforms of vascular endothelial growth factor-A, and it is undergoing evaluation as a treatment for neovascular age-related macular degeneration (nAMD). Novartis has indicated that regulatory submissions for brolucizumab may occur in December 2018.Citation62 In October 2018, Novartis released 96-week results from the Phase 3 HAWK (NCT02307682) and HARRIER (NCT02434328) studies that reaffirmed positive 48-week findings.Citation63 The two studies included more than 1,800 patients in comparing the efficacy and safety of intravitreal injections of 6 mg brolucizumab or 3 mg brolucizumab (HAWK study only) versus 2 mg aflibercept in patients with nAMD. The primary efficacy endpoint of the studies, non-inferiority to aflibercept (EYLEA®) in mean change in best-corrected visual acuity (BCVA) at week 48, was met. The 96-week results indicate patients administered brolucizumab maintained robust visual gains, with mean change in BCVA of 5.9 letters for brolucizumab 6 mg versus 5.3 letters for aflibercept in the HAWK study, and 6.1 letters versus 6.6 letters, respectively, in the HARRIER study. Superior reductions in retinal fluid and central subfield thickness (CST) demonstrated at 48 weeks were reaffirmed at 96 weeks. The percentage of patients with nAMD that had intra-retinal fluid and/or sub-retinal fluid was 24% for brolucizumab 6 mg vs. 37% for aflibercept in HAWK (p = 0.0001) and 24% vs. 39%, respectively, in the HARRIER study (P < 0.0001). Absolute reductions in CST from baseline were -175 µm for brolucizumab 6 mg versus -149 µm for aflibercept in HAWK (p = 0.0057) and -198 µm versus -155 µm, respectively, in the HARRIER study (P < 0.0001).
Eptinezumab (Alder Biopharmaceuticals, Inc.)
Like the marketed products Emgality and Ajovy, eptinezumab targets CGRP and it is for migraine prevention. In November 2018, Alder announced that they are on track to complete a BLA submission in the first quarter of 2019 that will include chemistry, manufacturing, and controls processes; positive results from a pharmacokinetic study intended to support the comparability evaluation of the clinical supply for eptinezumab and its planned commercial supply; data from eptinezumab’s PROMISE 1 (NCT02559895) and PROMISE 2 (NCT02974153) Phase 3 clinical studies; and long-term safety data.Citation64,Citation65 The PROMISE 1 and PROMISE 2 studies evaluated the effects of eptinezumab in episodic migraine patients (n = 888) or chronic migraine patients (n = 1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies. Alder announced one-year results from the PROMISE 1 study in June 2018, which indicated that, following the first quarterly infusion, episodic migraine patients treated with 300 mg eptinezumab experienced 4.3 fewer monthly migraine days (MMDs) from a baseline of 8 MMDs, compared to 3.2 fewer MMDs for placebo from baseline (p = 0.0001). At one year after the third and fourth quarterly infusions, patients treated with 300 mg eptinezumab experienced further gains in efficacy, with a reduction of 5.2 fewer MMDs compared to 4.0 fewer MMDs for placebo-treated patients.Citation66 In addition, ~31% of episodic migraine patients achieved, on average per month, 100% reduction of migraine days from baseline compared to ~ 21% for placebo. New 6-month results from the PROMISE 2 study were also released in June 2018.Citation67 These results indicated that, after the first quarterly infusion, chronic migraine patients dosed with 300 mg of eptinezumab experienced 8.2 fewer MMDs, from a baseline of 16 MMDs, compared to 5.6 fewer MMDs for placebo from baseline (p < .0001). A further reduction in MMDs was seen following a second infusion; 8.8 fewer MMDs for patients dosed with 300 mg compared to 6.2 fewer MMDs for those with placebo. In addition, ~ 21% of chronic migraine patients achieved, on average, 100% reduction of MMDs from baseline compared to 9% for placebo after two quarterly infusions of 300 mg of eptinezumab.
Teprotumumab (Horizon Pharma)
This human IgG1 mAb targets insulin-like growth factor-1 receptor. Teprotumumab is in development for moderate-to-severe thyroid eye disease, which is commonly associated with Grave’s disease (hyperthyroidism). Data from the Phase 3 OPTIC study (NCT03298867) is expected in the second quarter of 2019, and a BLA submission is expected mid-2019.Citation68 In the OPTIC study, participants receive 8 infusions of teprotumumab or placebo every 3 weeks for a total of 21 weeks. Teprotumumab (10 mg/kg) is administered on Day 1, with 20 mg/kg administered for the remaining 7 infusions. The primary endpoint at week 24 is the proptosis responder rate, defined as the percentage of participants with >2 mm reduction in study eye without deterioration (≥2 mm increase) of proptosis in the fellow eye. An open-label extension study (NCT03461211; OPTIC-X) is enrolling by invitation. Teprotumumab was granted Fast Track, Breakthrough Therapy and Orphan Drug designations by FDA.
Crizanlizumab (SEG101; Novartis)
Sickle cell disease, caused by mutations in the gene encoding hemoglobin subunit β, is characterized by production of red blood cells that are misshapen, prone to hemolysis and can occlude the vasculature, causing pain.Citation69 P-selectin found on endothelial cells and platelets participates in cell-cell interactions that contribute to the pathogenesis of vaso-occlusion and sickle cell-related pain crises. Crizanlizumab, a humanized mAb, binds P-selectin and blocks these interactions. Post-hoc analysis of data from the Phase 2 SUSTAIN study (NCT01895361) showed that more patients treated with crizanlizumab did not experience a vaso-occlusive crisis (VOC) compared to those treated with placebo (35.8% vs 16.9%), specifically patients with a history of 2–10 VOCs in the previous year.Citation70 Crizanlizumab was granted Orphan Drug designation in the US and EU for the treatment of sickle cell-related pain crises. Novartis anticipates submitting a BLA in 2019.Citation71
Satralizumab (SA237; Chugai Pharmaceutical Co., Ltd.)
This humanized anti-interleukin-6 (IL-6) receptor mAb is undergoing development as a treatment for neuromyelitis optica spectrum disorder (NMOSD), which is a rare autoimmune disease that affects the central nervous system. The constant and variable regions of satralizumab were engineered to give the molecule longer plasma half-life. In October 2018, Chugai announced that positive results from the Phase 3 SAkuraSky Study (NCT02028884) of were presented at the 2018 Congress of European Committee for Treatment and Research in Multiple Sclerosis held October 10–12, 2018, in Berlin, Germany. In this study, satralizumab (120 mg) or placebo was added to baseline therapy (azathioprine, mycophenolate mofetil and/or corticosteroids). Both treatments were administered SC to patients at week 0, 2, and 4, then subsequent treatment was continued at 4-week intervals. Satralizumab with immunosuppressive therapy significantly reduced the risk of relapse by 62% (hazard ratio = 0.38 [95% confidence interval: 0.16–0.88], p = 0.0184 [stratified log-rank test]) in patients with NMOSD including anti-aquaporin-4 (AQP4) antibody positive (AQP4 Ab positive) and negative (AQP4 Ab negative) patients, achieving the primary endpoint of time to first protocol-defined relapse in the double-blind period. The proportion of patients relapse free at weeks 48 and 96 was 88.9% and 77.6% with satralizumab and 66.0% and 58.7% with placebo, respectively.Citation72 Satralizumab has been granted US and EU orphan designations for the treatment of NMOSD. According to the Roche pipeline listing, an application submission for satralizumab is anticipated in 2019.
Tanezumab (Pfizer Inc., Eli Lilly and Company)
Therapeutics that block nerve growth factor (NGF) have the potential to ameliorate pain associated with various disorders.Citation73 The humanized anti-NGF IgG2 antibody tanezumab has been studied extensively in late-stage clinical studies of patients with osteoarthritis pain, cancer pain, and chronic low back pain. In July 2018, Pfizer and Lilly announced positive results from a 16-week Phase 3 study (NCT02697773) evaluating the efficacy and safety of SC administration of tanezumab compared to placebo in patients with osteoarthritis of the knee or hip.Citation74 In this study, 698 patients were randomized into one of 3 study arms, and they then received injections once every eight weeks. Patients in arm 1, 2 or 3 received two doses of placebo, two doses of tanezumab 2.5 mg, or one dose of tanezumab 2.5 mg followed by one dose of tanezumab 5 mg eight weeks later, respectively. Patients who received two doses of tanezumab experienced a statistically significant improvement in pain, physical function and the patients’ overall assessment of their osteoarthritis compared to those receiving placebo. Tanezumab was granted FDA’s Fast Track designation for tanezumab for the treatment of osteoarthritis pain and chronic low back pain. Lilly has indicated that a BLA for tanezumab for osteoarthritis pain may potentially be submitted in 2019.Citation75
Antibodies to watch in 2019: cancer indications
As of November 2018, 33 novel antibody therapeutics were in late-stage clinical studies for cancer indications (). Antibody therapeutics for solid tumors clearly predominated, with less than 20% of the total developed solely for hematological malignancies. Of the 33 product candidates, polatuzumab vedotin may enter regulatory review by the end of 2018, and at least 7 (isatuximab, spartalizumab, MOR208, oportuzumab monatox, TSR-042, enfortumab vedotin, ublituximab) may enter regulatory review in 2019.
Table 4. Investigational monoclonal antibodies in late-stage clinical studies for cancer indications.
Polatuzumab vedotin (DCDS4501A, RG7596; Roche)
In collaboration with Seattle Genetics, Roche is developing polatuzumab vedotin, which is composed of a humanized anti-CD79b IgG1 antibody conjugated to the antimitotic agent monomethyl auristatin E (MMAE). The antibody’s target is highly expressed on B cells of patients with lymphoma. Polatuzumab vedotin was granted FDA’s Breakthrough Therapy designation, EMA’s PRIME designation, and US and EU Orphan Drug designations for diffuse large B-cell lymphoma (DLBCL). Roche has indicated that, based on positive clinical data from a randomized Phase 2 study (NCT02257567/GO29365),Citation76 the company anticipates an accelerated submission for polatuzumab vedotin in DLBCL in 2018.Citation77 The Phase 2 NCT02257567 study evaluated polatuzumab vedotin administered by IV infusion in combination with standard doses of bendamustine (B) and rituximab (R) or obinutuzumab in patients with relapsed or refractory follicular lymphoma (FL) or diffuse DLBCL. Study results indicating that, at a median follow-up of 15 months, rates of PFS and complete response were similar among FL patients. In contrast, the combination of polatuzumab vedotin and BR significantly increased all efficacy outcomes, compete response (p = 0.012), median PFS (p < 0.0001), and median overall survival (p = 0.0008), in the DLBCL group. The combination of polatuzumab vedotin with R-CHP protocol (rituximab, cyclophosphamide, doxorubicin and prednisone) versus R-CHOP (rituximab-cyclophosphamide, doxorubicin, vincristine and prednisone) in DLBCL patients is currently being investigated in the Phase 3 POLARIX (NCT03274492) study. The primary endpoint is PFS. Secondary outcome measures include PFS, compete response and overall survival. The estimated primary completion date of the study is December 2019.
Isatuximab (SAR650984; Sanofi)
This anti-CD38 IgG1 chimeric mAb is being evaluated as a treatment for patients with multiple myeloma (MM). Isatuximab in combination with chemotherapy is being evaluated in three Phase 3 studies (ICARIA, IKEMA, and IMROZ) of MM patients. Sanofi expects results from ICARIA by Q1 2019; positive clinical data may allow marketing application submissions for isatuximab in 2019.Citation78 In the ICARIA study (NCT02990338), the effects of isatuximab in combination with pomalidomide and low-dose dexamethasone are being compared to those of only the chemotherapy drugs in patients with refractory or relapsed and refractory MM.Citation79 The primary endpoint is PFS. Key secondary endpoints include overall response rate and overall survival. The IKEMA (NCT03275285) and IMROZ (NCT03319667) studies are evaluating isatuximab with other chemotherapy combinations (carfilzomib/dexamethasone and bortezomib/lenalidomide/dexamethasone, respectively) in MM patients. Isatuximab was granted US and EU Orphan Drug designations for the treatment of MM.
Spartalizumab (PDR001, Novartis)
This humanized IgG4 mAb binds PD-1 with sub-nanomolar affinity and blocks interaction with PD-L1/PD-L2, thus preventing PD-1-mediated inhibitory signaling and leading to T-cell activation. Spartalizumab is being evaluated in the randomized, double-blind, placebo-controlled Phase 3 COMBI-i study (NCT02967692) evaluating the safety and efficacy of spartalizumab combined with dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) versus matching placebo in combination with dabrafenib and trametinib in previously untreated patients with BRAF V600–mutant unresectable or metastatic melanoma. Determination of dose-limiting toxicities, changes in PD-L1 levels and CD8+ cells in the tumor microenvironment, and PFS are the primary endpoints of the study. Key secondary endpoints are overall survival, overall response rate and duration of response. The estimated primary completion date of the study is July 2019. Novartis plans to submit marketing applications for PDR001 in combination with trametinib + dabrafenib for the treatment of metastatic BRAF V600+ melanoma in 2019.Citation80 Spartalizumab is undergoing evaluation in Phase 2 studies as a treatment for other cancers, including neuroendocrine tumors, nasopharyngeal carcinoma, non-small cell lung cancer, triple-negative breast cancer, hepatocellular carcinoma and colorectal cancer. Spartalizumab was granted US Orphan Drug designation for the treatment of neuroendocrine tumors.Citation81
MOR208 (MorphoSys)
Formerly known as Xmab®5574, MOR208 is a humanized Fc-engineered mAb directed against CD19. The Fc domain contains 2 amino acid substitutions, S239D and I332E, that enhance cytotoxicity by increasing the affinity for activatory FcγRIIIa on effector cells. MOR208 has been shown to induce lysis of leukemia cells that is mediated by natural killer cells.Citation82 The mAb was granted EU and US Orphan Drug designations for the treatment of chronic lymphocytic leukemia (CLL), as well as FDA’s Breakthrough Therapy and Fast Track designations for DLBCL. MOR208 is under clinical investigation as a treatment option in combination with other anti-cancer agents for both of these hematological malignancies. In March 2018, MorphoSys reported results from the Phase 2 L-MIND study (NCT02399085), which evaluated MOR208 combined with lenalidomide in patients with relapsed or refractory DLBCL.Citation83 A total of 81 patients enrolled in the study, with 68 available for efficacy assessment at the time of data cut-off. With a median observation time of 8.3 months, the data showed an overall response rate of 49%, and a compete response in 31% of the patients. The preliminary PFS rate at 12 months was 50.4% (95% confidence interval 40–67%) and the preliminary median PFS had not been reached (95% confidence interval: 4.3 months-not reached). In November 2018, MorphoSys indicated that they are in discussions with the FDA to evaluate possible paths to market, including the possibility of an expedited regulatory submission and potential approval based primarily on the L-MIND study.Citation84 MOR208 with bendamustine versus rituximab with bendamustine is being evaluated in the Phase 2/3 B-MIND study (NCT02763319) of patients in patients with relapsed or refractory DLBCL. The primary endpoint of the study is PFS and the primary completion date is March 2020.
Oportuzumab monatox (VB4-845, Vicinium™; Sesen Bio)
This immunotoxin is composed of a recombinant humanized antibody scFv targeting epithelial cell adhesion molecule (EpCAM) conjugated to Pseudomonas aeruginosa exotoxin A.Citation85 Once bound to EpCAM expressed by cancer cells, oportuzumab monatox is internalized into the cytoplasm, where it induces apoptosis. Oportuzumab monatox was granted US and EU Orphan Drug designations in 2005, and FDA’s Fast Track designation for non-muscle invasive bladder cancer (NMIBC) that is unresponsive to treatment with bacillus Calmette-Guérin (BCG) in August 2018.Citation86 The efficacy and tolerability of intravesical Vicinium™ is being evaluated in the open-label, multicenter Phase 3 VISTA study (NCT02449239) of NMIBC patients previously treated with BCG. Enrollment was completed in March 2018 with a total of 133 patients with high-grade NMIBC that is either carcinoma in situ (CIS) or papillary with or without CIS, who have been previously treated with BCG. The primary endpoint is the complete response rate in patients with CIS with or without papillary disease. In an analysis assessing pooled CIS patients (n = 77), Vicinium treatment resulted in a complete response rate of 42% at three months.Citation87 Sesen Bio expects to provide a six-month update from the VISTA study in December 2018, and is on track to report 12-month VISTA study data in mid-2019.Citation88 Positive results could allow a BLA submission by the end of 2019.
TSR-042 (TESARO, Inc.)
TESARO is developing anti-PD-1 mAb TSR-042 as a treatment for several types of cancers. In October 2018, TESARO announced results from a Phase 1 dose escalation and cohort expansion study (GARNET; NCT02715284), which is intended to support a BLA submission to the FDA in 2019.Citation89 The GARNET study is evaluating TSR-042 in patients with advanced solid tumors who have limited available treatment options. In the ongoing cohort expansion portion of GARNET, patients are administered TSR-042 at a dose of 500 milligrams every 3 weeks for the first 4 cycles, and 1000 milligrams every 6 weeks thereafter in four cohorts: microsatellite instability high (MSI-H) endometrial cancer, MSI-H non-endometrial cancer, microsatellite-stable endometrial cancer and non-small cell lung cancer. Among the 25 patients with MSI-H endometrial cancer who had at least one post-baseline tumor assessment, one had a complete response and 12 had partial responses (including 1 unconfirmed response) by immune-related response evaluation criteria in solid tumors (irRECIST) criteria.Citation89 Of the 13 responses, 12 are ongoing (92%), including three patients with partial responses who have thus far received over 60 weeks of treatment with TSR-042. Three additional patients (12%) had stable disease. TSR-042 is also being evaluated in the Phase 3 FIRST study (NCT03602859), which is comparing platinum-based therapy with TSR-042 and niraparib versus standard of care platinum-based therapy as first-line treatment of Stage III or IV non-mucinous epithelial ovarian cancer.
Enfortumab vedotin (ASG22ME; Seattle Genetics, Inc./Astellas Pharma Inc.)
This ADC directed against nectin-4 is undergoing evaluation as a treatment for locally advanced or metastatic urothelial cancer. FDA has granted enfortumab vedotin Breakthrough Therapy designation for this disease. Enfortumab vedotin as monotherapy was evaluated at escalating doses in a Phase 1 study (NCT02091999) of 81 patients with locally advanced or metastatic urothelial cancer. The results of the clinical study showed that, of 71 patients evaluated for response, 41% had an objective response, including three (4%) complete responses and 26 (37%) partial responses. Disease control was achieved in 51 patients (72%), defined as the sum of patients achieving a complete response, partial response or stable disease.Citation90 Data from the study supported the start of the pivotal single-arm, open-label, multicenter Phase 2 EV-201 study (NCT03219333), which is evaluating the safety and efficacy of enfortumab vedotin in patients with locally advanced or metastatic urothelial cancer who previously received a checkpoint inhibitor (PD-L1 or PD-1) and either previously received platinum-containing chemotherapy (Cohort 1) or are platinum-naïve and cisplatin-ineligible (Cohort 2). The primary endpoint is the objective response rate, and secondary key outcomes measures include duration of response, disease control rate, and PFS. Seattle Genetics and Astellas expect to report top-line data from the EV-201 study in the first quarter of 2019. Positive data from this study could serve as the basis for a BLA submission under the FDA’s accelerated approval pathway.Citation91 Enfortumab vedotin is also under evaluation in the global Phase 3 EV-301 study (NCT03474107) in the same indication. The EV-301 study is intended to support a broader global registration strategy and to serve as the confirmatory randomized trial in the US. The estimated primary completion date is September 2021.
Ublituximab (TG Therapeutics, Inc.)
The glyco-engineered anti-CD20 antibody ublituximab is currently under clinical investigation in a total of 5 late-stage clinical studies in cancer (CLL, non-Hodgkin’s lymphoma) and non-cancer (multiple sclerosis) indications, with the most advanced being the studies for cancer. TG Therapeutics is awaiting pivotal data from all of them. In hematological malignancies, TG Therapeutics is conducting three Phase 3 studies exploring the efficacy of ublituximab in combination with other anti-cancer agents. The UNITY-CLL Phase 3 study (NCT02612311) evaluated the combination of ublituximab and TGR-1202, a PI3K delta inhibitor, versus anti-CD20 obinutuzumab plus chlorambucil in untreated and previously treated CLL patients. The primary and secondary outcome measures are PFS and overall response rate, respectively. TG Therapeutics had announced plans to prepare and potentially submit a marketing application by the end of 2018, but, in September 2018, TG Therapeutics reported that an interim analysis of the overall response rate in UNITY-CLL trial ‘could not be conducted at this time as the data were not sufficiently mature to conduct the analysis.’Citation92 The company will now focus on PFS data, with a readout anticipated in 2019. Another Phase 3 study (NCT02301156) is comparing ublituximab in combination with Ibrutinib to Ibrutinib alone in previously treated CLL patients. The primary outcome measures are overall response rate and PFS. The estimated primary completion date is June 2019. The Phase 2/3 UNITY-NHL (NCT02793583) study is evaluating ublituximab in combination with TGR-1202 with or without bendamustine in 500 patients with previously treated non-Hodgkin’s lymphoma. Overall response rate is the primary outcome measure and the estimated primary completion date is May 2019.
Two Phase 3 studies (ULTIMATE 1, NCT03277261 and ULTIMATE 2, NCT03277248) are evaluating the efficacy and safety of ublituximab compared to teriflunomide in 440 patients with relapsing multiple sclerosis. For both studies, the primary outcome measure is annualized relapse rate and the estimated primary completion date is March 2021.
Outlook for the near future
Medical care of patients will likely be strongly influenced by the increased number and variety of antibody therapeutics that may be approved in the near future. This is especially the case for cancer patients, who may soon have a substantially larger number of antibody immune checkpoint modulators and ADCs available to them. Of the 33 antibody therapeutics currently in late-stage clinical development for cancer, 11 modulate immune checkpoints and 8 are ADCs. Of the immune checkpoint modulators, ‘antibodies to watch’ include anti-CTLA4 tremelimumab (AstraZeneca), anti-PD-1 spartalizumab (Novartis), anti-PD-1 BCD-100 (Biocad), and radiolabeled anti-B7-H3 omburtamab (Y-mAbs Therapeutics), which are all being evaluated in clinical studies with primary completion dates in late 2018 and in 2019. Looking further into the future, results from studies of antibodies targeting immune checkpoints other than CTLA4, PD-1 and PD-L1 are eagerly anticipated. Results of the Phase 3 study (NCT02951156) of the CD137/4-1BB agonist utomilumab (PF-05082566, Pfizer) in combination regimens in patients with relapsed or refractory diffuse large B-cell lymphoma may be available near the study’s primary completion date of March 2020. In addition, results of the Phase 2/3 study (NCT03470922) of anti-LAG3 relatlimab (Bristol-Myers Squibb) combined with nivolumab versus nivolumab in patients with previously untreated metastatic or unresectable melanoma may be released near the study’s primary completion date of July 2020.
ADC ‘antibodies to watch’ include mirvetuximab soravtansine (ImmunoGen), trastuzumab duocarmazine (Synthon BioPharmaceuticals), and depatuxizumab mafodotin (Abbvie). Mirvetuximab soravtansine, an ADC that targets α-folate receptor 1, is undergoing evaluation in the Phase 3 FORWARD I study (NCT02631876) of patients with folate receptor alpha-positive advanced epithelial ovarian cancer, primary peritoneal cancer or fallopian tube cancer. This study has a primary completion date in November 2018, and top-line results from the study are expected in the first half of 2019. The safety and efficacy of anti-HER2 trastuzumab duocarmazine (Synthon BioPharmaceuticals) is being evaluated in the Phase 3 TULIP study (NCT03262935) of patients with human epidermal growth factor receptor 2 (HER2)-positive unresectable locally advanced or metastatic breast cancer. This study has a primary completion date in May 2019. Trastuzumab duocarmazine was granted a Fast Track designation for treating patients diagnosed with HER2-positive metastatic breast cancer that has progressed during or after at least two HER2-targeting treatment regimens for locally advanced or metastatic disease, or progressed during or after [ado-]trastuzumab emtansine treatment. The anti-epidermal growth factor receptor ADC depatuxizumab mafodotin is being evaluated as a treatment for glioblastoma in two Phase 3 studies, NCT03419403 and NCT02573324, with primary completion dates in September 2019 and March 2020, respectively. In addition, anti-DLL3 rovalpituzumab tesirine (AbbVie) is undergoing evaluation in three Phase 3 studies of small cell lung cancer (SCLC) patients, TAHOE (NCT03061812), MERU (NCT03033511) and M16-292 (NCT03334487), with primary completion dates in September 2019, November 2019, and September 2020. However, AbbVie announced in March 2018 that they will not seek accelerated approval for rovalpituzumab tesirine in third-line relapsed/refractory SCLC based on the magnitude of effect across multiple parameters in the Phase 2 TRINITY study.Citation93
We anticipate that a sufficient number of the ~ 225 antibody therapeutics in Phase 2 studies will transition to late-stage studies to keep the total number in the range of 50–65 in 2019 and beyond. For example, the Phase 3 IMAGINE study (NCT03744910) to evaluate the safety and efficacy of anti-IL-6 clazakizumab for the treatment of chronic active antibody-mediated rejection in kidney transplant recipients is due to start in March 2019, and Phase 3 studies to investigate the efficacy and safety of UB-421 monotherapy as substitution for stable antiretroviral therapy in HIV-1 infected adults (NCT03149211) and UB-421 in combination with optimized background therapy regimen in patients with multi-drug resistant HIV-1 infection (NCT03164447) are due to start in July and September 2019, respectively. We look forward to documenting progress made with these and other ‘antibodies to watch’ in the next installment of this article series.
Abbreviations
| ADC | = | antibody-drug conjugate |
| AE | = | adverse events |
| ARR | = | annualized relapse rate |
| ART | = | antiretroviral therapy |
| aTTP | = | acquired thrombotic thrombocytopenic purpura |
| BCG | = | bacillus Calmette-Guérin |
| BLA | = | biologics license application |
| C5 | = | complement component 5 |
| CCR4 | = | CC chemokine receptor 4 |
| CGRP | = | calcitonin gene-related peptide |
| CHMP | = | Committee for Medicinal Products for Human Use |
| CIS | = | carcinoma in situ |
| CLL | = | chronic lymphocytic leukemia |
| CR | = | complete response |
| CSCC | = | cutaneous squamous cell carcinoma |
| CTCL | = | cutaneous T-cell lymphoma |
| DLBCL | = | diffuse large B-cell lymphoma |
| EC | = | European Commission |
| EMA | = | European Medicines Agency |
| EpCAM | = | epithelial cell adhesion molecule |
| EU | = | European Union |
| FDA | = | US Food and Drug Administration |
| FL | = | follicular lymphoma |
| HAE | = | hereditary angioedema |
| HCL | = | hairy cell leukemia |
| HIV | = | human immunodeficiency virus |
| HLH | = | hemophagocytic lymphohistiocytosis |
| IV | = | intravenous |
| mAb | = | monoclonal antibody |
| MF | = | mycosis fungoides |
| MM | = | multiple myeloma |
| MMD | = | monthly migraine days |
| MSI | = | microsatellite instability |
| nAMD | = | neovascular age-related macular degeneration |
| NDA | = | new drug application |
| NGF | = | nerve growth factor |
| NMIBC | = | non-muscle invasive bladder cancer |
| NMOSD | = | neuromyelitis optica spectrum disorder |
| NMPA | = | National Medical Products Administration |
| PD-1 | = | programmed cell death 1 protein |
| PD-L1 | = | programmed death ligand 1 |
| PFS | = | progression-free survival |
| PNH | = | paroxysmal nocturnal hemoglobinuria |
| SC | = | subcutaneous |
| scFv | = | single-chain variable fragment |
| SCLC | = | small cell lung cancer |
| SS | = | Sézary syndrome |
| US | = | United States |
| XLH | = | X-linked hypophosphatemia |
Disclosure Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Mrinalini (Mini) Muralidharan for preparation of the figures, Vandna Prasad Rath, Hanson Wade, for providing access to the Beacon Targeted Therapies database, and Rob Jones, Craic Computing LLC, for providing access to the Therapeutic Antibody Database.
Related Research Data
References
- The Royal Swedish Academy of Sciences. Scientific background on the Nobel Prize in Chemistry 2018: directed evolution of enzymes and binding proteins. https://www.nobelprize.org/uploads/2018/10/advanced-chemistryprize-2018.pdf
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–1317. doi:10.1126/science.4001944.
- Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249(4967):386–390.
- Frenzel A, Schirrmann T, Hust M. Phage display-derived human antibodies in clinical development and therapy. MAbs. 2016;8(7):1177–1194. doi:10.1080/19420862.2016.1212149.
- Nixon AE, Sexton DJ, Ladner RC. Drugs derived from phage display: from candidate identification to clinical practice. MAbs. 2014;6(1):73–85. doi:10.4161/mabs.27240.
- The Royal Swedish Academy of Sciences. The Nobel Prize in Physiology or Medicine 2018. NobelPrize.org. Nobel Media AB 2018. Fri. [ accessed 2018 Oct 26]. https://www.nobelprize.org/prizes/medicine/2018/summary/.
- Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992;11(11):3887–3895.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736.
- Donini C, D’Ambrosio L, Grignani G, Aglietta M, Sangiolo D. Next generation immune-checkpoints for cancer therapy. J Thorac Dis. 2018;10(Suppl 13):S1581–S1601. doi:10.21037/jtd.2018.02.79.
- Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23(9):1073–1078. doi:10.1038/nbt0905-1073.
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32(1):40–51. doi:10.1038/nbt.2786.
- Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. 2018;10(2):183–203. doi:10.1080/19420862.2018.1415671.
- Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9(2):167–181. doi:10.1080/19420862.2016.1269580.
- Reichert JM. Antibodies to watch in 2016. MAbs. 2016;8(2):197–204. doi:10.1080/19420862.2015.1125583.
- Reichert JM. Antibodies to watch in 2015. MAbs. 2015;7(1):1–8. doi:10.4161/19420862.2015.988944.
- Reichert JM. Antibodies to watch in 2014. MAbs. 2014;6(1):5–14. doi:10.4161/mabs.27333.
- Reichert JM. Which are the antibodies to watch in 2013? MAbs. 2013;5(1):1–4. doi:10.4161/mabs.22976.
- Reichert JM. Which are the antibodies to watch in 2012? MAbs. 2012;4(1):1–3. doi:10.4161/mabs.4.1.18719.
- Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3(1):76–99.
- Reichert JM. Antibodies to watch in 2010. MAbs. 2010;2(1):84–100.
- US Food and Drug Administration. FDA approves novel preventive treatment for migraine. May 17, 2018 press release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm608120.htm?utm_campaign=05172018_PR_FDA%20approves%20migraine%20prevention%20treatment&utm_medium=email&utm_source=Eloqua.
- Teva Pharmaceutical Industries Ltd. Teva announces U.S. approval of AJOVY™ (fremanezumab-vfrm) injection, the first and only anti-CGRP treatment with both quarterly and monthly dosing for the preventive treatment of migraine in adults. September 14, 2018 press release. http://ir.tevapharm.com/phoenix.zhtml?c=73925&p=irol-newsArticle&ID=2367592.
- Dodick DW, Silberstein SD, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, Grozinski-Wolff M, Yang R, Ma Y, Aycardi E. Effect of fremanezumab compared with placebo for prevention of episodic migraine: a randomized clinical trial. JAMA. 2018;319(19):1999–2008. doi:10.1001/jama.2018.4853.
- Lilly E and Company. Lilly’s Emgality™ (galcanezumab-gnlm) receives U.S. FDA approval for the preventive treatment of migraine in adults. September 27, 2018 press release. https://investor.lilly.com/news-releases/news-release-details/lillys-emgalitytm-galcanezumab-gnlm-receives-us-fda-approval.
- Kyowa Hakko Kirin Co. Ltd, Kyowa Kirin International PLC, Ultragenyx Pharmaceutical Inc. Kyowa Kirin and Ultragenyx Announce Crysvita® (burosumab) receives conditional marketing authorisation in Europe for the treatment of X–linked hypophosphatemia in children. February 23, 2018 press release. www.kyowa-kirin.com/news_releases/2018/pdf/e20180223_01.pdf.
- Insogna KL, Briot K, Imel EA, Kamenický P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S, et al., AXLES 1 Investigators. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. J Bone Miner Res. 2018 Aug;33(8):1383–1393. doi:10.1002/jbmr.3475.
- US Food and Drug Administration. FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia. April 17, 2018 press release. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm604810.htm
- Shire plc. Shire announces FDA approval of TAKHZYRO™ (lanadelumab-flyo), a first-of-its-kind mAb preventive treatment for hereditary angioedema. August 23, 2018 press release. https://www.shire.com/en/newsroom/2018/august/4fhgmy
- Sanofi. Cablivi™ (caplacizumab) approved in Europe for adults with acquired thrombotic thrombocytopenic purpura (aTTP). September 3, 2018 press release. http://www.news.sanofi.us/2018-09-03-Cablivi-TM-caplacizumab-approved-in-Europe-for-adults-with-acquired-thrombotic-thrombocytopenic-purpura-aTTP
- Scully M, Cataland S, Peyvandi F, Coppo P, Knoebl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, et al. Results of the randomized, double-blind, placebo-controlled, phase 3 Hercules study of caplacizumab in patients with acquired thrombotic thrombocytopenic purpura. 59th Annual Meeting of the American Society of Hematology; 2017 Dec 12, Atlanta, GA. https://www.ablynx.com/uploads/events/ca854140-58c3-4357-9695-4b81c1996c75-12dec2017_caplacizumabherculesash_final.pdf
- US Food and Drug Administration. FDA approves treatment for two rare types of non-Hodgkin lymphoma. August 8, 2018 press release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm616176.htm
- Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs. 2012;4(4):419–425. doi:10.4161/mabs.20996.
- Pastan I, Onda M, Weldon J, Fitzgerald D, Kreitman R. Immunotoxins with decreased immunogenicity and improved activity. Leuk Lymphoma. 2011;52(Suppl 2):87–90. doi:10.3109/10428194.2011.573039.
- Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Karlin L, Robak T, Gladstone DE, le Coutre P, Dietrich S, Gotic M, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia. 2018;32(8):1768–1777. doi:10.1038/s41375-018-0210-1.
- Regeneron Pharmaceuticals, Inc. FDA approves Libtayo® (cemiplimab-rwlc) as first and only treatment for advanced cutaneous squamous cell carcinoma. September 28, 2018 press release. https://investor.regeneron.com/news-releases/news-release-details/fda-approves-libtayor-cemiplimab-rwlc-first-and-only-treatment
- US Food and Drug Administration. FDA approves new HIV treatment for patients who have limited treatment options. March 6, 2018 press release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm599657.htm
- Emu B, Fessel J, Schrader S, Kumar P, Richmond G, Win S, Weinheimer S, Marsolais C, Lewis S. Phase 3 study of ibalizumab for multidrug-resistant HIV-1. N Engl J Med. 2018;379(7):645–654. doi:10.1056/NEJMoa1711460.
- US Food and Drug Administration. TROGARZO™ (ibalizumab-uiyk) prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761065lbl.pdf
- Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaçi D, Nograles K, Mehta A, Cichanowitz N, Li Q, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017 Jul 15;390(10091):276–288. doi:10.1016/S0140-6736(17)31279-5.
- Blauvelt A, Reich K, Papp KA, Kimball AB, Gooderham M, Tyring SK, Sinclair R, Thaçi D, Li Q, Cichanowitz N, et al. Safety of tildrakizumab for moderate-to-severe plaque psoriasis: pooled analysis of three randomized controlled trials. Br J Dermatol. 2018;179(3):615–622. doi:10.1111/bjd.16724.
- US Food and Drug Administration. FDA approves first treatment specifically for patients with rare and life-threatening type of immune disease. November 20, 2018 press release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm626263.htm.
- Immunomedics. Corporate overview. September 2018. https://www.immunomedics.com/wp-content/uploads/2018/09/Corporate-Presentation-September-2018.pdf
- Alexion Pharmaceuticals. FDA accepts priority review of ALXN1210 as a treatment for patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) in the US. August 20, 2018 press release. http://ir.alexion.com/news-releases/news-release-details/fda-accepts-priority-review-alxn1210-treatment-patients
- Alexion Pharmaceuticals. Alexion announces positive top-line results showing successful phase 3 clinical study of ALXN1210 in complement inhibitor treatment-naïve patients with Paroxysmal Nocturnal Hemoglobinuria (PNH). March 15, 2018 press release. https://news.alexion.com/press-release/product-news/alexion-announces-positive-top-line-results-showing-successful-phase-3-cl.
- AbbVie. AbbVie submits biologics license application to U.S. FDA for investigational treatment risankizumab for moderate to severe plaque psoriasis. April 25, 2018 press release. https://news.abbvie.com/news/abbvie-submits-biologics-license-application-to-us-fda-for-investigational-treatment-risankizumab-for-moderate-to-severe-plaque-psoriasis.htm.
- Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, Papp KA, Sofen H, Puig L, Foley P, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–661. doi:10.1016/S0140-6736(18)31713-6.
- Amgen and UCB. Amgen and UCB provide update on regulatory status of EVENITY™ (romosozumab) in the US. July 16, 2017. https://www.amgen.com/media/news-releases/2017/07/amgen-and-ucb-provide-update-on-regulatory-status-of-evenity-romosozumab-in-the-us/.
- Amgen and UCB. Amgen and UCB resubmit biologics license application (BLA) for EVENITY™ (romosozumab) to the US FDA. July 12, 2018 press release. https://www.amgen.com/media/news-releases/2018/07/amgen-and-ucb-resubmit-biologics-license-application-bla-for-evenity-romosozumab-to-the-us-fda/.
- Zhang H, Deng M, Lin P, Liu J, Liu C, Strohl WR, Wang S, Ho M. Frontiers and opportunities: highlights of the 2nd annual conference of the Chinese antibody society. Antib Ther. 2018 Sep;1(2):65–74. doi:10.1093/abt/tby009.
- Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, Zhang Y, Zhou X, Wang Z, Wang Y, et al. The binding of an anti-PD-1 antibody to FcγRΙ has a profound impact on its biological functions. Cancer Immunol Immunother. 2018;67(7):1079–1090. doi:10.1007/s00262-018-2160-x.
- Beigene Ltd. BeiGene announces acceptance of new drug application for anti-PD-1 antibody tislelizumab in Hodgkin’s lymphoma in China. August 31, 2018 press release. http://ir.beigene.com/phoenix.zhtml?c=254246&p=irol-newsArticle&ID=2365642.
- Innovent Biologics (Suzhou) Co. Ltd. Innovent biologics announces the study result of its anti-PD-1 antibody in Hodgkin lymphoma May 17, 2018 press release. http://innoventbio.com/en/#/news/102.
- LSK BioPharma, Jiangsu Hengrui Medicine. LSK Biopharma and Jiangsu Hengrui medicine announce global clinical collaboration to evaluate the combination of anti-angiogenesis and immuno-oncology therapy for patients with advanced Hepatocellular Carcinoma (HCC). October 21, 2018 press release. http://lskbiopharma.com/lsk-biopharma-and-jiangsu-hengrui-medicine-announce-global-clinical-collaboration-to-evaluate-the-combination-of-anti-angiogenesis-and-immuno-oncology-therapy-for-patients-with-advanced-hepatocellular/.
- Wang C, Liu Y, Nie J, Shen L, Yang Q, Han W Safety and efficacy of decitabine-primed anti-PD-1 (SHR-1210) treatment in patients with relapsed/refractory classical Hodgkin lymphoma. 2018 American Society of Clinical Oncology meeting abstract. https://meetinglibrary.asco.org/record/162336/abstract.
- Zhou C, Gao G, Wu F, Chen X, Li W, Xiong A, Su C, Cai W, Ren S, Jiang T, et al. A phase Ib study of SHR-1210 plus apatinib for heavily previously treated advanced non-squamous non-small cell lung cancer (NSCLC) patients. 2018 American Society of Clinical Oncology meeting abstract. https://meetinglibrary.asco.org/record/160803/abstract.
- Xu JM, Zhang Y, Jia R, Yue CY, Chang L, Liu RR, Zhang G, Zhao CH, Zhang YY, Chen CX, et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric or esophagogastric junction cancer: an open-label, dose escalation and expansion study. Clin Cancer Res. 2018 Oct 22;2018:2484. pii: clincanres. doi:10.1158/1078-0432.CCR-18-2484.
- Fang W, Yang Y, Ma Y, Hong S, Lin L, He X, Xiong J, Li P, Zhao H, Huang Y, et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 2018 Oct;19(10):1338–1350. doi:10.1016/S1470-2045(18)30495-9.
- Huang J, Xu B, Mo H, Zhang W, Chen X, Wu D, Qu D, Wang X, Lan B, Yang B, et al. Safety, activity, and biomarkers of SHR-1210, an anti-PD-1 antibody, for patients with advanced esophageal carcinoma. Clin Cancer Res. 2018;24(6):1296–1304. doi:10.1158/1078-0432.CCR-17-2439.
- Mo H, Huang J, Xu J, Chen X, Wu D, Qu D, Wang X, Lan B, Wang X, Xu J, et al. Safety, anti-tumour activity, and pharmacokinetics of fixed-dose SHR-1210, an anti-PD-1 antibody in advanced solid tumours: a dose-escalation, phase 1 study. Br J Cancer. 2018;119(5):538–545. doi:10.1038/s41416-018-0100-3.
- Scurci I, Martins E, Hartley O. CCR5: established paradigms and new frontiers for a ‘celebrity’ chemokine receptor. Cytokine. 2018;109:.81–93. doi:10.1016/j.cyto.2018.02.018.
- CytoDyn Inc. CytoDyn’s PRO 140 (leronlimab) HIV monotherapy trial results show 92% responder’s rate at 700 mg dose. November 13, 2018 press release. https://www.cytodyn.com/media/press-releases/detail/297/cytodyns-pro-140-leronlimab-hiv-monotherapy-trial.
- Novartis. New analysis of Novartis phase III brolucizumab (RTH258) data reinforces superior reduction of retinal fluid, a key marker of disease activity in nAMD. September 22, 2018 press release. https://www.novartis.com/news/media-releases/new-analysis-novartis-phase-iii-brolucizumab-rth258-data-reinforces-superior-reduction-retinal-fluid-key-marker-disease-activity-namd.
- Novartis. Two-year data for Novartis brolucizumab reaffirm superiority versus aflibercept in reducing retinal fluid in patients with nAMD. October 27, 2018 press release. https://www.novartis.com/news/media-releases/two-year-data-novartis-brolucizumab-reaffirm-superiority-versus-aflibercept-reducing-retinal-fluid-patients-namd.
- Alder Biopharmaceuticals. Alder BioPharmaceuticals® reports third quarter 2018 financial and operating results. November 5, 2018 press release. https://investor.alderbio.com/news-releases/news-release-details/alder-biopharmaceuticalsr-reports-third-quarter-2018-financial.
- Alder Biopharmaceuticals. Transforming the prevention treatment paradigm for migraine patients. November 5, 2018 corporate presentation. https://investor.alderbio.com/index.php/encrypt/files?file=nasdaq_kms/assets/2018/11/06/3-08-52/Corporate%20Deck%20November%205th%20FINAL%20FINAL.pdf&file_alias=18416.
- Alder Biopharmaceuticals. Alder BioPharmaceuticals® presents new one-year data for eptinezumab from PROMISE 1 phase 3 trial demonstrating long-term efficacy in episodic migraine. June 29, 2018 press release. https://investor.alderbio.com/news-releases/news-release-details/alder-biopharmaceuticalsr-presents-new-one-year-data-eptinezumab.
- Alder Biopharmaceuticals. Alder BioPharmaceuticals® presents new six-month data for eptinezumab demonstrating improvement in efficacy in PROMISE 2 phase 3 trial for chronic migraine. June 29, 2018 press release. https://investor.alderbio.com/news-releases/news-release-details/alder-biopharmaceuticalsr-presents-new-six-month-data.
- Horizon Pharma. An overview of Thyroid Eye Disease (TED) and teprotumumab clinical data. October 4, 2018 corporate presentation. http://ir.horizon-pharma.com/static-files/c153d2c9-fae6-4e3b-ab18-7e8485e2c2d4.
- Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, et al. Sickle cell disease. Nat Rev Dis Primers. 2018 Mar 15;4:18010. doi:10.1038/nrdp.2018.10.
- Kutlar A, Kanter J, Liles DK, Alvarez OA, Cançado RD, Friedrisch JR, Knight-Madden JM, Bruederle A, Shi M, Zhu Z, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol. 2018 Oct 8. doi:10.1002/ajh.25308.
- Novartis. Novartis analysis shows crizanlizumab (SEG101) increased the number of patients free of sickle cell pain crises vs placebo during SUSTAIN study. October 9, 2018 press release. https://www.novartis.com/news/media-releases/novartis-analysis-shows-crizanlizumab-seg101-increased-number-patients-free-sickle-cell-pain-crises-vs-placebo-during-sustain-study
- Chugai Pharmaceutical Co., Ltd. Chugai presents results from phase III study of satralizumab in NMOSD at ECTRIMS 2018. October 15, 2018 press release. https://www.chugai-pharm.co.jp/english/news/detail/20181015120001_561.html.
- Denk F, Bennett DL, McMahon SB. Nerve growth factor and pain mechanisms. Annu Rev Neurosci. 2017;40:.307–325. doi:10.1146/annurev-neuro-072116-031121.
- Pfizer Inc., Eli Lilly and Company. Pfizer and Lilly announce positive top-line results from phase 3 trial of tanezumab for the treatment of Osteoarthritis (OA) pain. July 18, 2018 press release. https://www.pfizer.com/news/press-release/press-release-detail/pfizer_and_lilly_announce_positive_top_line_results_from_phase_3_trial_of_tanezumab_for_the_treatment_of_osteoarthritis_oa_pain.
- Eli Lilly and Company. Form 10-Q quarterly report for the quarter ended June 30, 2018. https://www.sec.gov/Archives/edgar/data/59478/000005947818000188/lly-6302018x10q.htm.
- Sehn LH, Kamdar M, Herrera AF, McMillan A, Flowers C, Kim WS, Kim TM, Ozcan M, Trněný M, Demeter J, et al. Adding polatuzumab vedotin (POLA) to bendamustine and rituximab (BR) treatment improves survival in patients with relapsed/refractory DLBCL: results of a phase 2 clinical trial. 23rd Congress of the European Hematology Association, 2018 June 16; Stockholm, Sweden. https://www.primeoncology.org/app/uploads/hematology-updates-stockholm-2018-dlbcl-s802-sehn.pdf
- Roche. Roche virtual late stage pipeline event 2018. September 13, 2018 corporate presentation. https://www.roche.com/dam/jcr:8142e2f9-9525-4a8f-ad1f-d5f605e36991/en/irp20180913.pdf
- Sanofi. Q3 2018 results. October 31, 2018 corporate presentation, slides 38, 41. https://www.sanofi.com/-/media/Project/One-Sanofi-Web/Websites/Global/Sanofi-COM/Home/en/investors/docs/Q-R/Q3_2018_Final_Slides.pdf?la=fr&hash=1746EC206B1D3AEB873C251D8512A5F62C28AE10
- Richardson PG, Attal M, Campana F, Le-Guennec S, Hui AM, Risse ML, Corzo K, Anderson KC. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma: ICARIA phase III study design. Future Oncol. 2018 May;14(11):1035–1047. doi:10.2217/fon-2017-0616.
- Novartis AG Q2 2018 Results. July 18, 2018 investor presentation, slides 36, 37, 81. https://www.novartis.com/sites/www.novartis.com/files/q2-2018-ir-presentation.pdf.
- Novartis. Q2 results confirm full year guidance. Strong pipeline results underpin potential of several highly innovative products. July 18, 2017 press release. https://www.novartis.com/news/media-releases/q2-results-confirm-full-year-guidance-strong-pipeline-results-underpin-potential.
- Kellner C, Zhukovsky EA, Pötzke A, Brüggemann M, Schrauder A, Schrappe M, Kneba M, Repp R, Humpe A, Gramatzki M, et al. The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients. Leukemia. 2013 Jul;27(7):1595–1598. doi:10.1038/leu.2012.373.
- Morphosys. MorphoSys reports updated data from L-MIND study of MOR208 plus lenalidomide in aggressive lymphoma (r/r DLBCL). March 13, 2018. https://www.morphosys.com/media-investors/media-center/morphosys-reports-updated-data-from-l-mind-study-of-mor208-plus.
- Morphosys. MorphoSys AG announces third quarter 2018 results. November 5, 2018. https://www.morphosys.com/media-investors/media-center/morphosys-ag-announces-third-quarter-2018-results.
- Biggers K, Scheinfeld N. VB4-845, a conjugated recombinant antibody and immunotoxin for head and neck cancer and bladder cancer. Curr Opin Mol Ther. 2008;10(2):176–186.
- Sesen Bio. Sesen Bio announces vicinium granted fast track designation by FDA for treatment of non-muscle invasive bladder cancer. August 09, 2018 press release. https://sesenbio.gcs-web.com/news-releases/news-release-details/sesen-bio-announces-vicinium-granted-fast-track-designation-fda.
- Sesen Bio. Phase 3 registration trial for non-muscle invasive bladder cancer achieves 42% complete response rate at three months in carcinoma in situ patients. May 21, 2018 press release. https://sesenbio.gcs-web.com/news-releases/news-release-details/phase-3-registration-trial-non-muscle-invasive-bladder-cancer.
- Sesen Bio. Sesen Bio reports third quarter 2018 financial results and planned VISTA trial readouts. November 8, 2018 press release. https://sesenbio.gcs-web.com/news-releases/news-release-details/sesen-bio-reports-third-quarter-2018-financial-results-and.
- TESARO, Inc. TESARO announces data presentations at ESMO 2018 congress. October 20, 2018. http://ir.tesarobio.com/news-releases/news-release-details/tesaro-announces-data-presentations-esmo-2018-congress-0.
- Seattle Genetics, Inc. Seattle Genetics and Astellas announce updated enfortumab vedotin phase 1 data in metastatic urothelial cancer at 2017 ASCO annual meeting. June 5, 2017 press release. http://investor.seattlegenetics.com/news-releases/news-release-details/seattle-genetics-and-astellas-announce-updated-enfortumab.
- Seattle Genetics, Inc. Seattle Genetics and Astellas announce progress in enfortumab vedotin urothelial cancer clinical development program. October 25, 2018 press release. http://investor.seattlegenetics.com/news-releases/news-release-details/seattle-genetics-reports-third-quarter-2018-financial-results.
- TG Therapeutics. TG therapeutics announces update regarding UNITY-CLL phase 3 trial. September 25, 2018 press release. http://ir.tgtherapeutics.com/news-releases/news-release-details/tg-therapeutics-announces-update-regarding-unity-cll-phase-3.
- AbbVie. AbbVie announces results from phase 2 study evaluating rovalpituzumab tesirine (Rova-T) for third-line treatment of patients with DLL3-expressing relapsed/refractory small cell lung cancer. March 22, 2018 press release. https://news.abbvie.com/news/abbvie-announces-results-from-phase-2-study-evaluating-rovalpituzumab-tesirine-rova-t-for-third-line-treatment-patients-with-dll3-expressing-relapsedrefractory-small-cell-lung-cancer.htm.