
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Preclinical studies of PD-L1 and CTLA-4 blockade have relied heavily on mouse syngeneic tumor models with intact immune systems, which facilitate dissection of immunosuppressive mechanisms in the tumor microenvironment. Commercially developed monoclonal antibodies (mAbs) targeting human PD-L1, PD-1, and CTLA-4 may not demonstrate cross-reactive binding to their mouse orthologs, and surrogate anti-mouse antibodies are often used in their place to inhibit these immune checkpoints. In each case, multiple choices exist for surrogate antibodies, which differ with respect to species of origin, affinity, and effector function. To develop relevant murine surrogate antibodies for the anti-human PD-L1 mAb durvalumab and the anti-human CTLA-4 mAb tremelimumab, rat/mouse chimeric or fully murine mAbs engineered for reduced effector function were developed and compared with durvalumab and tremelimumab. Characterization included determination of target affinity, in vivo effector function, pharmacokinetic profile, and anti-tumor efficacy in mouse syngeneic tumor models. Results showed that anti–PD-L1 and anti–CTLA-4 murine surrogates with pharmacologic properties similar to those of durvalumab and tremelimumab demonstrated anti-tumor activity in a subset of commonly used mouse syngeneic tumor models. This activity was not entirely dependent on antibody-dependent cellular cytotoxicity, antibody-dependent cellular phagocytosis effector function, or regulatory T-cell depletion, as antibodies engineered to lack these features showed activity in models historically sensitive to checkpoint inhibition, albeit at a significantly lower level than antibodies with intact effector function.
Introduction
Monoclonal antibodies (mAbs) that block programmed cell death protein 1 (PD-1), PD-1 ligand (PD-L1), or cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) to enhance anti-tumor T-cell activity have shown promising clinical efficacy in multiple tumor types.Citation1 This success has led to the approval of anti–PD-1 and anti–PD-L1 antibodies by the U.S. Food and Drug Administration (FDA) for melanoma, non–small-cell lung cancer (NSCLC), small-cell lung cancer, triple-negative breast cancer, renal cell carcinoma, Merkel cell carcinoma, classical Hodgkin lymphoma, head and neck squamous cell carcinoma, urothelial carcinoma, cervical cancer, hepatocellular carcinoma, gastric cancer, and microsatellite instability–high or mismatch repair–deficient cancers (MSI-H/dMMR), including colorectal cancerCitation2 (). Likewise, anti–PD-1 and anti–PD-L1 antibodies used in combination with anti–CTLA-4 antibodies have shown signs of additive clinical benefit in melanoma, renal cell carcinoma, MSI-H/dMMR cancers, and NSCLC and have the potential to change the standard of care in these malignancies.Citation2
Table 1. Characteristics of FDA-approved anti-human PD-1, PD-L1, and CTLA-4 mAbs
The FDA-approved anti–PD-1 antibodies nivolumab (Opdivo) and cemiplimab (Libtayo) are fully human antibodies, pembrolizumab (Keytruda) is a humanized antibody, and all are of the immunoglobulin G4 (IgG4) isotype (). This isotype choice is designed to minimize effector functions such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity.Citation15 Amelioration of effector functions is important because anti–PD-1 mAbs with these functions run the risk of depleting activated anti-tumor T cells expressing PD-1 within the tumor microenvironment or peripheral blood of patients with cancer.Citation16,Citation17 Anti–PD-1 antibodies block the interaction of PD-1 (CD279) with both of its ligands, PD-L1 (CD274, B7-H1) and PD-L2 (CD273, B7-DC).
Similar to anti–PD-1 mAbs, clinically approved anti–PD-L1 mAbs block the interaction of PD-L1 with PD-1. However, in contrast to anti–PD-1 mAbs, anti–PD-L1 mAbs also block the interaction of PD-L1 with CD80 while leaving the PD-1–PD-L2 interaction intact.Citation18 The currently approved anti–PD-L1 antibodies durvalumab (Imfinzi), atezolizumab (Tecentriq), and avelumab (Bavencio) are either fully human (durvalumab, avelumab) or humanized (atezolizumab) antibodies of the IgG1 isotypeCitation2 (). Each differs, however, in its fragment crystallizable (Fc) domain. Avelumab has an unmodified IgG1 Fc domain and is therefore capable of mediating effector functions, such as ADCC, against target cells that express PD-L1.Citation19–25 Both durvalumab and atezolizumab have engineered Fc domains that dramatically lower their affinities for Fc gamma receptor (FcγR) binding, thus minimizing their potential for effector functions such as ADCCCitation8,Citation10,Citation26 (). In the case of durvalumab, this action is represented by a three–amino acid change,Citation8 and for atezolizumab, a single amino acid substitutionCitation10 in the Fc domain.
The anti–CTLA-4 mAb ipilimumab is an unmodified, fully human IgG1, and the anti–CTLA-4 mAb tremelimumab is an unmodified, fully human IgG2 isotype antibody (). As expected, ipilimumab is capable of mediating ADCC and ADCP of CTLA-4–expressing target cells in vitro.Citation27–29 In contrast, tremelimumab does not induce natural killer (NK) cell–mediated ADCC, because the IgG2 Fc has low affinity for CD16/FcγRIII.Citation30,Citation31 Data have shown that surrogate anti-mouse CTLA-4 mAbs that mediate regulatory T-cell (Treg) depletion are highly active in certain syngeneic tumor models compared with mAbs that lack effector function.Citation32–34 These findings led to the hypothesis that the anti-tumor activity of anti–CTLA-4 mAbs is dependent on effector function.Citation18,Citation33,Citation34 However, the relevance of Treg depletion for the clinical activity of ipilimumab and tremelimumab has been called into question because a lack of intratumoral Treg depletion has been observed in melanoma cancer patients treated with these antibodies.Citation30,Citation31,Citation35
Mouse syngeneic tumor models have been used for many years to better understand the in vivo mechanisms of action of anti–PD-1, anti–PD-L1, and anti–CTLA-4 mAbs. These models have the advantage of harboring a fully intact immune system in which the effect of immune modulation can be carefully studied. However, because none of the approved anti-human mAbs against PD-1, PD-L1, and CTLA-4 listed in , with the exception of avelumab and atezolizumab, cross-react with the mouse orthologs of their human target, their use is irrelevant in unmodified mouse syngeneic tumor models. Murine surrogates of these antibodies that bind their mouse targets have therefore been developed and are widely used in studies of anti-tumor immunity.
Despite the availability and utility of commercially available murine surrogate checkpoint blockade antibodies, the choice of surrogate must be judicious to ensure the highest chance of clinical translatability. Some of the antibody characteristics that are important to match between a clinical anti-human drug antibody and a surrogate antibody are target affinity, epitope, potency of receptor-ligand blockade for antagonist antibodies, and the relative potential for effector functions such as ADCC and ADCP. The characteristics of the ideal surrogate would qualitatively and quantitatively equal those of its human counterpart. Because anti-human PD-1, anti-human PD-L1, and anti-human CTLA-4 mAbs that are approved or in clinical development differ in these characteristics, it is important to develop murine surrogate antibodies with properties that are similar to those of these particular anti-human antibodies whose pharmacology is to be mimicked in mouse models.
Commercially available murine surrogate antibodies for PD-L1 and CTLA-4 have immunoglobulin isotypes that either do not resemble those of durvalumab or tremelimumab in their potential for effector function or are not intended for in vivo use (, S1 Table). Moreover, commercially available anti-mouse PD-L1 mAbs are not typically derived from mice and contain antibody sequences from other species, such as rats or rabbits (S1 Table). For this reason, antidrug antibodies (ADAs) may develop after repeated administration in mice, which may be seen even in murine chimeras.Citation36 In contrast, ADA formation has not been commonly observed in human clinical trials with durvalumab or tremelimumabCitation13,Citation37,Citation38 (). Because of these undesirable features, we developed a rat/mouse chimeric anti–PD-L1 surrogate antibody, termed “clone 80,” with anti–PD-L1 binding and lack of effector function similar to that of durvalumab, through a de novo hybridoma screening campaign. Likewise, we engineered a murine surrogate anti–CTLA-4 antibody to more closely resemble the binding and effector potential of tremelimumab. Each was assessed for pharmacokinetic (PK) properties, anti-tumor efficacy, and pharmacodynamic activity in mouse syngeneic tumor models.
Results
Characterization of anti-human PD-L1 and CTLA-4 mAbs
The properties of anti-human PD-L1 and CTLA-4 mAbs were characterized with respect to affinity, biological activity, and effector function to enable selection and evaluation of appropriate mouse surrogate mAbs. The values for 50% receptor saturation under equilibrium binding conditions (EC50s) that represent bivalent (avid) antibody binding for durvalumab, atezolizumab, and avelumab to human PD-L1 on the surface of activated primary human T cells, PD-L1–expressing human Jurkat T cells, and human tumor cell lines were in the range of 10–180 pM ( and S1 Fig). Durvalumab showed EC50s (mean ± standard deviation) of 27 ± 4.6 for binding to primary human CD3 T cells, 10 ± 5.7 for PD-L1–overexpressing Jurkat T cells, 86 ± 22 for MDA MB-231 human breast cancer, and 51 ± 15 pM for A498 human lung cancer cell lines. An affinity equilibrium dissociation constant (KD) of 831 pM was found for durvalumab binding to recombinant human PD-L1, as determined by surface plasmon resonance (SPR) measurement (). The EC50s of the anti–CTLA-4 mAb tremelimumab were 0.80 ± 0.42 nM for human CTLA-4 on the surface of CTLA-4–overexpressing Jurkat human T cells and 4.3 ± 4.6 nM for human CTLA-4–expressing Chinese hamster ovary (CHO) cells ( and S1 Fig). Ipilimumab demonstrated EC50s of 2.9 ± 2.1 nM and 2.3 ± 0.61 nM, respectively, for these cells ( and S1 Fig). The KD for binding of recombinant human CTLA-4 was 2.14 ± 0.97 nM for tremelimumab and 4.42 ± 1.01 nM for ipilimumab ().
Table 2. Apparent binding EC50s of anti-human or anti-mouse PD-L1 and CTLA-4 mAbs to target-expressing cells
Table 3. Binding affinities of anti-mouse and anti-human PD-L1 and CTLA-4 mAbs to recombinant protein
The biological activity of each anti-human PD-L1 and anti-human CTLA-4 mAb was measured in Jurkat luciferase reporter systems (S2 Fig). The anti–PD-L1 mAbs demonstrated human PD-L1 neutralization (EC50s) of approximately 300–400 pM (). Likewise, the anti-human CTLA-4 mAbs showed potencies for blocking the CTLA-4–CD80 interaction and inducing reporter activity, with EC50s of 13 nM for tremelimumab and 32 nM for ipilimumab (). These biological activity values, which reflected the relative potencies of the antibodies, maintained trends that were similar to those found for the cell based binding EC50s described above, suggesting that the binding potency of antibodies to targets on cells is positively correlated to their subsequent biological function.
Table 4. Reporter bioassay data, ADCC, and ADCP activity of anti-human PD-L1 and CTLA-4 mAbs
The potential for ADCC by the human anti–PD-L1 and anti–CTLA-4 mAbs was determined by incubating isolated primary human NK cells together with PD-L1–expressing human tumor cells or human CTLA-4–expressing CHO cells, respectively. Consistent with previous reports, we found that atezolizumab did not mediate NK cell–mediated killing, and minimal-to-moderate ADCC (<15% specific activity) was observed for durvalumabCitation10,Citation26 whereas avelumab demonstrated this activityCitation19−13 (S3 Fig, ). Likewise, ipilimumab demonstrated NK cell ADCC but tremelimumab did not, consistent with previously published results.Citation28,Citation30,Citation31 The ability of the mAbs to mediate phagocytosis of target-expressing cells was measured by the ability of in vitro–derived macrophages to engulf target cells in the presence of antibody. Minimal ADCP of ES2 cells that endogenously express PD-L1 was observed for durvalumab but not for atezolizumab, but avelumab did mediate antibody-mediated phagocytosis (S4 Fig, ). None of the anti–PD-L1 mAbs demonstrated macrophage-mediated cell killing of non-phagocytosed cells, as determined by uptake of dead cell stain (S4 Fig). As reported elsewhere,Citation27,Citation29 we confirmed that both tremelimumab and ipilimumab induced ADCP of CTLA-4–expressing cells in vitro, with ipilimumab demonstrating greater potency (S4 Fig, ). Likewise, both antibodies demonstrated the ability to mediate macrophage-mediated killing of non-phagocytosed target cells, again with ipilimumab demonstrating superior potency.
Generation and characterization of the anti-mouse PD-L1 surrogate antibody clone 80
Together with existing clinical data, the preclinical data presented here and in previously published reportsCitation26,Citation38 suggest that the ideal murine surrogate for durvalumab would be an antibody of relatively high affinity that lacks effector function and demonstrates a relatively low incidence of ADA formation. Therefore, we sought to generate an anti-mouse PD-L1 surrogate mAb with these properties.
To generate candidates for anti-mouse PD-L1 mAb surrogates, rats were immunized with a recombinant mouse PD-L1–human Fc chimeric fusion protein, and hybridomas were generated. A total of 3,405 hybridoma clones secreting IgG were selected and screened in two binding assays (binding to mouse PD-L1 and non-binding to mouse PD-L2–human Fc). Positive clones were further characterized in three epitope competition inhibition assays, including blockade of interactions between mouse PD-L1 and mouse PD-1, between mouse PD-L1 and mouse CD80, and between mouse PD-L1 and the commercially available rat anti–PD-L1 clone 10 F.9G2 (referred to here as “clone 10 F.9G2”) mAbs, using homogeneous time-resolved fluorescence resonance energy transfer (HTRF) (S5 Fig). Of the 136 antibodies identified from the primary high-throughput binding assays, 125 were purified and tested in the three competition HTRF assays, using an 11-point titration as single replicates. Thirty-six showed weak or no inhibition in at least one of the three competition assays. In total, 85 hits were identified that bound natively expressed mouse PD-L1 on Renca cells (and not the closely related mouse PD-L2 protein or human Fc tag; data not shown), showed inhibition in receptor-ligand competition assays with mouse PD-1 and mouse CD80, and competed for binding with the mouse PD-L1–positive control clone 10 F.9G2 (). For the 85 hits identified, the amino acid sequences of the variable heavy (VH) and light (VL) chains showed high sequence diversity, based on VH and VL complementarity-determining region 3 (CDR3) sequences, with only 13% of the clones having identical HCDR3 and LCDR3 pairings. The CDR3 regions typically are principle drivers of antibody paratope-epitope recognition. Of these same 85 hits, four were selected for further testing in additional binding and competition assays based on their activity in screening assays and sequence diversity information. Each were confirmed as highly potent competitors of mouse PD-1–mouse PD-L1 and mouse CD80–mouse PD-L1 receptor-ligand inhibitors, having 50% inhibitory concentrations (IC50s) of 10–25 pM, and all competed for the same epitope as clone 10 F.9G2 and bound to mouse tumor cells expressing mouse PD-L1 (S2 Table). The four antibodies in the final panel also had highly diverse variable-chain sequences ().
Figure 1. Characterization of antibodies identified from a hybridoma screen in biochemical competition and cell binding assays. Hybridoma clone testing in the (a) mouse PD-L1–mouse CD80, (b) mouse PD-L1–mouse PD-1, and (c) mouse PD-L1–mAb 10 F.9G2 competition assays. (d) Of the inhibitory antibodies, 85 bound mouse PD-L1–positive Renca cells by flow cytometry with geomean fluorescence above the negative control. The dotted lines represent geomean fluorescence binding for the rat IgG2b control antibody (lower panel) and the 10 F.9G2-positive control mAb tested at 20 µg/mL (upper panel). The four IgGs denoted by red triangles (clones 17, 37, 56, and 58) were subcloned into recombinant expression vectors and were expressed and purified for further characterization in vivo. (e) Amino acid sequences of the VH and VL regions of these four clones. Statistical P values indicate one-way analysis of variance with the Dunnet posttest. FW, framework
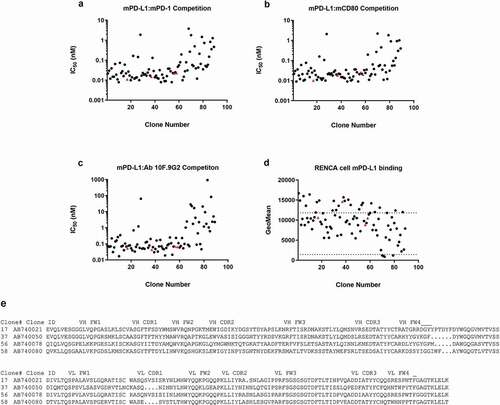
Based on the mouse PD-L1 binding, mouse PD-1 and mouse CD80 receptor-ligand blocking, and antibody purification characteristics, the anti-mouse PD-L1 antibody AB740080 was taken forward for further Fc engineering and characterization. The rat VL of clone AB740080 was grafted onto the mouse constant kappa light chain, and the rat VH of clone AB740080 was grafted on to the mouse IgG1 backbone (CH1-CH3), which contains a single amino acid substitution (D265A) designed to minimize binding of the antibody to mouse FcγRs and negate Fc-mediated effector function.Citation39 This rat/mouse chimeric antibody was thereafter referred to as “clone 80.”
To determine whether the affinity of clone 80 for mouse PD-L1 was comparable to those of the anti–human PD-L1 mAbs and clone 10 F.9G2, we measured the binding affinity of clone 80 for recombinant mouse PD-L1 and determined the EC50s for natively expressed mouse PD-L1 on the surface of murine tumor cell lines by flow cytometry. The KD of clone 80 binding to mouse PD-L1 was 2.8 nM, whereas that of clone 10 F.9G2 was 1.9 nM (). The EC50 for native mouse PD-L1 on the surface of mouse tumor cell lines was 270 pM for EMT6 tumor cells and 2,500 pM for CT26 cells (). Based on these results, clone 80 appeared to have a binding potency for recombinant PD-L1 and native PD-L1 expressed on cell surfaces that was approximately 3.4- and 10-fold lower, respectively, than that of durvalumab. This was the most potent anti-mouse PD-L1 mAb isolated from the hybridoma screen, and so was progressed to in vivo PK and anti-tumor efficacy studies in combination with anti-mouse CTLA-4 mAbs, as described in the following section.
Generation and characterization of an anti-mouse CTLA-4 tremelimumab surrogate antibody
To generate an anti–CTLA-4 mAb with reduced potential for effector function, the antigen-binding fragment (Fab) sequence of the anti-mouse CTLA-4 mIgG2b clone 9D9 mAbCitation33 (referred to here as “mIgG2b clone 9D9”) was synthesized and inserted into an expression vector for production of mouse IgG1 Fc isotype variants. Previous studies have demonstrated that the mouse IgG2b isotype of anti-mouse CTLA-4 mAbs possesses effector function and is capable of depleting cells expressing high levels of CTLA-4 within tumors but not within tumor-draining lymph nodes or spleen, whereas the mIgG1 isotype (referred to here as “mIgG1 clone 9D9”) lacks this function.Citation32–34 After purification, the surrogate anti-mouse CTLA-4 mAbs were characterized to determine EC50s for binding to native mouse CTLA-4 expressed on CHO cells and to recombinant mouse CTLA-4. As expected, the two isotypes demonstrated comparable apparent EC50s of 342 ± 194 pM for the mIgG2b isotype and 743 ± 452 pM for the mIgG1 isotype (). For binding to recombinant mouse CTLA-4, affinity KDs were determined to be 10.1 ± 0.58 nM for the mIgG2b isotype and 18.9 ± 0.928 nM for the mIgG1 isotype. The binding affinity for mIgG1 clone 9D9 was relatively comparable to that for tremelimumab (KD ratio, 8.8). Based on this and the murine isotype having minimal effector function potential, mIgG1 clone 9D9 was progressed to in vivo studies.
In vivo PK of durvalumab and tremelimumab murine surrogates
To further elucidate the PK characteristics of the anti-mouse PD-L1 surrogate mAbs, two PK studies, one using a single dose and one a multiple dose, were completed in CD1 mice. In the single-dose study, a single intravenous administration of clone 80 or clone 10 F.9G2 was administered (). Serum concentration data from multiple animals were used to estimate PK parameters (). A comparison of the PK for the 10-mg/kg single intravenous doses indicated that the maximum observed concentration for clone 80 (1,490 μg/mL) was greater than that for clone 10 F.9G2 (671 µg/mL), driven by a smaller terminal volume of distribution (10.9 and 21.3 mL/kg, respectively). Clone 80 had a slower rate of clearance than clone 10 F.9G2 (0.111 and 0.390 mL/h/kg, respectively) and as a result, a longer terminal half-life (68.3 and 37.9 h, respectively). A single 1-mg/kg dose of clone 80 was also administered and was eliminated almost three-fold more rapidly than the 10-mg/kg dose (0.273 and 0.111 mL/h/kg, respectively), suggesting nonlinear PK. Because a 1-mg/kg dose of clone 10 F.9G2 was not tested, it was not possible to determine whether nonlinear PK would be observed at that dose.
Table 5. PK parameters from the single-dose study*
Figure 2. PK profiling of mAbs. In vivo PK profiles from (a) a single-dose study and (b) a multiple-dose study of anti-mouse PD-L1 clone 80 mIgG1 D265A, anti-mouse PD-L1 clone 10 F.9G2, and anti-mouse mIgG1 CTLA-4. Measured serum mAb concentrations are indicated on the y-axis
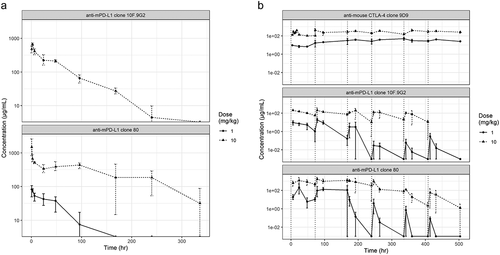
Figure 2. (Continued)
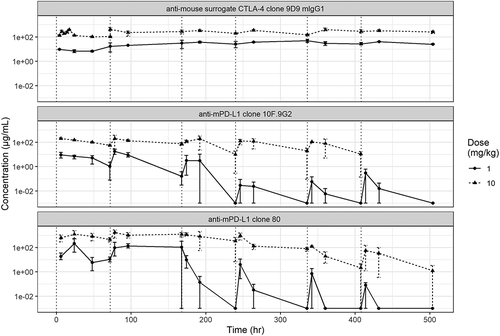
In the second, multiple-dose PK study, 1- and 10-mg/kg doses of clone 80, clone 10 F.9G2, and mIgG1 clone 9D9 were administered twice weekly to CD1 mice (). Serial PK samples from multiple mice were used to estimate PK parameters for each antibody and dose level (). Of the three antibodies tested, clone 80 had the greatest exposure in terms of average concentration and area under the concentration-time curve for each dose interval (AUCτ), with an exposure of up to eight-fold greater than those of clone 10 F.9G2 and mIgG1 clone 9D9 for the 1-mg/kg dose (AUCτ of 4,940, compared with 431 and 656 µg/mL*h, respectively) and six-fold greater for the 10-mg/kg dose (AUCτ of 61,000, compared with 9,360 and 10,900 µg/mL*h, respectively). Clone 80 also had the slowest clearance rate, but did not have the longest terminal half-life of all the antibodies tested. The mIgG1 clone 9D9 had the longest terminal half-life, which was five-fold longer than those of clone 80 and clone 10 F.9G2. The terminal half-lives in the multiple-dose study were generally shorter than those in the single-dose studies, which is probably indicative of enhanced clearance due to ADAs. The longer half-life of mIgG1 clone 9D9 resulted in accumulation that was greater than two-fold those of clone 80 and clone 10 F.9G2. Accumulation was not observed for clone 80 or clone 10 F.9G2; instead, enhanced elimination was observed after four to six doses, probably owing to ADAs in mouse sera at these time points. Confirmatory assays would be needed to verify the presence of ADAs.
Table 6. PK parameters from the multiple-dose study*
In vivo anti-tumor activity of clone 80 and anti–CTLA-4 murine surrogates of various effector function potentials
After determination of the PK properties of clone 80, the in vivo anti-tumor efficacy of the antibody in mouse syngeneic tumor models was tested as monotherapy and in combination with either effector function–deficient (mIgG1clone 9D9) or enabled (mIgG2b clone 9D9) anti-mouse CTLA-4 mAbs. The efficacy varied in tumor models, as drug-mediated anti-tumor effects were observed in some models (CT26 colon, EMT6 breast, and MCA205 fibrosarcoma), but were minimal to absent in others (, –c, S6 and S7 Fig, S3 Table). As expected, mixed responses (stronger tumor control in some mice relative to others) were observed within each syngeneic model. In the immunogenic EMT6 breast cancer model, strong monotherapy activity was observed for clone 80 and for both the mIgG1 and mIgG2b isotypes of clone 9D9, with complete responses commonly observed (, , S6 Fig). Combination of clone 80 with either isotype of clone 9D9 likewise produced strong activity in this model, as most mice achieved complete anti-tumor responses. Moderate activity was observed for clone 80 monotherapy in both the CT26 colon and MCA205 sarcoma models, with few complete responses (, c, , S6 Fig). In these models, monotherapy with the mIgG2b isotype of clone 9D9 produced moderate to strong activity, whereas minimal activity was observed for the mIgG1 isotype. Combination of clone 80 with either isotype of clone 9D9 in the CT26 model showed additive anti-tumor activity. For the MCA205 model, combination of clone 80 with the mIgG1 isotype of clone 9D9 demonstrated additive activity, whereas additive activity of clone 80 combined with the mIgG2b isotype could not be assessed due to the high percentage of responses to monotherapy with the mIgG2b isotype in this model (, , S6 Fig).
Table 7. Complete response rate of anti-mouse PD-L1 clone 80 and anti-mouse CTLA-4 mAbs across eight mouse syngeneic tumor models
Figure 3. Anti-tumor efficacy and pharmacodynamic profiling of mAbs. Shown are in vivo efficacy and pharmacodynamic effects of anti-mouse PD-L1 and CTLA-4 surrogates in mouse syngeneic tumor models that are sensitive to checkpoint inhibition. (a–c) Kaplan-Meier survival curves of mice engrafted with EMT6, CT26, or MCA205 tumors and treated with anti–PD-L1 or anti–CTLA-4 mAbs. (d–g) Pharmacodynamic activity of monotherapy or combination anti-PD–L1 plus anti–CTLA-4 therapy in EMT6, CT26, and MCA205 models as measured by CD8 or conventional CD4 T-cell proliferation (Ki67 positivity). (h–j) Proportion of FoxP3+ Tregs in tumors, tumor-draining lymph nodes, and spleens of mice engrafted with CT26 tumors
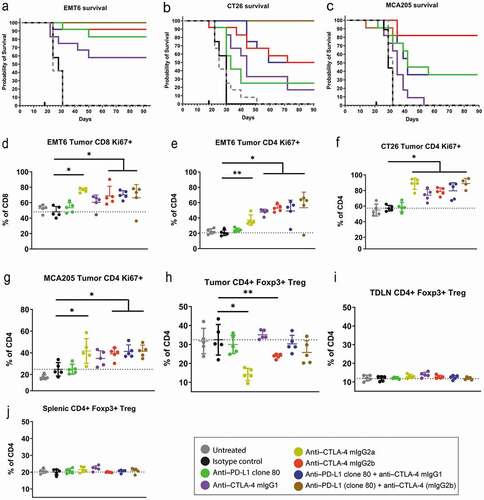
In line with the anti-tumor efficacy observed in the CT26, EMT6, and MCA205 models, pharmacodynamic activity was observed for the anti–CTLA-4 mAbs administered as monotherapy and in combination with the anti–PD-L1 mAbs. The presence of proliferating CD4 or CD8 T cells in tumors, as measured by Ki67-positive cells, was significantly higher for the mIgG2b isotype of clone 9D9 administered alone or in combination with clone 80 than that for the isotype control group (–g). Proliferation or induction of infiltration of proliferating cells induced by monotherapy with mIgG1 clone 9D9 was also numerically higher (although not statistically significant) in the EMT6 and MCA205 models and was statistically significantly higher in the CT26 model than in the isotype control group. Likewise, combination of the mIgG1 or mIgG2b isotype of clone 9D9 with clone 80 induced proliferation or infiltration of proliferating cells that was numerically higher than in monotherapy controls, although the differences did not reach statistical significance.
Other studies have reported that depletion of CTLA-4–expressing Tregs may be required for the activity of anti-mouse CTLA-4 antibodies in mouse syngeneic tumor models.Citation33,Citation34 We therefore examined Treg levels within tumor and spleen after treatment with clone 80, the mIgG1 or mIgG2b isotype of clone 9D9, or combinations thereof in the mouse EMT6, CT26, and MCA205 syngeneic models that showed response to these therapies. Consistent with previous reports, the mIgG2b isotype of clone 9D9, as well as the effector-capable mIgG2a isotype, significantly reduced tumor Treg percentages 3 days after drug treatment without affecting levels in tumor-draining lymph nodes or spleen (–j). In contrast, the mIgG1 isotype did not alter Treg percentages in any of these tissues at 3 or 10 days after treatment, consistent with the relative lack of effector function for this isotype. These data, which showed a complete lack of Treg depletion in models where mIgG1 clone 9D9 showed monotherapy or combination therapy activity, suggest that intratumoral Treg depletion augmented the activity of CTLA-4 antibodies in some mouse tumor models, but is not an absolute requirement for anti-tumor efficacy.
Discussion
Commercially available, receptor-ligand blocking antibodies for mouse PD-L1 and CTLA-4 are often used in anti-tumor studies to assess their mechanisms of action and anti-tumor efficacy. These reagents are surrogates for their anti-human PD-L1 and CTLA-4 counterparts, and the intention of these studies is generally to generate or test hypotheses regarding targeting PD-L1 or CTLA-4 that could be translated to the human clinical setting. If this is the intention, then the murine surrogates ought to resemble their anti-human counterparts as closely as possible. With this in mind, we generated anti-mouse PD-L1 and anti-mouse CTLA-4 surrogate mAbs that were similar to durvalumab and tremelimumab, respectively, in their binding and effector functions and tested them for their PK profiles and efficacy in mouse syngeneic tumor models.
To generate candidates for anti-mouse PD-L1 mAb surrogates for durvalumab, rat hybridomas were produced and antibodies screened for mouse PD-L1 binding and ability to block mouse PD-L1 binding to mouse PD-1 and mouse CD80. From this screen, the top candidate was identified and grafted to a mouse IgG1 lacking effector potential to produce “clone 80.” This anti-mouse PD-L1 surrogate was engineered to eliminate effector function (ADCC and ADCP) and resembles durvalumab in this regard. The relative affinity of the antibody for mouse PD-L1 was less than that of durvalumab for human PD-L1, and some degree of epitope difference may exist between clone 80 and durvalumab. However, with its potent blockade of both mouse PD-1 and mouse CD80 binding to mouse PD-L1, Fc engineering to limit effector function, and in vivo activity, clone 80 has been identified as an appropriate murine surrogate for durvalumab. Of note, although clone 10 F.9G2 has similar binding and receptor-ligand binding properties, it differs from clone 80 in having effector function potential as a rat IgG2b. This ability to mediate effector function is not desired for murine surrogates of durvalumab, due to the minimal potential of these antibodies to mediate effector function in humans. In this case, clone 10 F.9G2 may be a better anti-mouse PD-L1 surrogate for avelumab, which as a human IgG1 isotype has demonstrated in vitro effector function.Citation19–25 As an alternative to a de novo hybridoma antibody approach, we could have modified existing surrogate antibodies, such as 10 F.9G2, to the mouse IgG1 isotype with a D265A mutation; however, since the protein sequence of 10 F.9G2 was not available to us at the time of surrogate generation, we did not take this approach. Instead, we took advantage of the availability of the protein sequence of anti-mouse CTLA-4 clone 9D9 for anti-mouse CTLA-4 generation and generated the tremelimumab surrogate from existing 9D9 sequence information.
Because durvalumab and tremelimumab have a low propensity to develop ADA responses in patients treated with these immunotherapies,Citation37,Citation38 the ideal murine surrogates would also infrequently generate ADAs in mice. For single doses of clone 80 and clone 10 F.9G2, PK parameters were consistent with those of an IgG in mouse. Clone 80 had generally more favorable PK properties; the 10-mg/kg dose was eliminated three-fold more slowly than the equivalent dose of clone 10 F.9G2. The 1-mg/kg dose of clone 80 was eliminated at a greater rate than the 10-mg/kg dose, which could be indicative of target-mediated drug disposition. Because the 1-mg/kg dose of clone 10 F.9G2 was not tested, it is not possible to predict whether nonlinearity would be present. In the multiple-dose study, drug concentrations dropped considerably between the fourth and sixth doses for both clone 80 and clone 10 F.9G2. These large drops in serum drug concentrations are consistent with enhanced clearance from ADA responses. Due to sample volume limitations, it was not possible to perform ADA confirmatory assays, but the timing and behavior of the enhanced clearance was a strong indicator of the presence of ADAs. Both clone 80 and clone 10 F.9G2 contain substantial numbers of rat amino acids; rat variable sequence is present on clone 80 and unspecified rat sequences are present in clone 10 F.9G2. The ADA responses were probably triggered by these rat CDR sequences, as murinization of clone 80, by engrafting the rat CDRs onto mouse framework regions, had little impact on PK properties or ADA responses observed in vivo (data not shown).
These data show that caution must be exercised in predicting long-term drug exposure when using species-unrelated immunoglobulins in mice. Although these surrogates are efficacious in some mouse anti-tumor models, the efficacy probably results from anti-tumor effects created by receptor-ligand blockade that was present before precipitous drops in serum drug levels (apparent ADA response). Therefore, potentially ADA-generating murine surrogates must be used with caution in models where chronic administration is required beyond a time when ADAs may form. In these studies, it is prudent to measure drug exposure in treated animals to verify target coverage during in vivo experiments. Likewise, given these limitations, there is a need to generate fully mouse anti-target surrogate mAbs that may not generate the ADAs we have observed for the rat/mouse chimeric anti-mouse PD-L1 clone 80 and the rat anti-mouse PD-L1 clone 10 F.9G2.
Despite the apparent appearance of ADAs for clone 80, we verified the anti-tumor activity of the durvalumab and tremelimumab surrogate antibodies when administered as monotherapy and in combination in syngeneic mouse tumor models. The results showed that clone 80 was effective when given as monotherapy or in combination with mIgG1 clone 9D9 in mouse models known to demonstrate sensitivity to checkpoint inhibitors. Use of a Treg-depleting anti–CTLA-4 surrogate mAb (mIgG2b clone 9D9) produced superior anti-tumor activity compared with a non–Treg-depleting tremelimumab surrogate (mIgG1 clone 9D9). However, the activity of mIgG1 clone 9D9 as monotherapy and in combination with clone 80 suggests that, although Treg depletion may augment the activity of CTLA-4 blockade in these mouse models, it is not an absolute requirement for effective anti-tumor immunity, as has been suggested by others.Citation18,Citation33,Citation34,Citation40–42 Given the apparent lack of Treg depletion observed for anti–CTLA-4 mAbs in human clinical trials,Citation30,Citation31,Citation35 we propose that the non–Treg-depleting, anti-mouse CTLA-4, mIgG1 clone 9D9 mAb is the appropriate surrogate to mimic the mechanism of action of tremelimumab. Although a D265A mutation could be added to the Fc domain of an anti-mouse CTLA-4 mAb to further ensure lack of effector function, we found that the mIgG1 did not lead to Treg depletion, as has been previously reported for the mIgG1 and mIgG1-D265A isotypes of clone 9D9,Citation33 and thus chose not to further modify mIgG1 clone 9D9.
In summary, we generated anti-mouse surrogate antibodies for durvalumab and tremelimumab that were more similar to their human counterparts than commercially available antibodies. Our finding of poor exposure for clone 80 and clone 10 F.9G2 upon repeat dosing suggests that ADAs are likely to form and highlights the need for the development of fully mouse anti-PD-L1 surrogate mAbs that lack this attribute. The finding that non-depleting anti-mouse CTLA-4 surrogate antibodies demonstrate efficacy in murine syngeneic tumor models shows that Treg depletion is not an absolute requirement for efficacy and likewise suggests that the anti-mouse CTLA-4 mIgG1 clone 9D9 mAb is an appropriate surrogate for the anti-human CTLA-4 drug tremelimumab.
Materials and methods
Use of laboratory animals
All animal experiments were conducted in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care under the guidelines of the Institutional Animal Care and Use Committee and appropriate animal research approvals.
Immunization for anti-mouse PD-L1 mAb
Female Sprague–Dawley rats (6–8 weeks of age; Charles River Laboratories) were injected subcutaneously with 20 µg of recombinant mouse PD-L1–Fc fusion protein (R&D Systems, catalog no. 1019-B7-100) in Freund complete adjuvant (Sigma-Aldrich). Three subsequent subcutaneous booster injections of 20 µg of recombinant mouse PD-L1–Fc protein in Freund incomplete adjuvant (Sigma-Aldrich) were administered at 7, 14, and 22 days after the initial priming. Two days after the fourth booster injection, a final injection of 20 µg of recombinant mouse PD-L1–Fc in phosphate-buffered saline (PBS) was administered intraperitoneally. Four days after the last injection, rats were euthanized and lymph nodes were harvested.
Hybridoma generation
Cells were isolated from lymph nodes by mechanical disruption with a gentleMACS dissociator (Miltenyi) and were fused with SP2/0 myeloma cells (American Type Culture Collection), using a Legacy ECM2001 electrofuser (BTX Molecular Delivery Systems). After electrofusion, cells were resuspended in semisolid selection media containing fluorescein isothiocyanate (FITC)–conjugated anti-rat IgG mAbs (CloneMatrix concentrate; Genetix, catalog #K8510), Dulbecco modified Eagle medium (DMEM) powder (Gibco), 20% fetal bovine serum (SAFC Biosciences), 2% GlutaMAX (Gibco), 1% sodium pyruvate (Sigma-Aldrich), 1% penicillin-streptomycin (Gibco), 10% hybridoma cloning factor (Roche), 2% oxaloacetate-pyruvate-insulin (Sigma-Aldrich), 2% hypoxanthine-azaserine (Sigma-Aldrich), 11 µg/mL goat anti-rat IgG-FITC (Jackson ImmunoResearch, catalog no. 115–095-003), 4 µg/mL goat anti-rat IgG1-FITC (Novus Biologicals, catalog no. NB7119), and 4 µg/mL goat anti-rat IgG2b-FITC (Novus Biologicals, catalog no. NB7129). Medium containing cells was divided into Nunc OmniTrays (Thermo Fisher Scientific), and cells were grown at 37°C under a 7.5% CO2–enriched atmosphere. At 13–16 days after fusion, IgG-secreting hybridomas were detected and picked into 96-well plates (Costar) containing medium consisting of DMEM, 20% fetal calf serum, 2% GlutaMAX (Gibco), 1% penicillin-streptomycin, 10% hybridoma cloning factor, 2% oxaloacetate-pyruvate-insulin, and 2% hypoxanthine and thymidine (Sigma-Aldrich), using a ClonePix robot (Molecular Devices). Hybridomas were grown for 3–7 days before supernatant was harvested from each well with a MiniTrak robot (PerkinElmer).
Complementary DNA preparation and variable-chain sequencing
Hybridoma messenger RNA (mRNA) for VH and VL regions were isolated with Oligo (dT)25 Magnetic Beads (Novagen) in combination with a Kingfisher 96 automated magnetic separator (Thermo Fisher Scientific). Contaminating SP2/0 MOPC abVκ mRNA was removed by targeted digestion with RNAse H (NEB) at 37°C for 1 h. Purified mRNA was transcribed to complementary DNA by using Superscript III reverse transcriptase (Invitrogen) at 44°C for 1 h and then tailed with poly(G) by incubation with excess deoxyguanosine triphosphate and terminal transferase (New England Biolabs) at 37°C for 1 h. VH and VL genes were amplified with Taq polymerase (Thermo Fisher Scientific) in separate reactions, using oligo(dC)25 and specific primers to either the CH1 or kappa constant domain. Chain termination method was used to sequence the polymerase chain reaction product.
IgG purification
Selected hybridomas were cultured in serum-free medium (HL-1 [Lonza], 2% HybER-Zero [Statins Serum Institute], 2% GlutaMAX [Gibco]) for 10 days at 37°C under a 7.5% CO2–enriched atmosphere. IgG was purified from the culture supernatants using either ProPlus or a Phytip column containing CaptureSelect Multi-Species Resins (Phynexus) on a MiniTrak robot (PerkinElmer). Absorbance of purified material was read using an EnVision plate reader (PerkinElmer) at 280 nm, and IgG concentration was determined using rat IgG isotype calibration curves.
Converting IgGs to mouse IgG1 D265A
Clones were converted to a mouse IgG1 D265A antibody format essentially as described by Persic et al.,Citation43 with the following modifications. An oriP fragment was included in the expression vectors to facilitate use with CHO transient cells and to allow episomal replication. The VH domain was cloned into a vector containing the mouse heavy-chain constant domains and regulatory elements to express whole IgG1 heavy chain in mammalian cells. This constant region contained the D265A mutation, resulting in a mouse IgG1 with reduced effector function, as previously described.Citation44 Similarly, the VL domain was cloned into a vector for the expression of the mouse light-chain (kappa) constant domains and regulatory elements to express whole IgG light chain in mammalian cells. To obtain IgGs, the heavy- and light-chain IgG–expressing vectors were transfected into CHO transient mammalian cells.Citation45 IgGs were expressed and secreted into the medium. Harvested media were filtered before IgG purification from culture supernatant, using AKTAxpress (GE Healthcare) and 1-mL MabSelect Sure columns (GE Healthcare). IgGs were eluted from the columns with 0.1 M sodium citrate (pH 3.0) and buffer exchanged into PBS with PD10 columns (GE Healthcare). The concentration of the antibodies was determined spectrophotometrically, using extinction coefficients based on the individual amino acid sequences of the IgGs.Citation46 Integrity and purity of purified IgGs were determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and high-pressure liquid chromatography–size exclusion chromatography, and endotoxin levels were quantified by using a LAL Kinetic QCL Kit (Lonza). Purified IgGs all had a purity of greater than 95% and endotoxin levels of less than 0.5 endotoxin units per mg.
Flow cytometry binding experiments with hybridoma supernatants
To determine the binding of antibody to mouse PD-L1 in hybridoma supernatants, Renca mouse renal carcinoma cells were incubated with either 10-fold diluted supernatants from hybridoma cultures, 20 µg/mL rat anti-mouse PD-L1 IgG2b antibody (clone 10 F.9G2) (Bio X cell, catalog no. BE0101), or rat IgG2b control antibody (AstraZeneca) for 1 h at 4°C. After washing, cells were resuspended in 2.5 µg/mL Alexa Fluor® 488–conjugated goat anti-rat IgG secondary antibody (Invitrogen, catalog no. A-11006) and incubated for a further 20 min at 4°C. Immunolabeled Renca cells were fixed in 3.7% formaldehyde and analyzed with a BD FACSCanto II instrument (BD Biosciences). Data analysis was performed using FlowJo software (Becton Dickinson).
HTRF competition assays
Biochemical assays with HTRF technologyCitation47 were established to measure the binding of mouse PD-L1 to mouse PD-1, mouse CD80, and clone 10 F.9G2. Recombinant mouse PD-1 Fc chimera protein (R&D Systems, catalog #1021-PE-100), recombinant mouse B7-1/CD80 Fc chimera His-tag protein (R&D Systems, catalog #740-B1-100), and purified anti-mouse PD-L1 clone 10 F.9G2 (BioLegend, catalog #124302) were conjugated with DyLight-650 (Innova Biosciences) according to the manufacturer’s directions. PD-L1 Fc was conjugated with europium cryptate trisbipyridine–N-hydroxysuccinimide (Cisbio) according to the manufacturer’s directions, and the cryptate conjugate assay dilution factor was determined by comparing the fluorescence (at 590 nm) with the supplied calibrator. For the mouse PD-L1–PD-1 competition assay, the final assay mixture consisted of 0.5 nM DyLight-650–conjugated mouse PD-1, a 1:7,500 dilution of europium cryptate–labeled mouse PD-L1, and 25% (vol/vol) hybridoma supernatant in a total reaction volume of 10 µL diluted in 1× assay buffer (50 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], pH 7.4; 400 mM potassium fluoride; and 0.1% [wt/vol] bovine serum albumin [BSA]) in a 384-well plate (Corning). Nonspecific binding was defined with 150 nM clone 10 F.9G2. For the mouse PD-L1–CD80 competition assay, the final assay mixture consisted of 0.5 nM DyLight-650–conjugated mouse CD80, a 1:7,500 dilution of europium cryptate–labeled mouse PD-L1, and 25% (vol/vol) hybridoma. For the clone 10 F.9G2–mouse PD-L1 competition assay, the final assay mixture consisted of 0.04 nM DyLight-650–conjugated clone 10 F.9G2, a 1:7,500 dilution of europium cryptate–labeled mouse PD-L1, and 25% (vol/vol) hybridoma supernatant. The plates were sealed and incubated overnight at room temperature before the fluorescence was measured on an EnVision plate reader (PerkinElmer), using an excitation filter of 320 nm (75-nm bandwidth) and emission filters of 665 nm (7.5-nm bandwidth) and 590 nm (20-nm bandwidth). Time-resolved fluorescence (TRF) was measured using a delay time of 70 μs and an integration time of 500 μs with 100 flashes. The raw data were initially analyzed with the equation
and were then expressed as percent delta function, using the equation
where the nonspecific binding (NSB) wells were used for the negative controls. The percent specific binding was calculated from the percent delta function using the equation
For determination of IC50, purified IgG samples were serially diluted into the assay over 11 points. The data were then analyzed using Prism, version 5.01 for Windows (GraphPad Software), with the four-parameter equation
where x is the logarithm of the sample concentration and Y is the percent inhibition.
HTRF direct binding assays
Biochemical assays using HTRF technology were established to measure direct binding of antibodies to recombinant mouse PD-L1 Fc chimera and recombinant mouse PD-L2 Fc chimera (R&D Systems, catalog nos. 1019-B7-100 and 1022-PL-100). Conjugations with europium cryptate trisbipyridine–N-hydroxysuccinimide (Cisbio) were performed according to the manufacturer’s directions, and the cryptate conjugate assay dilution factor was determined by comparing the fluorescence (at 590 nm) with the supplied calibrator. For assay of direct binding to mouse PD-L1, the final assay mixture consisted of 5 nM biotin goat anti-rat IgG (Jackson ImmunoResearch, catalog no. 112–065-003), 8 nM streptavidin-XLent! (Cisbio), a 1:7,500 dilution of europium cryptate–labeled PD-L1, and 2.5% (vol/vol) hybridoma supernatant in a total reaction volume of 10 µL diluted in 1× assay buffer (50 mM HEPES, pH 7.4; 400 mM potassium fluoride; 0.1% BSA [wt/vol]) in a 384-well plate (Corning). Clone 10 F.9G2, 1 nM, was used as a positive control, and 1 nM NIP228 control mAb was used to define nonspecific binding. For assay of the direct binding to mouse PD-L2, the final assay mixture consisted of 5 nM biotin goat anti-rat IgG, 8 nM streptavidin-XLent!, a 1:8,000 dilution of europium cryptate–labeled PD-L2, and 2.5% (vol/vol) hybridoma supernatant in a total reaction volume of 10 µL in 1× assay buffer (50 mM HEPES, pH 7.4; 400 mM potassium fluoride; 0.1% [wt/vol] BSA). Anti-mouse PD-L2 antibody TY25, 1 nm (eBioscience), was used as a positive control, and 1 nM NIP228 control mAb was used to define nonspecific binding. The plates were sealed and incubated overnight at room temperature before the fluorescence was measured on an EnVision plate reader (PerkinElmer), using an excitation filter of 320 nm (75-nm bandwidth) and emission filters of 665 nm (7.5-nm bandwidth) and 590 nm (20-nm bandwidth). TRF was measured using a delay time of 70 μs and an integration time of 500 μs with 100 flashes. The raw data were initially analyzed using the equation
and was then expressed as percent delta function by using the equation
where the NSB wells were used for the negative controls. The percent specific binding was calculated from the percent delta function values by using the equation
SPR affinity measurements
All SPR assays were performed using a Biacore T200 biosensor instrument (GE Healthcare) at 25°C with a carboxymethyl Series S Sensor chip CM5 (GE Healthcare) and assay buffer consisting of HBS-EP+ Buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM ethylenediaminetetraacetic acid [EDTA], and 0.05% (vol/vol) Surfactant P20, pH 7.4) (GE Healthcare).
The His-tagged recombinant proteins human PD-L1 (catalog no. 10084-H08H), human CTLA-4 (catalog no. 11159-H08H), and mouse CTLA-4 (catalog no. 50503-M08H) were purchased from Sino Biological. For human anti–PD-L1 and mouse anti–CTLA-4 binding assays, mouse anti-histidine–tagged antibody (Bio-Rad, catalog no. MCA1396) was diluted to 20 µg/mL in 10 mM sodium acetate, pH 5.0, and immobilized by amine coupling chemistry to two flow cells of a sensor chip. Approximately 7,000 response units of protein was immobilized on both flow cells. His-tagged recombinant human PD-L1 protein (R&D Systems, catalog no. 9049-B7-100) at 2 µg/mL or recombinant mouse CTLA-4 protein (R&D Systems, catalog no. 434-CT-200/CF) at 0.125 µg/mL was captured at a low density only on the experimental flow cell, and the second flow cell served as the reference surface. A dilution series of the human anti–PD-L1 or mouse anti–CTLA-4 antibody was then passed over the protein surface at 30 µL/min. The human anti–PD-L1 antibody dilution series was 30, 10, 3, 3, 1, 1, and 0.37 nM, and association and dissociation data were collected for 180 s and 900 s, respectively. The mouse anti–CTLA-4 antibody dilution series was 200, 67, 22, 7.4, and 2.5 nM, and association and dissociation data were collected for 120 s and 180 s, respectively. Both flow cells were regenerated with a 35-s injection of 20 mM HCl. The data were subtracted from the reference flow cell and a buffer cycle to remove the effects of nonspecific binding and were then fit to a 1:1 or bivalent binding model, using Biacore Evaluation software (GE Healthcare). Both models fit well, having low chiCitation2 values and similar Rmax values, but KD values from the bivalent model are reported here to account for potential avidity effects in the assay.
For the human anti–CTLA-4 binding assays, approximately 500 response units of protein A (Thermo Fisher Scientific) was immobilized to two flow cells of a Series S Sensor Chip CM5 (GE Healthcare) by amine coupling. The human anti–CTLA-4 antibody was captured only on the experimental flow cell, and the second flow cell served as the reference surface. The human anti–CTLA-4 antibodies were diluted to approximately 1 µg/mL and injected over 30 s at 10 µl/min. The human CTLA-4 protein dilution series (100, 33, 11, 3.7, and 1.2 nM) was injected using an association time of 180 s and a dissociation time of 180 s. Both surfaces were regenerated with a 40-s injection of glycine, pH 1.7, after each assay cycle. The data were subtracted by the reference flow cell and a buffer cycle to account for the effects of nonspecific binding and were then fit to a 1:1 binding model, using Biacore Evaluation software (GE Healthcare).
For mouse and rat anti–mouse PD-L1 binding assays, His-tagged recombinant protein murine PD-L1 (catalog no. 9048-B7-100) was purchased from R&D Systems. Rabbit anti-murine IgG antibody (catalog no. BR-1008-38; GE) was diluted to 30 µg/mL in 10 mM sodium acetate, pH 5.0, and immobilized by amine coupling chemistry to two flow cells of a sensor chip. Approximately 4,500 response units of protein was immobilized on both flow cells. Anti-murine PD-L1 mAb at 25 nM was captured only on the experimental flow cell, and the second flow cell served as the reference surface. A dilution series of the murine–PD-L1 was then passed over the mAb and reference surfaces at 75 µL/min. The murine–PD-L1 dilution series was 150, 50, 16.7, 5.6, 1.9, 0.6, 0.2, and 0.06 nM, and association and dissociation data were collected for 120 s and 900 s, respectively. Both flow cells were regenerated with a 120-s injection of 10 mM glycine, pH 1.7. The data were subtracted from the reference flow cell, and a buffer cycle was used to remove the effects of nonspecific binding, bulk shift, injection noise, and drift. The data were then fit to a 1:1 binding model, using Biacore Evaluation software (GE Healthcare).
Generation of anti-mouse CTLA-4 mAbs
The mouse anti-mouse CTLA4 antibody clone 9D9 was originally generated by immunization of human CTLA-4 transgenic mice in the laboratory of James Allison (University of Texas, MD Anderson Cancer Center, Houston, TX) as described previously.Citation33 The mIgG1 and mIgG2b isotypes of clone 9D9 were generated by subcloning of VH and VL sequences from the original 9D9 clone into expression vectors, which resulted in the production of the mIgG1 and mIgG2b isotypes of clone 9D9. Briefly, the 9D9 VH domain was cloned into a vector containing the mouse IgG1 or IgG2a heavy-chain constant domain and regulatory elements to express whole IgG heavy chain in mammalian cells. Similarly, the 9D9 VL domain was cloned into a vector for the expression of the mouse light chain (kappa) constant domains and regulatory elements to express whole IgG light chain in mammalian cells. Expression and purification were carried out as described above.
Cell lines and tissue culture reagents and conditions
Cell lines used in binding, biological and tumor engraftment studies included CHO, CT26 mouse colon cancer, EMT6 mouse breast cancer, Renca mouse renal cancer, 4T1 mouse breast cancer, B16F10 mouse melanoma, LL2 mouse lung cancer, MC38 mouse colon cancer, EO771 mouse breast cancer, Raji human B cells, Jurkat human T cells, H441 human lung cancer, MDA MB-231 human breast cancer, and ES2 human ovarian cancer cells, all obtained from the American Type Culture Collection. MCA205 mouse fibrosarcoma cells were obtained from the Providence Cancer Center, Oregon Health and Science University. CT26, EMT6, Renca, MCA205, Jurkat, H441, MDA MB-231, and ES2 cells were cultured in RPMI 1640 complete medium containing glutamine and glucose supplements (catalog no. A1049101; Life Technologies) plus 10% (vol/vol) heat-inactivated newborn calf serum and incubated in a humidified tissue culture incubator at 37°C with 5% CO2 before being used in binding or biological assays. CHO cells were cultured in DMEM plus 10% newborn calf serum and 1% nonessential amino acids, and Raji B cells were cultured in RPMI 1640 medium plus GlutaMAX (Gibco), 10% newborn calf serum, and 100 mM sodium pyruvate, before being used in reporter bioactivity assays.
mAbs
Rat anti-mouse PD-L1 mAb clone 10 F.9G2 and mouse anti-mouse CTLA-4 mIgG2B clone 9D9 were purchased from Bio X Cell (catalog nos. BE0101 and BE0164). The anti-human PD-L1 mAbs atezolizumab (lot no. 3166643; Tecentriq) and avelumab (lot no. AU020120; Bavencio) and the anti-human CTLA-4 mAb ipilimumab (lot no. 930944; Yervoy) were purchased from Blue Door Pharma. These antibodies were stored at 4°C until use and before their expiration dates, as recommended in their package inserts. The anti-human PD-L1 mAbs durvalumab (Imfinzi) and tremelimumab and mIgG2a clone 9D9 were produced and purified at AstraZeneca. These antibodies were stored at – 80°C until use.
Cell-based flow cytometry binding assays
Cultured cell lines were removed from cell culture plastic with 0.05% trypsin-EDTA (Thermo Fisher Scientific), suspended in cold culture medium, and centrifuged to pellet cells. Primary human CD3 T cells were obtained from sodium heparin anti-coagulated whole blood from healthy human donors through negative selection of peripheral mononuclear blood cells (PBMCs), using Robosep automated cell separation (StemCell Technologies). Human CD3 T cells were cultured in tissue culture plates coated overnight at 4°C with 2 µg/mL each anti-human CD3 (clone OKT3; BioLegend, catalog no. 317326) and anti-human CD28 (clone 28.2; BioLegend, catalog no. 302934) to activate T cells and upregulate PD-L1. After counting, 1 × 105 activated human T cells, PD-L1–expressing Jurkat T cells, and cultured tumor cells were then bound to test mAbs in flow cytometry buffer (2% neonatal calf serum in PBS, pH 7.2, without CaCitation2+ and Mg;Citation2+ Thermo Fisher Scientific) for 4 h at 4°C. After washes, bound primary antibody was detected with either Alexa Fluor 647–labeled goat anti-human IgG or goat anti-mouse IgG secondary antibody (Thermo Fisher Scientific), as appropriate for the primary antibody species, by incubation at 10 µg/mL in flow cytometry buffer containing 5 µg/mL propidium iodide for 20 min at 4°C. After further washes, bound secondary antibody was detected by flow cytometry on an LSR II cytometer (BD Biosciences) and gating on live (propidium iodide–negative) cells, using FlowJo software (BD Biosciences). The apparent (avidity-driven) EC50s for each antibody were determined after background subtraction (secondary antibody binding only), using nonlinear regression analysis (log[mAb] vs. mean fluorescence intensity [variable slope/four parameter fit]) in Prism, version 7.1 for Windows (GraphPad Software).
ADCP and macrophage cytotoxicity assay
PBMCs were isolated from whole blood obtained from healthy donors in the AstraZeneca blood donor program, using lymphocyte separation medium (MP Biomedicals) density centrifugation, and primary human monocytes were isolated with the EasySep Human Monocyte Enrichment Kit without CD16 depletion (StemCell Technologies). Monocytes were subsequently cultured in RMPI 1640 medium containing 4.5 g/L glucose and L-glutamine (catalog no. A1049101; Thermo Fisher Scientific), supplemented with 10% newborn calf serum (complete RPMI 10 medium; Thermo Fisher Scientific) and 100 ng/mL recombinant human macrophage colony-stimulating factor (R&D Systems, catalog no. 216-MC-025), for 6 days in a humidified tissue culture chamber at 37°C with 5% CO2 to generate in vitro–derived macrophages. Cells were removed from tissue culture plastic with Accutase (StemCell Technologies) and labeled with CellTrace Violet, using the CellTrace Violet Cell Proliferation Kit (Thermo Fisher Scientific) according to the manufacturer’s recommended protocol. Subsequently, 4 × 104 cells were seeded in sterile, flat-bottomed, 96-well tissue culture plates overnight in complete RPMI 10 medium without human macrophage colony-stimulating factor. Human ES2 ovarian carcinoma cells expressing PD-L1, or CHO cells engineered to express human CTLA-4, were trypsinized, suspended in complete RPMI 10 medium, pelleted, and labeled with carboxyfluorescein succinimidyl ester (CFSE) using a CellTrace CFSE Cell Proliferation Kit for flow cytometry (Thermo Fisher Scientific) according to the manufacturer’s protocol. After labeling, 4 × 104 cells were added per well (macrophage-to-target cell ratio, 1:1) for ADCP assays with anti–PD-L1 mAb (for ES2 cells) or anti–CTLA-4 mAb (for CTLA-4–expressing CHO cells). CFSE-labeled target cells were incubated for 4 h with macrophages labeled with CellTrace Violet (Thermo Fisher Scientific) to allow macrophage phagocytosis and killing of target cells. After 4 h of incubation, cells were removed from culture wells with Accutase (StemCell Technologies); collected in a separate round-bottomed, 96-well plate; washed with PBS; and suspended in PBS containing Live-Dead Fixable Far Red Dead Cell Stain (Thermo Fisher Scientific) for 30 min at room temperature in the dark. Cells were subsequently washed in flow cytometry buffer (PBS plus 2% newborn calf serum) and fixed with 4% (wt/vol) formalin in PBS. Cells were analyzed by flow cytometry on an LSR II cytometer (BD Biosciences) and events were analyzed with FlowJo software (BD Biosciences). Phagocytosed target cells were defined as the percentage of total cells with dual CellTrace Violet and CFSE positivity (S3 Fig), which we had previously verified as macrophage-engulfed target cells by immunofluorescence microscopy. Target cells that were killed directly by macrophages in the absence of phagocytosis were identified as nonviable (positive for Live-Dead Fixable Far Red Dead Cell staining), CFSE-labeled target cells that did not demonstrate a signal indicative of phagocytosed target cells when labeled with CellTrace Violet (Thermo Fisher Scientific).
ADCC assay
Primary human NK cells were purified from heparin-anticoagulated whole human blood after PBMC isolation, using an EasySep human NK cell isolation kit (StemCell Technologies). After isolation, cells were suspended in warm complete RPMI medium (RPMI 1640 medium, 10% fetal bovine serum, 1× penicillin-streptomycin), and 2 × 105 effector NK cells were added to each well of round-bottomed, non–tissue culture–treated, 96-well plates (catalog no. 3788; Costar). CHO cells expressing CTLA-4 and ES2 human ovarian cancer cells expressing endogenous PD-L1 were labeled with DiO dye, using Vybrant DiO cell labeling solution (Thermo Fisher Scientific), and 2 × 104 DiO-labeled target cells were added to each well for an effector-to-target ratio of 10:1. Control wells included unlabeled target cells, DiO-labeled target cells alone, NK effector cells alone, and NK cells plus DiO-labeled target cells. Anti–PD-L1, anti–CTLA-4, and control mAbs were diluted in complete medium and added to wells containing effector and target cells, and plates were incubated for 18 h to allow for target cell killing. Nonviable DiO-positive target cells were detected by propidium iodide uptake after flow cytometry analysis on an LSR II flow cytometer, and data were analyzed with FlowJo software (BD Biosciences). Specific NK cell activity was determined by subtracting mean values from wells containing NK cells plus DiO-labeled target cells in the absence of drug and was graphed versus the log drug concentration, using Prism version 7.1 for Windows (GraphPad Software) to assess dose-dependent activity.
Anti-human PD-L1 reporter cell–based bioassay
The biological activity of anti–PD-L1 antibodies was measured with a reporter gene bioassay. CHO cells expressing anti-CD3 single-chain variable fragment from clone OKT3 and human PD-L1 were prepared to 0.2 × 106 cells per mL in cell culture medium (DMEM, 10% FBS, 1% nonessential amino acids), and 50 µL (1 × 104 cells) was added to tissue culture–treated, white-walled, 96-well assay plates. The assay plates were incubated at 37°C with 5% CO2 in a humidified tissue culture incubator for 3 h. An 11-point dilution series of the anti–PD-L1 antibodies was prepared in assay medium (RPMI 1640 medium, 10% FBS). Jurkat human T-lymphocyte cells expressing PD-1 and an NFAT (nuclear factor of activated T cells) luciferase reporter were prepared to 2.0 × 106 cells per mL in assay medium. One hundred microliters of diluted antibody and 100 µL of Jurkat cells were mixed in 96-well preincubation plates and incubated at 37°C for 2 h. Fifty microliters of the antibody-Jurkat mixture was added to the CHO cells in the assay plates, which were then incubated at 37°C with 5% CO2 in a humidified tissue culture incubator for 18 h. Steady-Glo luciferase substrate (Promega) was added to the assay plates and incubated according to the manufacturer’s instructions. The amount of luminescence proportional to T-cell activity was quantified with a luminometer.
Anti–CTLA-4 reporter cell–based bioassay
The biological activity of the anti–CTLA-4 antibodies was measured with a reporter gene bioassay. Raji human B-lymphocyte cells were prepared to 0.33 × 106 cells per mL in cell culture medium (RPMI 1640 medium, GlutaMAX [Gibco], 10% FBS, 100 mM sodium pyruvate) containing 50 pg/mL staphylococcal enterotoxin E (SEE) and incubated for 60 min with shaking. An 11-point dilution series of anti–CTLA-4 antibodies was prepared in cell culture medium. Jurkat human T-lymphocyte cells that had been engineered to stably express human CTLA-4, and a luciferase reporter gene under the control of the transcription factor NFκB (nuclear factor kappa-B), were prepared to 1.33 × 106 cells per mL in a preincubated Raji cell suspension. Anti–CTLA-4 antibodies and the Jurkat-Raji-SEE mixture was then added to 96-well assay plates at a 1:1 ratio. The plates were incubated at 37°C with 5% CO2 in a humidified tissue culture incubator for 19 h and then allowed to cool for 10 min at room temperature. An equal volume of Steady-Glow luciferase substrate (Promega) was added to the assay plates, which were incubated for 1.5 h at room temperature. The amount of luminescence that was proportional to the Jurkat reporter cell activity was quantified with a luminometer.
PK analysis
PK studies were performed in female CD1 mice (Envigo and Charles River). Blood samples for the multiple-dose PK study were collected from the retroorbital sinuses of mice into serum separator tubes (Prepared Microtubes, Z-Gel; Sarstedt), except for the final collection, which was collected via cardiac puncture at the time of euthanasia. For the single-dose PK study, blood was collected from the tail veins of mice, with one final collection via cardiac puncture. Mice were bled before administration of the test article and after intravenous dosing per body weight with clone 80, clone 10 F.9G2, or mIgG1 clone 9D9. All serum samples were stored at – 80°C until analyzed by PK enzyme-linked immunosorbent assay (ELISA) for the multiple-dose study or by electrochemiluminescence (ECL) (Meso Scale Discovery Diagnostics) for the single-dose study.
For analysis of clone 80 in serum by ELISA, half-well ELISA plates (Costar, catalog no. 3690; VWR) were coated overnight at 4°C with 30 µL of 1-µg/mL recombinant mouse PD-L1 (catalog no. 1019-B7; R&D Systems) in PBS. Plates were blocked in 4% milk–PBS for 1 h at room temperature and then washed four times with 100 µL of PBS. Standards or diluted samples at 30 µL per well were incubated for 2 h at room temperature with shaking on an orbital shaker. After four washes in PBS, 30 µL of horseradish peroxidase (HRP)–conjugated goat anti-mouse IgG Fc (0.8 mg/mL stock; Southern Biotech, catalog #1033-01) per well, at a dilution of 1:15,000 in 1% milk–PBS, was added to the plate and incubated for 1 h at room temperature. Plates were washed four times with PBS and then developed with 50 µL of tetramethylbenzadine substrate (Life Technologies) for 3–5 min. To terminate the tetramethylbenzadine reaction, 25 µL of 2 N sulfuric acid was added to each well, and plates were read on an M5 spectrophotometer (Molecular Devices) at 450 nm absorbance.
For analysis of clone 80 in serum by electrochemiluminescence, MSD Standard Bind 96-well Sector plates (Meso Scale Discovery) were coated overnight at 4°C with recombinant mouse PD-L1 (R&D Systems) in PBS. After washes in PBS plus 0.1% Tween 20 (PBS-Tween), plates were blocked for 1 h at room temperature with PBS plus 1% BSA (PBS-BSA). After washes in PBS-Tween, 12.5 µL of PBS-BSA was added to each well, followed by 12.5 µL of standards or diluted samples. Plates were subsequently incubated for 2 h at room temperature with shaking on an orbital shaker to bind free anti-mouse PD-L1 mAb. After three washes in PBS-Tween, 25 µL of biotin-SP AffiniPure goat anti-mouse IgG, Fc fragment specific (Jackson ImmunoResearch Laboratories, catalog no. 115–065-008), per well at 1 µg/mL in PBS-BSA containing 2% normal goat serum was added to the plate and incubated with shaking for 2 h at room temperature. Plates were washed three times with PBS-Tween, and then 25 µL of Streptavidin Sulfo-TAG at 0.5 μg/mL in PBS-BSA (Meso Scale Discovery) was added and incubated for 1 h at room temperature. After three washes in PBS-Tween, the electrochemiluminescence signal was detected by using 150 μL per well of 2× read buffer T and an MSD Sector imager (Meso Scale Discovery).
PK analysis of clone 10 F.9G2 was conducted by ELISA and electrochemiluminescence methods similar to that used to detect clone 80, with the modifications described here. For ELISA, plates were coated with 30 µL of 2.5 µg/mL anti-rat IgG Fc capture antibody (catalog no. 112–005-071; Jackson ImmunoResearch) per well in PBS, and captured clone 10 F.9G2 was detected with goat anti-rat light-chain HRP (0.8 mg/mL stock, catalog no. 112–035-175; Jackson ImmunoResearch) at a 1:13,000 dilution in 1% milk–PBS. For electrochemiluminescence, biotinylated goat anti-rat IgG at a concentration of 1 µg/mL was used in place of biotin-SP AffiniPure goat anti-mouse IgG to detect rat IgG (10 F.9G2).
The mIgG1 isotype of clone 9D9 was measured by ELISA, using plates coated with 30 µL of 2.5 µg/mL anti-His antibody (catalog no. AB9136; Abcam) per well in PBS, and then bound to 30 µL of 2 µg/mL His-tagged recombinant mouse CTLA-4 (Sino Biological) per well to allow for capture of free mouse anti–CTLA-4 mAb in serum. HRP goat anti-mouse light chain was used at a 1:2,000 dilution in 1% milk–PBS to detect bound mIgG1 clone 9D9, but otherwise the protocol remained as described for the clone 80 ELISA.
PK parameters were estimated by noncompartmental analysis in Phoenix WinNonlin software, version 8.1 (Certara). PK figures were produced using the ggplot2 package in R studio (R project, R studio version 1.1.463, R version 3.6.1).
Pharmacodynamic assays
Tumors, regional tumor-draining lymph nodes, and spleens were removed from mice after the beginning of drug treatments (20 mg/mL each for anti–PD-L1 and anti–CTLA-4) and prepared for flow cytometry assessment. Briefly, tumors were cut into small pieces with a scalpel and digested for 30 min at 37°C in a collagenase solution containing 200 U/mL type 3 collagenase (catalog no. LS004208; Worthington Biochemical Corporation), 0.3 mM CaCl2 (Sigma Aldrich), and 1× DNase 1 (Sigma Aldrich) in 1× Hanks buffered saline solution (Thermo Fisher Scientific). After tissue digestion, cells were strained through a 70-µm filter, washed in culture medium, and suspended in flow cytometry buffer (PBS plus 2% newborn calf serum), and cell viability and number were determined. Splenocytes and immune cells from tumor-draining lymph nodes were isolated by physical disruption of spleens or lymph nodes through a 100-µm filter. Removal of red blood cells from splenocytes was accomplished using 1× RBC Lysis Buffer (BioLegend) for 3 min at room temperature. After further filtering and suspension in flow cytometry buffer, splenocyte and lymph node immune-cell viability and number were determined. Cells from tumors, spleens, or tumor-draining lymph nodes were analyzed by immunophenotyping flow cytometry after live/dead viability staining with the Live/Dead Fixable Aqua Kit (Thermo Fisher Scientific) and incubated with a cocktail of anti-mouse antibodies consisting of BV786 anti-CD45 (catalog no. 564225; BD Biosciences), eBioscience APC-780 anti-CD8 antibodies (catalog no. 47–0081-82; Thermo Fisher Scientific), BUV395 anti-CD4 antibodies (catalog no. 563790; BD Biosciences), Alexa Fluor 488 anti-CD25 antibodies (catalog no. 102017; BioLegend), and PerCP anti-CD69 antibodies (catalog no. 104522; BioLegend), and after fixation and permeabilization with FoxP3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent (catalog no. 00–5521, eBioscience; Thermo Fisher Scientific) according to the manufacturer’s protocol (eBioscience PE anti-FoxP3, catalog no. 25–5773-82, Thermo Fisher Scientific; APC anti-granzyme B, catalog no. 515405, BioLegend; and eBioscience efluor 450 Ki67, catalog no. 48–5698-82, Thermo Fisher Scientific).
In vivo tumor growth inhibition studies
Syngeneic mouse colon cancer (CT26, MC38), breast cancer (EMT6, 4T1, EO771), melanoma (B16F10), renal cell cancer (RENCA), lung cancer (LL/2 lewis lung), or fibrosarcoma (MCA205) tumor cells were engrafted in 6- to 8-week-old female Balb/C (CT26, EMT6, 4T1, RENCA) or female C57BL/6 (B16F10, LL/2 lewis lung, MC38, EO771, MCA205) mice (Envigo). Cells were implanted subcutaneously in the flank at 5 × 105 (CT26, RENCA, LL/2 lewis lung) or 2.5 × 105 (B16F10, MC38, MCA205) cells per site. EMT6 and 4T1 breast tumors were established in the mammary fat pads of female Balb/c mice at 1 × 105 cells per site and EO771 in the fat pads of female C57BL/6 mice at 2.5 × 105 cells per site. Tumor growth was monitored over the course of the study, and tumor volume was calculated as
When the tumors reached 100–150 mm3, mice were randomly assigned into groups with equal median tumor volumes, using the deterministic randomization method built into the Study Director software package (Studylog Systems). Mice were then either left untreated or were administered intraperitoneal injections of 20 mg/kg twice a week for four doses of clone 80, mIgG1 clone 9D9, a combination of each, or an isotype control antibody (NIP228 mouse IgG1 D265A) at the indicated doses. Mouse body weight and tumor measurements were determined twice weekly for the duration of the study. Mice were euthanized for evidence of significant disease burden (weight loss of more than 20%, hind limb paralysis, extreme lethargy, or tumor mass exceeding 2,000 mm3), which was considered the survival endpoint for Kaplan-Meier analysis.
Statistical analyses
Statistical significance of differences between pharmacodynamic changes in CD4, CD8, or Tregs observed in the CT26, EMT6, and MCA205 tumor models was calculated by one-way analysis of variance followed by Dunnett correction for multiple comparisons, using Prism, version 8.0.0 for Windows (GraphPad Software). Statistical analysis of survival differences between treatment groups in animal studies was performed using the log-rank (Mantel-Cox) test, also in Prism software.
Abbreviations
Supplemental Material
Download MS Word (5 MB)Acknowledgments
We thank Andrew Weinberg of the Providence Cancer Center and Oregon Health and Science University School of Medicine, Portland, Oregon, for the kind gift of MCA205 mouse fibrosarcoma cells. Likewise, we would like to thank Kim Rosenthal within Antibody Development and Protein Engineering at AstraZeneca for generating SPR binding data and analysis for the anti-mouse PD-L1 mAbs clone 80 and 10F.9G2.
Disclosure statement
All authors are or were employees of AstraZeneca at the time this work was performed and may have stock and/or stock options or interests in AstraZeneca.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–18. doi:10.1126/science.aar4060.
- Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39. doi:10.1016/j.intimp.2018.06.001.
- van Vugt M, de Greef R, Freshwater T, Mangin E, van Aarle F, Kondic A. Immunogenicity of pembrolizumab in patients with advanced melanoma and non-small-cell lung cancer: pooled results from KEYNOTE-001, 002, 006, and 010. Abstract 3063. J Clin Oncol. 2016;34:3063. doi:10.1200/JCO.2016.34.15_suppl.3063.
- U.S. Food and Drug Administration. Keytruda® (pembrolizumab) prescribing information. [accessed 2020 Dec 14]. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf.
- U.S. Food and Drug Administration. Opdivo® (nivolumab) prescribing information. [accessed 2020 Dec 14]. https://packageinserts.bms.com/pi/pi_opdivo.pdf.
- Agrawal S, Statkevich P, Bajaj G, Feng Y, Saeger S, Desai DD, Park JS, Waxman IM, Roy A, Gupta M. Evaluation of immunogenicity of nivolumab monotherapy and its clinical relevance in patients with metastatic solid tumors. J Clin Pharmacol. 2017;57:394–400. doi:10.1002/jcph.818.
- U.S. Food and Drug Administration. Libtayo® (cemiplimab) prescribing information. [accessed 2020 Dec 14]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761097s000lbl.pdf
- Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008;64:700–04. doi:10.1107/S0907444908007877.
- U.S. Food and Drug Administration. Imfinzi® (durvalumab) prescribing information. [accessed 2020 Dec 14]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761069s000lbl.pdf.
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al.. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–67. doi:10.1038/nature14011.
- U.S. Food and Drug Administration. Tecentriq® (atezolizumab) prescribing information. [accessed 2020 Dec 14]. https://www.gene.com/download/pdf/tecentriq_prescribing.pdf.
- U.S. Food and Drug Administration. Bavencio® (avelumab) prescribing information. https://www.emdserono.com/us-en/pi/bavencio-pi.pdf.
- Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, Narwal R, Steele K, Gu Y, Karakunnel JJ, et al.. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17:299–308. doi:10.1016/S1470-2045(15)00544-6.
- U.S. Food and Drug Administration. Yervoy® (ipilimumab) prescribing information. [accessed 2020 Dec 14]. https://packageinserts.bms.com/pi/pi_yervoy.pdf.
- Davies AM, Sutton BJ. Human IgG4: a structural perspective. Immunol Rev. 2015;268:139–59.
- Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, et al.. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22:433–38. doi:10.1038/nm.4051.
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al.. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–59. doi:10.1172/JCI73639.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–86. doi:10.1158/2159-8290.CD-18-0367.
- Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, Schlom J. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3:1148–57. doi:10.1158/2326-6066.CIR-15-0059.
- Donahue RN, Lepone LM, Grenga I, Jochems C, Fantini M, Madan RA, Heery CR, Gulley JL, Schlom J. Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti-PD-L1 monoclonal antibody. J Immunother Cancer. 2017;5:20. doi:10.1186/s40425-017-0220-y.
- Fenerty KE, Padget M, Wolfson B, Gameiro SR, Su Z, Lee JH, Rabizadeh S, Soon-Shiong P, Hodge JW. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J Immunother Cancer. 2018;6:133. doi:10.1186/s40425-018-0445-4.
- Fujii R, Friedman ER, Richards J, Tsang KY, Heery CR, Schlom J, Hodge JW. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. 2016;7:33498–511. doi:10.18632/oncotarget.9256.
- Jochems C, Hodge JW, Fantini M, Tsang KY, Vandeveer AJ, Gulley JL, Schlom J. ADCC employing an NK cell line (haNK) expressing the high affinity CD16 allele with avelumab, an anti-PD-L1 antibody. Int J Cancer. 2017;141:583–93. doi:10.1002/ijc.30767.
- Julia EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 anti-PD-L1 immune checkpoint inhibitor, triggers NK cell-mediated cytotoxicity and cytokine production against triple-negative breast cancer cells. Front Immunol. 2018;9:2140. doi:10.3389/fimmu.2018.02140.
- Khanna S, Thomas A, Abate-Daga D, Zhang J, Morrow B, Steinberg SM, Orlandi A, Ferroni P, Schlom J, Guadagni F, et al.. Malignant mesothelioma effusions are infiltrated by CD3(+) T cells highly expressing PD-L1 and the PD-L1(+) tumor cells within these effusions are susceptible to ADCC by the anti-PD-L1 antibody avelumab. J Thorac Oncol. 2016;11:1993–2005. doi:10.1016/j.jtho.2016.07.033.
- Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, Watkins A, Mullins S, Chodorge M, Andrews J, et al.. Identification and characterization of MEDI4736, an antagonistic anti-PD-L1 monoclonal antibody. Cancer Immunol Res. 2015;3:1052–62. doi:10.1158/2326-6066.CIR-14-0191.
- Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, et al.. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 2018;33:649–63. doi:10.1016/j.ccell.2018.02.010.
- Laurent S, Queirolo P, Boero S, Salvi S, Piccioli P, Boccardo S, Minghelli S, Morabito A, Fontana V, Pietra G, et al.. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J Transl Med. 2013;11:108. doi:10.1186/1479-5876-11-108.
- Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–45. doi:10.1073/pnas.1417320112.
- Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, Sazegar H, Seja E, Villanueva A, Gomez-Navarro J, et al.. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res. 2011;17:4101–09. doi:10.1158/1078-0432.CCR-11-0407.
- Khan S, Burt DJ, Ralph C, Thistlethwaite FC, Hawkins RE, Elkord E. Tremelimumab (anti-CTLA4) mediates immune responses mainly by direct activation of T effector cells rather than by affecting T regulatory cells. Clin Immunol. 2011;138:85–96. doi:10.1016/j.clim.2010.09.011.
- Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C, et al.. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS One. 2016;11:e0161779. doi:10.1371/journal.pone.0161779.
- Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi:10.1158/2326-6066.CIR-13-0013.
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce Vargas F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al.. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–710. doi:10.1084/jem.20130579.
- Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, Sharma P. Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory T cells (Tregs) in human cancers. Clin Cancer Res. 2019;25:1233–38. doi:10.1158/1078-0432.CCR-18-0762.
- Bruggemann M, Winter G, Waldmann H, Neuberger MS. The immunogenicity of chimeric antibodies. J Exp Med. 1989;170:2153–57. doi:10.1084/jem.170.6.2153.
- Baverel PG, Dubois VFS, Jin CY, Zheng Y, Song X, Jin X, Mukhopadhyay P, Gupta A, Dennis PA, Ben Y, et al.. Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin Pharmacol Ther. 2018;103:631–42. doi:10.1002/cpt.982.
- Davda J, Declerck P, Hu-Lieskovan S, Hickling TP, Jacobs IA, Chou J, Salek-Ardakani S, Kraynov E. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J Immunother Cancer. 2019;7:105. doi:10.1186/s40425-019-0586-0.
- Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, Petry F, Ravetch JV, Nishimura S, Izui S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. 2008;181:6664–69. doi:10.4049/jimmunol.181.9.6664.
- Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, Devenport M, Lazarski CA, Zhang P, Wang X, et al.. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018;28:416–32. doi:10.1038/s41422-018-0011-0.
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–25. doi:10.1084/jem.20082492.
- Tang F, Du X, Liu M, Zheng P, Liu Y. Anti-CTLA-4 antibodies in cancer immunotherapy: selective depletion of intratumoral regulatory T cells or checkpoint blockade?. Cell Biosci. 2018;8:30. doi:10.1186/s13578-018-0229-z.
- Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene. 1997;187:9–18. doi:10.1016/S0378-1119(96)00628-2.
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–46. doi:10.1038/74704.
- Daramola O, Stevenson J, Dean G, Hatton D, Pettman G, Holmes W, Field R. A high-yielding CHO transient system: coexpression of genes encoding EBNA-1 and GS enhances transient protein expression. Biotechnol Prog. 2014;30:132–41. doi:10.1002/btpr.1809.
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–23. doi:10.1002/pro.5560041120.
- Mathis G. Probing molecular interactions with homogeneous techniques based on rare earth cryptates and fluorescence energy transfer. Clin Chem. 1995;41:1391–97. doi:10.1093/clinchem/41.9.1391.