
ABSTRACT
LIGHT is a member of the tumor necrosis factor superfamily, which has been claimed to mediate anti-tumor activity on the basis of cancer cures observed in immunocompetent mice bearing transgenic LIGHT-expressing tumors. The preclinical development of a LIGHT-based therapeutic has been hindered by the lack of functional stability exhibited by this protein. Here, we describe the cloning, expression, and characterization of five antibody-LIGHT fusion proteins, directed against the alternatively spliced extra domain A of fibronectin, a conserved tumor-associated antigen. Among the five tested formats, only the sequential fusion of the F8 antibody in single-chain diabody format, followed by the LIGHT homotrimer expressed as a single polypeptide, yielded a protein (termed “F8-LIGHT”) that was not prone to aggregation. A quantitative biodistribution analysis in tumor-bearing mice, using radio-iodinated protein preparations, confirmed that F8-LIGHT was able to preferentially accumulate at the tumor site, with a tumor-to-blood ratio of ca. five to one 24 hours after intravenous administration. Tumor therapy experiments, performed in two murine tumor models (CT26 and WEHI-164), featuring different levels of lymphocyte infiltration into the neoplastic mass, revealed that F8-LIGHT could significantly reduce tumor-cell growth and was more potent than a similar fusion protein (KSF-LIGHT), directed against hen egg lysozyme and serving as negative control of irrelevant specificity in the mouse. At a mechanistic level, the activity of F8-LIGHT was mainly due to an intratumoral expansion of natural killer cells, whereas there was no evidence of expansion of CD8 + T cells, neither in the tumor, nor in draining lymph nodes.
Abbreviations: CTLA-4: Cytotoxic T-lymphocytes-associated protein 4; EGFR: Epidermal growth factor receptor; HVEM: Herpesvirus entry mediator; IFNγ: Interferon-gamma; LIGHT: Lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T lymphocytes; LTβR: Lymphotoxin beta receptor; NF-κB: Nuclear factor “kappa-light-chain-enhancer” of activated B cells; NK: Natural killer cells; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; TNF: Tumor necrosis factor.
Introduction
Immunotherapy of cancer is gaining momentum based on the realization that a subset of patients may achieve durable responses upon treatment with immunotherapeutic agents.Citation1 Immune checkpoint inhibitors (e.g., antibodies directed against PD-1, PD-L1 and CTLA-4) have shown activity against different types of cancer and are widely used in the clinical practice.Citation2–4Cytokines represent a complementary class of therapeutic proteins that modulate the activity of the immune system, and engineered cytokine products are increasingly being used (alone or in combination) for oncological applications.Citation5–7 Cytokines are typically active in patients at very low doses (often at less than 1 mg) and have a narrow therapeutic window. Research efforts have been dedicated to the development of more selective types of cytokine-based therapeutics with improved activity and/or safety.
Tumor-targeting antibody-cytokine fusions (also called “immunocytokines”) represent a promising class of anticancer agents, and some of these products have moved to advanced clinical trials.Citation8A preferential accumulation of the cytokine payload in the neoplastic mass may help boost the activity of tumor-resident T cells and natural killer (NK) cells locally,Citation9resulting in lower systemic toxicity.Citation10,Citation11 In a number of comparative preclinical studies, tumor-homing immunocytokines were substantially more active than fusions based on antibodies of irrelevant specificity, even though this difference may be less drastic for long-lived IgG-based products.Citation12–16,Citation17
LIGHT, which is an acronym for Lymphotoxin-like, exhibits Inducible expression and competes with HSV Glycoprotein D for binding to Herpesvirus entry mediator, a receptor expressed on T lymphocytes, is a member of the tumor necrosis factor (TNF) superfamily expressed on a number of immune cells, including immature dendritic cells and activated T lymphocytes.Citation18 The membrane-anchored form of LIGHT can be cleaved by proteases between residues L81 and I82, resulting in a functional soluble form.Citation19 This cytokine binds to two receptors: Herpesvirus entry mediator (HVEM), which is expressed on T cells, NK cells and dendritic cells, and Lymphotoxin beta receptor (LTβR), which is expressed mainly on non-lymphoid cells.Citation20 Upon homotrimerization and interaction with HVEM, LIGHT activates the NF-κB pathway leading to activation and stimulation of target lymphocytes, whereas signaling through LTβR leads to expression of chemokines and adhesion molecules involved in lymphoid organs organization and maintenance.Citation18,Citation21,Citation22 Transgenic expression of LIGHT on tumor cells has been shown to mediate a potent anti-tumor effect in mice.Citation19,Citation23 In two independent studies, LIGHT expression resulted in a massive increase of CD8 + T cells infiltration, which played a central role in the tumor rejection process, as demonstrated by depletion experiments.Citation19,Citation23
A fusion of murine LIGHT with the F8 antibody (specific to the alternatively-spliced extra domain A (EDA) of fibronectin, a conserved tumor-associated antigen) has previously been described, using the antibody in single-chain variable fragment (scFv) format and relying on the ability of LIGHT to form stable non-covalent homotrimers.Citation24 The resulting fusion protein was homogeneous in biochemical characterization assays and maintained binding ability in vitro. However, in contrast to the results obtained with fusions of the F8 antibody with both murine and human TNF (which showed selective uptake at the tumor site with excellent biodistribution results), the fusion of scFv(F8) and murine LIGHT exhibited poor uptake at the tumor site and rapid clearance from the body.Citation24 It has previously been reported that members of the TNF superfamily may gain stability when expressed as a single polypeptide, with linkers connecting the three monomeric subunits.Citation25,Citation26
Here, we describe the cloning, expression and characterization of five novel fusion proteins, featuring murine LIGHT expressed as a single polypeptide (comprising the three monomeric subunits) as cytokine payload and the F8 antibody in different formats as tumor-targeting agent. Of the five fusion proteins that were produced and purified, only the use of the F8 antibody in single-chain diabody format resulted in a product with adequate biochemical and immunological properties. The fusion protein preferentially accumulated in tumor lesions and mediated potent anticancer activity, which was mainly depended on NK cells and which was not potentiated by PD-1 blockade.
Results
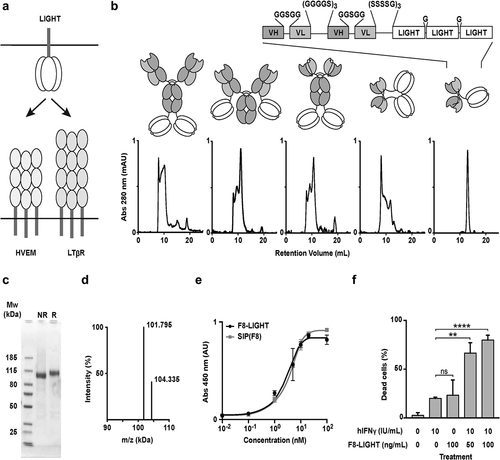
Expression and characterization of F8-LIGHT fusion proteins
LIGHT is a homotrimeric protein that can activate HVEM and LTβ receptors ().Citation13,Citation17 We cloned, expressed, and characterized five different versions of the F8 antibody, fused to murine homotrimeric LIGHT expressed as a single polypeptide. Specifically, we fused LIGHT at the C-terminus of the heavy or of the light chain of the F8 antibody in human IgG1 format, at the C-terminus of the F8 antibody in human scFv-Fc format and at the C-terminus of the F8 antibody in human single-chain diabody and diabody format. (). All products could be purified to homogeneity by protein A chromatography, as shown by SDS-PAGE analysis. However, a comparative evaluation of size-exclusion chromatography profiles revealed that only the single-chain diabody-based product (termed F8-LIGHT), ran as a single peak in gel filtration (), showed a single band in SDS-PAGE and a single peak in mass spectrometry, after PNGase F treatment (). F8-LIGHT was able to bind its cognate target antigen with high affinity in vitro, as shown by enzyme-linked immunosorbent assay (ELISA) () and was able to induce death of HT-29 cells in the presence of interferon (IFN) gamma (). When the cytotoxicity experiment on HT-29 cells was repeated with commercially available recombinant murine LIGHT as control, F8-LIGHT showed higher activity, possibly as a result of the increased stability of the LIGHT moiety expressed as a covalently linked homotrimer (Supplementary Figure 1).Citation25,Citation26 Dissolved in saline, the product was stable for months both at 4°C and at −80°C, even after repeated cycles of freeze-and-thaw (Supplementary Figure 2).
Figure 1. In vitro characterization of fusion proteins
F8-LIGHT selectively accumulate at tumor site in vivo
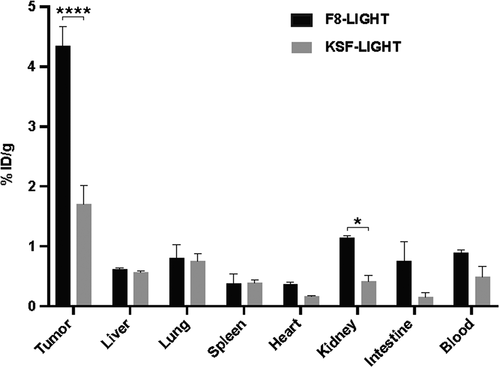
In order to test the in vivo targeting ability of our product, we performed a quantitative biodistribution study by intravenous injection of 125I-labeled F8-LIGHT. We used LIGHT linked to the KSF antibody (specific to hen egg lysozyme) as negative control of identical format (). After 24 h, about 4.5% of the injected dose of F8-LIGHT per gram of tissue was found in the tumor, with a tumor-to-blood ratio of 4.9. Similar to data previously reported for other antibody-cytokine fusion proteins, the transfection protocol used for transient gene expression procedures had an impact on biodistribution results (Supplementary Figure 3).Citation27,Citation28
Figure 2. Tumor targeting of F8-LIGHT in vivo.
F8-LIGHT delay progression of established murine tumors
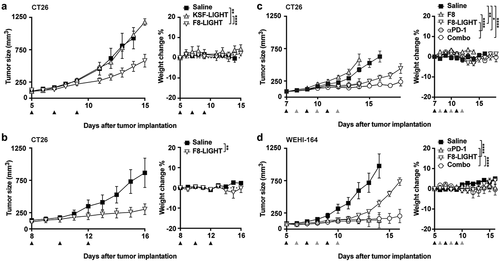
To evaluate the anti-tumor activity of F8-LIGHT, we performed a first therapy experiment in BALB/c mice bearing subcutaneous CT26 murine colon carcinoma, since forced expression of LIGHT in this model had previously shown the ability to induce complete tumor regression.Citation23 Treatment was initiated when tumors had reached a volume of about 100 mm3 and consisted in intravenous injection of 100 µg F8-LIGHT every other day, for a total of three injections. Treatment with F8-LIGHT induced tumor growth retardation, whereas the KSF-LIGHT fusion protein used as negative control gave profiles similar to the ones obtained in the saline treatment group. As no toxicity had been observed (), the dose was increased to 300 µg/injection, which was still well tolerated, but did not significantly improve anti-cancer activity (). In order to exclude a direct effect of the antibody portion of F8-LIGHT, we produced and characterized the F8 antibody in single-chain diabody format (Supplementary Figure 4) and included it as a control in a new therapy experiment. We also included a group to investigate the effect of F8-LIGHT combined with PD-1 blockade. F8 treatment did not show any detectable anti-tumor effect, whereas the combination of F8-LIGHT and anti-PD-1 antibody performed better than F8-LIGHT monotherapy. PD-1 treatment alone had an effect comparable to that of F8-LIGHT monotherapy until experimental day 14, when 3 out of five mice had to be euthanized because of tumor ulceration (). CT26 is an immunologically “hot” murine tumor that exhibits a rich infiltrate of lymphocytes.Citation29,Citation30 Flow cytometry experiments on saline-treated tumors showed that the proportion of T lymphocytes in CT26 tumors was higher than in another commonly studied BALB/c tumor, the WEHI-164 sarcoma model (Supplementary Figure 5a). Also, the percentage of CD8 + T cells specific to the AH1 rejection antigen was significantly higher in CT26 compared to WEHI-164 and, interestingly, to C51 colon carcinoma, where the proportion of CD8 + T cells in the tumor was comparable to CT26 (Supplementary Figure 5b). In order to study anticancer activity in a second immunocompetent mouse model, we treated WEHI-164 sarcomas, including a combination treatment with PD-1 blockade. Also in this case, F8-LIGHT monotherapy significantly inhibited tumor growth compared to saline. Combination treatment with an anti-PD-1 antibody led to tumor regression in all mice. Two of five animals enjoyed a complete and durable remission, but a similar activity was also observed in the PD-1 blockade monotherapy group, with three out of five animals experiencing complete tumor regression ().
Figure 3. Therapy experiments
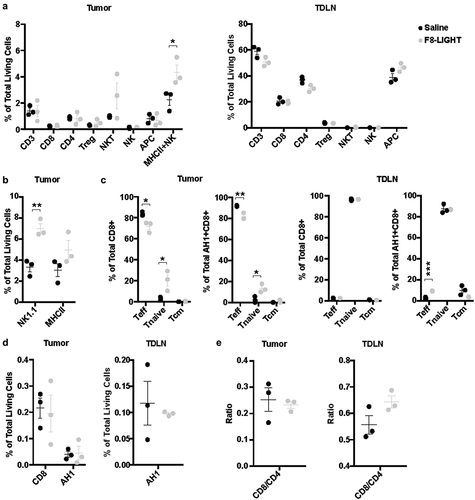
F8-LIGHT treatment increases NK cells but not CD8 + T cells infiltration in the tumor
Since previous studies had reported the ability of LIGHT to attract CD8 + T cells into the neoplastic mass, we treated WEHI-164-bearing mice with F8-LIGHT or saline and analyzed tumors and tumor-draining lymph nodes (harvested 48 hours after the last injection) by flow cytometry. In contrast to reports obtained by the transgenic expression of LIGHT by tumor cells, we did not observe any increase of CD8 + T cells in tumors from the F8-LIGHT treatment group compared to saline (). Instead, we found a significant increase of tumor-infiltrating cells positive for the NK cells marker NK1.1 (). Within the NK1.1 positive population, we could distinguish three different subpopulations: a CD3-negative (conventional NK cells), a CD3-positive (NKT cells) and a CD3-intermediate, MHC class II-positive population, which was the most abundant one (). Inspection of the phenotype composition of the CD8 + T cells revealed an increase in the proportion of CD62L+CD44low naïve CD8 + T cells infiltrating LIGHT-treated tumors (). The same feature was observed for the AH1-specific CD8 + T cell population in the tumor, whereas in the draining lymph node the situation was reversed, with an increased proportion of CD62L-CD44+ effector AH1-specific T cells after F8-LIGHT treatment (). No significant difference among the different therapy groups could be found in terms of abundance of CD3+, CD4+, MHC class II+ and AH1-specific CD8+ cells. No difference in CD8-to-CD4 ratios was observed in tumors or lymph nodes (, D-E). We repeated the analysis in mice bearing CT26. In this model, the population of CD8 + T cells infiltrating the tumor mass was significantly lower in F8-LIGHT-treated mice compared to the control group, supporting the hypothesis that anti-tumor effect of F8-LIGHT is not primarily mediated by these cells (Supplementary Figure 6).
Figure 4. Analysis of immune infiltrate
F8-LIGHT treatment mediated tumor growth retardation in both WEHI-164 and CT26 models. In WEHI-164-derived samples, however, the proportion of total living cells within the neoplastic mass (determined by 7-AAD staining on total events at time of analysis) was significantly higher in the F8-LIGHT group compared to saline, while leukocyte levels were similar (). When trying to identify the nature of living cells within the F8-LIGHT-treated neoplastic mass, we observed fewer tumor cells compared to the saline group, accompanied by the emergence of an abundant population of FSC-low and SSC-low particles (which we called SLE, for “small living events”) (). These SLEs stained negative for 7-AAD, CD3, CD4, CD8, NK1.1 and MHC class II (Supplementary Figure 8).
Figure 5. Analysis of tumor composition
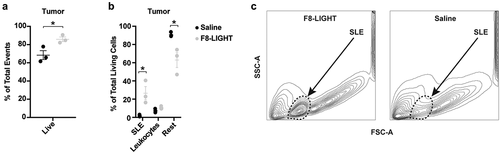
In a separate experiment, staining of lymph nodes cells with the TER-119 antibody suggested that the SLE population may correspond to erythrocytes, in view of size and granularity distribution. The reason why we detected this population in all F8-LIGHT samples and in none of the saline samples, despite the fact that all samples were processed in the exact same way, remains unknown.
Discussion
We have generated a novel fusion protein (termed F8-LIGHT), which was able to selectively deliver murine LIGHT to solid tumors, thanks to the binding properties of the F8 antibody, specific to the fibronectin EDA. This immunocytokine product was well behaved in biochemical assays and fully active in vitro, unlike other formats featuring murine LIGHT as module, described here or in previous studies.Citation31,Citation32 F8-LIGHT induced a specific tumor-growth retardation that was not observed for a similar fusion protein used as negative control of irrelevant specificity in the mouse (KSF-LIGHT). In spite of its potent anti-cancer activity, F8-LIGHT could not cure CT26 nor WEHI-164 tumors in mice, when used as a single agent.
The strongest evidence linking LIGHT to an anti-cancer activity had emerged from the use of tumor cells, which had been engineered to express large quantities of LIGHT and which were completely rejected by immunocompetent mice in a process that relied on CD8 + T lymphocytes.Citation19,Citation23,Citation33 Delivery of LIGHT to the tumor using the F8 antibody did not boost CD8 + T lymphocytes and did not mediate tumor rejection. This effect may be explained by differences in the experimental settings. The use of tumor-homing LIGHT fusion proteins may not reach in vivo concentrations of cytokine at the site of disease comparable to those achievable by genetic overexpression of LIGHT in tumor cells. Moreover, F8-LIGHT anchored the cytokine payload on the tumor extracellular matrix, which contained the fibronectin EDA, while transgenic tumor cells may display LIGHT on their surface.Citation19,Citation23,Citation34 The antibody-based delivery of cytokines to components of the modified extracellular matrix (e.g., splice variants of fibronectin or of tenascin-C) has been shown to be potently active for other immunomodulatory agents, including interleukin-2, interleukin-12 and tumor necrosis factor,Citation11,Citation14,Citation35 but may not be the best strategy to deliver LIGHT.
An epidermal growth factor receptor (EGFR)-targeted version of a mutant human LIGHT (EGFR-LIGHT) has recently been described.Citation32 The fusion protein cross-reacted with murine HVEM and LTβ receptors, as shown by flow cytometry analysis, and exhibited a potent antitumor activity in mice bearing small malignant lesions, but had limited effects in mice bearing more advanced tumors. Unlike what we observed for F8-LIGHT, EGFR-LIGHT treatment induced a strong infiltration of CD8 + T cells in the tumor mass. In a tumor model with a low natural level of immune infiltration, which did not respond to treatment with anti-PD-L1 or EGFR-LIGHT, combination with the two agents led to complete tumor eradication.Citation32
This study represents an example of a LIGHT-mutant delivered to a cellular-associated antigen (EGFR). It would be of interest to test if an equivalent of our F8-LIGHT, which features murine wild-type LIGHT and an antibody specific for a tumor-cell surface antigen instead of F8, behaves similarly.
Although many studies have focused on the effect of LIGHT on CD8+ lymphocytes,Citation19,Citation36 Fan et al. demonstrated the essential contribution of NK cells at an early phase of the LIGHT-induced anti-tumor response.Citation33 They showed that LIGHT was capable of activating and inducing the proliferation of NK cells and that the IFN gamma produced by these NK cells contributed to the subsequent activation of CD8+ cells. Treatment of tumor-bearing mice with F8-LIGHT led to an expansion of intratumoral NK cells, but not of CD8 + T cells.
In previous studies T cell infiltration was assessed 15–18 days after treatment initiation, whereas we analyzed immune infiltration 6 days after the first F8-LIGHT administration.Citation23,Citation32,Citation33 It is possible that CD8 + T cell expansion becomes evident only after a longer period of time. In our setting, mice usually reached criteria for humane endpoints around 10–14 days after the start of the treatment, making it challenging to perform similar analysis. Moreover, CD8 + T cell may require a prolonged period of exposure to LIGHT to infiltrate and expand in the tumor. In line with this hypothesis, infiltration of CD8 + T cells was increased in tumors stably expressing LIGHT, and in tumor treated with EGFR-LIGHT, which contains the IgG Fc-domain and is expected to have a longer half-life compared to our Fc-less F8-LIGHT.Citation19,Citation23,Citation32,Citation33
LIGHT is a member of the TNF superfamily. Our group and other researchers have previously studied the possibility of using tumor-homing antibodies for the selective delivery of TNF and related homotrimeric proteins.Citation17,Citation24,Citation25,Citation35,Citation37 TNF fusions exhibit an extremely selective tumor-targeting performance, possibly due to the vasoactive properties of their cytokine payload, while other superfamily members are difficult to deliver.Citation24 Besides falling in the second category in terms of tumor targeting ability, murine LIGHT has also been reported to be difficult to express as a recombinant.Citation31,Citation32,Citation38 In our hands, LIGHT showed selective accumulation into solid tumors only after extensive engineering of the protein format and of gene expression methods ( and Supplementary Figure 3). Similar features have recently been reported for other cytokine payloads, including interleukin-12 and interleukin-15.Citation27,Citation39,Citation40
Experimental tumors and neoplastic masses in patients can be defined as “hot” and as “cold,” based on the relative density of lymphocyte infiltration. The CT26 model, used in this study and in many other investigations, is considered a “hot” tumor, which readily responds to immunotherapy.Citation29,Citation30 By contrast, WEHI-164 fibrosarcoma (also used in our study) exhibits a lower level of lymphocyte infiltration (Supplementary Figure 5). Both models grow in BALB/c mice and in both models the antigenic peptide AH1 (derived from the gp70 envelope protein of the murine leukemia virus) can play a role for the tumor-rejection process.Citation41 Aberrantly expressed antigens (including retroviral gene products) can act as dominant tumor-rejection antigens in some settings.Citation42 Unlike what we had previously observed for other antibody-cytokine products,Citation11,Citation35,Citation43 treatment with F8-LIGHT did not substantially alter the tumor density of AH1-specific CD8 + T cells ().
A fully human analog of F8-LIGHT may represent a useful tool for the in vivo boosting of NK cell activity, but this agent may be suboptimal for selective activation of tumor-specific CD8 + T cells. The fibronectin EDA represents an ideal target for preclinical studies and for clinical translational activities, as the antigen is highly conserved from mouse to man. EDA is expressed in the majority of solid and hematological malignancies,Citation44,Citation45 while being virtually undetectable in normal adult tissues.Citation46
Materials and methods
Cell lines and animal models
CT26 colon carcinoma (ATCC CRL-2638), WEHI-164 fibrosarcoma (ATCC CRL-1751), F9 teratocarcinoma (ATCC CRL-1720), and HT-29 human adenocarcinoma cells (ATCC HTB-38) were obtained from ATCC, CHO-S cells from Invitrogen. Cells were handled according to supplier’s protocol and maintained as cryopreserved aliquots in liquid nitrogen. Authentication including check of post-freeze viability, growth properties and morphology, test for mycoplasma contamination, isoenzyme assay and sterility test were performed by the cell bank before shipment. Tumor-cell lines were kept in culture no longer than 3 weeks, CHO-S cells no longer than 4 weeks. Eight-weeks-old female BALB/c or 129/Sv mice were obtained from Janvier (France). All animal experiments were performed under a project license granted by the Veterinäramt des Kantons Zürich, Switzerland (04/2018).
Cloning, expression and biochemical characterization of fusion proteins
The DNA sequence encoding murine LIGHT extracellular domain (amino acids 87–239) in a single-chain format (in which three LIGHT subunits were genetically linked together by a Glycine codon) including an N-terminal (SSSSG)3-linker, was purchased from Eurofins genomics. The LIGHT gene was fused by PCR assembly to the C-terminal end of various formats of the F8 antibody via its 15 amino acids linker. The resulting genes were cloned into the mammalian vectors pcDNA3.1+ (for F8 in scFv-Fc and diabody formats) or pMM137 (for F8 in IgG format) by restriction enzymes digestion and ligation, followed by amplification in TG1 electrocompetent E. coli bacteria. pMM137 was kindly provided by Philochem AG and has been described elsewhere.Citation47 Fusion proteins were produced in CHO-S by transient gene expression as already described.Citation48,Citation49 Both “low density” (LD)Citation48 and “high density” (HD)Citation49 protocols were used. Proteins were purified to homogeneity by protein A affinity chromatography and characterized by size exclusion chromatography on a Äkta Pure FPLC system (GE Healthcare) with a Superdex S200 10/300 increase column (GE Healthcare) and by SDS-PAGE.
Mass spectrometry analysis of F8-LIGHT
The fusion protein was treated with glycerol-free PNGase F (NEB, P0705S) in non-denaturing reaction conditions, as indicated by the manufacturer, to remove N-linked glycans. The resulting protein was analyzed by liquid chromatography-mass spectrometry on a Waters Xevo G2-X2 Qtof instrument coupled to a Waters Acquity UPLC H-class system, using a 2.1 × 50 mm Acquity BEH300 C4 1.7 μm column (Waters).
Functional in vitro characterization of F8-LIGHT
Binding of the F8 moiety to its cognate antigen was tested by ELISA. Briefly, wells of an F96 Maxisorp nunc-immuno plate (Thermo Fisher) were coated with EDA and blocked with 2% milk powder in phosphate-buffered saline (PBS). F8-LIGHT and positive control F8 in small immune protein format (SIPF8) were added at different concentrations in wells, followed by incubation with polyclonal antibodies against human kappa light chains (Dako, A0192) and by HRP-conjugated protein A (GE Healthcare, NA9120V). Colorimetric reaction was started by adding BM Blue POD substrate (Roche, 11442066001) and quenched with 1 M H2SO4. Absorbance at 450 nm was measured with a Spectra Max Paradigm multimode plate reader (Molecular Devices). In vitro activity of the LIGHT moiety was tested with a cytotoxicity assay on HT-29 cells, which have been shown to be sensitive to LIGHT in the presence of human IFN gamma (hIFNγ).Citation50 Briefly, F8-LIGHT and recombinant hIFNγ (Biolegend, 570202) at given concentrations were added to wells containing 40000 HT-29 cells in McCoy’s 5A (Modified) Medium (Thermo Fisher, 16600082) supplemented with 10% fetal bovine serum (Thermo Fisher, 10270106) and 1x Antibiotic-Antimycotic (Thermo Fisher, 15240062). After incubation for 72 hours at 37°C, 5% CO2, cells were detached using Trypsin-EDTA solution (Thermo Fisher, 25200056), re-suspended in PBS containing 0.5% bovine serum albumin (BSA) and 2 mM EDTA and stained with 7-AAD (Biolegend, 420404) for 5ʹ at 4°C. The percentage of living cells was determined by flow cytometry analysis using a CytoFLEX S (Beckman Coulter). Data were analyzed with FlowJo software (FlowJo, LLC, version 10).
Quantitative biodistribution
In vivo targeting ability of F8-LIGHT was determined by quantitative biodistribution as already described.Citation34 Briefly, 1 × 107 F9 cells were subcutaneously injected in the right flank of 129/Sv mice. Tumor size was determined daily using the formula: ½ x (major diameter) x (minor diameter)2. When tumors reached a volume of 100–300 mm3, fusion proteins were labeled with Iodine-125, using Chloramine T. Radiolabeled protein was purified by size exclusion chromatography (PD-10 column, GE Healthcare, 17–0851-01) and injected in the lateral tail vein of tumor-bearing mice. Animals were sacrificed after 24 hours. Organs and tumors were harvested, weighed and radioactivity was determined using a Packard Cobra II Gamma Counter (GMI).
Tumor therapy experiments
Anti-tumor activity of F8-LIGHT was tested in two syngeneic murine models of cancer. CT26 and WEHI-164 tumors were implanted in the right flank of BALB/c mice by subcutaneous injection of 3 × 106 and 2.5 × 106 cells, respectively. Treatments were started when tumors reached a volume of about 100 mm3. The indicated dose of immunocytokines or the corresponding volume of saline, were administered by intravenous injection in the lateral tail vein, every other day for a total of three injections. The immune-checkpoint inhibitor anti-PD-1 (BioXCell, clone 29 F.1A12) was administered in the same way, at alternate days. Mice were inspected daily. Body weight was monitored, tumor was measured with a caliper and tumor size was determined using the formula: ½ x (major diameter) x (minor diameter)2. Mice with ulcerated hemorrhagic tumor, or with tumor bigger than 1500 mm3 where euthanized.
Antibody and reagents for flow cytometry
Fluorophore-conjugated antibodies against CD3 (clone 17A2), CD4 (clone GK1.5), CD8 (clone 53–6.7), NK1.1 (clone PK136), I-A/I-E (clone M5/114.15.2), CD62L (clone MEL-14), CD44 (clone IM7), and FoxP3 (clone MF-14), as well as 7-AAD and Zombie Red viability dies, were all purchased from BioLegend. AH1-loaded, PE-conjugated H-2Ld tetramers were obtained as already described.Citation43
Analysis of immune infiltrates
Immune infiltrates were analyzed in tumors and tumor-draining lymph nodes of mice bearing WEHI-164 sarcoma, 48 hours after the last injection of either saline or F8-LIGHT. Mice were euthanized and tumors, right axillary and right inguinal lymph nodes were harvested. Tumors were cut into small fragments and incubated in an orbital shaker at 37°C for 30ʹ in RPMI 1640 Medium (Thermo Fisher, 21875034) containing 1x Antibiotic-Antimycotic (Thermo Fisher, 15240062), 1 mg/mL Collagenase II (Thermo Fisher, 17101015) and 0.1 mg/mL DNAse I (Roche, 10104159001). Tumor cells suspensions were passed through a 70 μm cell strainer (Corning) and treated with Red Blood Cells Lysis buffer (Biolegend, 420301) following supplier’s recommendations. Lymph nodes were harvested and smashed on a 70 μm cell strainer (Corning) using the back of a syringe plunger. The resulting tumor and lymph nodes single-cell suspensions were washed in PBS, before incubation with staining reagents. Where appropriate, cells were stained with Zombie Red dye (diluted 1:500 in PBS) for 15ʹ at room temperature, followed by staining with antibodies and tetramers in FACS buffer (0.5% BSA, 2 mM EDTA in PBS) for 30ʹ at 4°C. Cells, which were not stained with Zombie Red, were stained with 7-AAD (diluted 1:100 in FACS buffer) for 5ʹ at 4°C. Intracellular staining with antibodies against FoxP3 was performed using eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher), following supplier’s protocol. Samples were analyzed with CytoFLEX S (Beckman Coulter) and data were processed using FlowJo software (FlowJo, LLC, version 10). A detailed description of the gating strategies can be found in the Supplemental materials (Supplementary Figure 7–9). Total of living cells in the tumor was calculated by subtracting dead cells and debris from the total number of recorded events.
Data analysis
Data were analyzed using Prism 7.0 (GraphPad Software, Inc.). Statistical significances of tumor therapy and flow cytometry data were determined with a regular two-way ANOVA test with Bonferroni posttest correction and with an unpaired, two-tailed t-test, respectively. Data represent means ± SEM. P < .05 was considered statistically significant.
Authors’ contributions
D.N. and M.S. planned and designed the study. M.S. performed the experiments. J.M. and P.M. helped with the biodistribution experiments. V.F. performed some experiments under the supervision of M.S. M.S. prepared the figures. D.N. and M.S. wrote the manuscript.
Consent for publication
The content of the manuscript and the supplemental material submitted for publication have been approved by all the authors and are not under consideration for publication elsewhere.
Competing interests
D.N. is co-founder and shareholder of Philogen SpA (Siena, Italy), a biotech company that owns the F8 antibody. The authors have no additional financial interests.
Ethics approval and consent to participate
Mice experiments were conducted under the ethical approval of the Veterinäramt des Kantons Zürich, Switzerland (License 04/2018).
Supplemental Material
Download MS Word (3.2 MB)Acknowledgments
D. N. gratefully acknowledges ETH Zürich, the Swiss National Science Foundation (Grant Nr. 310030_182003/1) and the European Research Council (ERC, under the European Union’s Horizon 2020 research and innovation program, grant agreement 670603) for financial support.
Data availability statement
Data and material are presented in the main text and supplementary information, raw data are available from the corresponding author on reasonable request.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–10. doi:10.1172/JCI83871.
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015; 372:2018–28
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015; 372:311–9
- Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999; 17:2105–16
- Golomb HM, Jacobs A, Fefer A, Ozer H, Thompson J, Portlock C, Ratain M, Golde D, Vardiman J, Burke JS, et al. Alpha-2 interferon therapy of hairy-cell leukemia: a multicenter study of 64 patients. Journal of Clinical Oncology 1986; 4:900–5 6 doi:10.1200/JCO.1986.4.6.900
- Solal-Celigny P, Lepage E, Brousse N, Reyes F, Haioun C, Leporrier M, Peuchmaur M, Bosly A, Parlier Y, Brice P, et al. Recombinant interferon alfa-2b combined with a regimen containing doxorubicin in patients with advanced follicular lymphoma. Groupe d'Etude des Lymphomes de l'Adulte. N Engl J Med 1993; 329:1608–14
- Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol. 2016;40:96–102. doi:10.1016/j.coi.2016.03.006.
- Amsen D, van Gisbergen K, Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. 2018;19:538–46. doi:10.1038/s41590-018-0114-2.
- Hutmacher C, Gonzalo Nunez N, Liuzzi AR, Becher B, Neri D. Targeted delivery of IL2 to the tumor stroma potentiates the action of immune checkpoint inhibitors by preferential activation of NK and CD8(+) T cells. Cancer Immunol Res. 2019;7:572–83. doi:10.1158/2326-6066.CIR-18-0566.
- Puca E, Probst P, Stringhini M, Murer P, Pellegrini G, Cazzamalli S, Hutmacher C, Gouyou B, Wulhfard S, Matasci M, et al. The antibody-based delivery of interleukin-12 to solid tumors boosts NK and CD8+T cell activity and synergizes with immune checkpoint inhibitors. Int J Cancer. 2020;146:2518–30. doi:10.1002/ijc.32603.
- Hemmerle T, Neri D. The antibody-based targeted delivery of interleukin-4 and 12 to the tumor neovasculature eradicates tumors in three mouse models of cancer. Int J Cancer. 2014;134:467–77. doi:10.1002/ijc.28359.
- Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A. 2015;112:3320–25. doi:10.1073/pnas.1416159112.
- Carnemolla B, Borsi L, Balza E, Castellani P, Meazza R, Berndt A, Ferrini S, Kosmehl H, Neri D, Zardi L, et al. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood. 2002;99:1659–65. doi:10.1182/blood.V99.5.1659.
- Lode HN, Xiang R, Duncan SR, Theofilopoulos AN, Gillies SD, Reisfeld RA. Tumor-targeted IL-2 amplifies T cell-mediated immune response induced by gene therapy with single-chain IL-12. Proc Natl Acad Sci U S A. 1999;96:8591–96. doi:10.1073/pnas.96.15.8591.
- Yoo EM, Trinh KR, Tran D, Vasuthasawat A, Zhang J, Hoang B, Lichtenstein A, Morrison SL. Anti-CD138-targeted interferon is a potent therapeutic against multiple myeloma. J Interferon Cytokine Res. 2015;35:281–91. doi:10.1089/jir.2014.0125.
- Zhang N, Sadun RE, Arias RS, Flanagan ML, Sachsman SM, Nien YC, Khawli LA, Hu P, Epstein AL. Targeted and untargeted CD137L fusion proteins for the immunotherapy of experimental solid tumors. Clin Cancer Res. 2007;13:2758–67. doi:10.1158/1078-0432.CCR-06-2343.
- Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh S-L, Nagata S, Ni J, Chen L. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol. 2000;164:4105–10. doi:10.4049/jimmunol.164.8.4105.
- Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, Fu Y-X, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. 2004;5:141–49. doi:10.1038/ni1029.
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998; 8:21–30
- Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, Browning JL, Sedgwick JD, Cyster JG. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. The Journal of experimental medicine 1999; 189:403–12
- Lu TT, Browning JL. Role of the lymphotoxin/LIGHT system in the development and maintenance of reticular networks and vasculature in lymphoid tissues. Front Immunol. 2014;5:47. doi:10.3389/fimmu.2014.00047.
- Qiao G, Qin J, Kunda N, Calata JF, Mahmud DL, Gann P, Fu YX, Rosenberg SA, Prabhakar BS, Maker AV. LIGHT Elevation Enhances Immune Eradication of Colon Cancer Metastases. Cancer research 2017; 77:1880–91
- Hemmerle T, Hess C, Venetz D, Neri D. Tumor targeting properties of antibody fusion proteins based on different members of the murine tumor necrosis superfamily. J Biotechnol. 2014;172:73–76. doi:10.1016/j.jbiotec.2013.12.010.
- Fellermeier S, Beha N, Meyer JE, Ring S, Bader S, Kontermann RE, Müller D. Advancing targeted co-stimulation with antibody-fusion proteins by introducing TNF superfamily members in a single-chain format. Oncoimmunology. 2016;5:e1238540. doi:10.1080/2162402X.2016.1238540.
- Richards DM, Marschall V, Billian-Frey K, Heinonen K, Merz C, Redondo Muller M, Sefrin JP, Schröder M, Sykora J, Fricke H. HERA-GITRL activates T cells and promotes anti-tumor efficacy independent of FcgammaR-binding functionality. J Immunother Cancer. 2019;7:191. doi:10.1186/s40425-019-0671-4.
- Venetz D, Hess C, Lin CW, Aebi M, Neri D. Glycosylation profiles determine extravasation and disease-targeting properties of armed antibodies. Proc Natl Acad Sci U S A. 2015;112:2000–05. doi:10.1073/pnas.1416694112.
- Muthing J, Kemminer SE, Conradt HS, Sagi D, Nimtz M, Karst U, Peter-Katalinić J. Effects of buffering conditions and culture pH on production rates and glycosylation of clinical phase I anti-melanoma mouse IgG3 monoclonal antibody R24. Biotechnol Bioeng. 2003;83:321–34. doi:10.1002/bit.10673.
- Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–89. doi:10.1097/01.cji.0000436722.46675.4a.
- Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol Res 2017; 5:29–41
- Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, Hertig S, Ambrose C, Tschopp J, Schneider P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem. 2006;281:13964–71. doi:10.1074/jbc.M601553200.
- Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, Fu Y-X. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. 2016;29:285–96. doi:10.1016/j.ccell.2016.02.004.
- Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, Sun Y, Fu Y-X. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. 2006;107:1342–51. doi:10.1182/blood-2005-08-3485.
- Villa A, Trachsel E, Kaspar M, Schliemann C, Sommavilla R, Rybak JN. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int J Cancer. 2008;122:2405–13. doi:10.1002/ijc.23408.
- Probst P, Kopp J, Oxenius A, Colombo MP, Ritz D, Fugmann T, Neri D. Sarcoma Eradication by Doxorubicin and Targeted TNF Relies upon CD8(+) T-cell Recognition of a Retroviral Antigen. Cancer research 2017; 77:3644–54
- Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu Y-X. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi:10.1016/j.immuni.2006.06.016.
- Hutt M, Fellermeier-Kopf S, Seifert O, Schmitt LC, Pfizenmaier K, Kontermann RE. Targeting scFv-Fc-scTRAIL fusion proteins to tumor cells. Oncotarget. 2018;9:11322–35.
- Del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukoc Biol. 2010;87:223–35. doi:10.1189/jlb.0809590.
- Pasche N, Wulhfard S, Pretto F, Carugati E, Neri D. The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin Cancer Res. 2012;18:4092–103. doi:10.1158/1078-0432.CCR-12-0282.
- Kaspar M, Trachsel E, Neri D. The antibody-mediated targeted delivery of interleukin-15 and GM-CSF to the tumor neovasculature inhibits tumor growth and metastasis. Cancer Res. 2007;67:4940–48. doi:10.1158/0008-5472.CAN-07-0283.
- Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci U S A 1996; 93:9730–5
- Laumont CM, Vincent K, Hesnard L, Audemard E, Bonneil E, Laverdure JP, Gendron P, Courcelles M, Hardy M-P, Côté C. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10:10. doi:10.1126/scitranslmed.aau5516.
- Probst P, Stringhini M, Ritz D, Fugmann T, Neri D. Antibody-based delivery of TNF to the tumor neovasculature potentiates the therapeutic activity of a peptide anticancer vaccine. Clin Cancer Res. 2019;25:698–709. doi:10.1158/1078-0432.CCR-18-1728.
- Rybak JN, Roesli C, Kaspar M, Villa A, Neri D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 2007;67:10948–57. doi:10.1158/0008-5472.CAN-07-1436.
- Schliemann C, Wiedmer A, Pedretti M, Szczepanowski M, Klapper W, Neri D. Three clinical-stage tumor targeting antibodies reveal differential expression of oncofetal fibronectin and tenascin-C isoforms in human lymphoma. Leuk Res. 2009;33:1718–22. doi:10.1016/j.leukres.2009.06.025.
- Schwager K, Kaspar M, Bootz F, Marcolongo R, Paresce E, Neri D, Trachsel E. Preclinical characterization of DEKAVIL (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis. Arthritis Res Ther 2009; 11:R142
- Putelli A, Kiefer JD, Zadory M, Matasci M, Neri D. A fibrin-specific monoclonal antibody from a designed phage display library inhibits clot formation and localizes to tumors in vivo. J Mol Biol. 2014;426:3606–18.
- Pasche N, Woytschak J, Wulhfard S, Villa A, Frey K, Neri D. Cloning and characterization of novel tumor-targeting immunocytokines based on murine IL7. J Biotechnol. 2011;154:84–92. doi:10.1016/j.jbiotec.2011.04.003.
- Rajendra Y, Kiseljak D, Baldi L, Hacker DL, Wurm FM. A simple high-yielding process for transient gene expression in CHO cells. J Biotechnol. 2011;153:22–26.
- Zhang MC, Liu HP, Demchik LL, Zhai YF, Yang DJ. LIGHT sensitizes IFN-gamma-mediated apoptosis of HT-29 human carcinoma cells through both death receptor and mitochondria pathways. Cell Res. 2004;14:117–24. doi:10.1038/sj.cr.7290210.