
ABSTRACT
Bispecific antibodies, engineered to recognize two targets simultaneously, demonstrate exceptional clinical potential for the therapeutic intervention of complex diseases. However, these molecules are often composed of multiple polypeptide chains of differing sequences. To meet industrial scale productivity, enforcing the correct quaternary assembly of these chains is critical. Here, we describe Chain Selectivity Assessment (CSA), a high-throughput method to rationally select parental monoclonal antibodies (mAbs) to make bispecific antibodies requiring correct heavy/light chain pairing. By deploying CSA, we have successfully identified mAbs that exhibit a native preference toward cognate chain pairing that enables the production of hetero-IgGs without additional engineering. Furthermore, CSA also identified rare light chains (LCs) that permit positive binding of the non-cognate arm in the common LC hetero-IgGs, also without engineering. This rational selection of parental mAbs with favorable developability characteristics is critical to the successful development of bispecific molecules with optimal manufacturability properties.
Introduction
Bispecifics represent an exciting new generation of large molecule therapeutics in a field currently dominated by canonical monospecific monoclonal antibodies (mAbs).1,Citation2 A defining feature of bispecifics is the ability to recognize two epitopes locating on the same or distinct targets. This dual-recognition capability expands the functionality of conventional mAbs, allowing for diverse applications such as recruiting immune cells to destroy tumor cells, crosslinking distinct cell surface receptors or enhancing tissue specificity.Citation1,Citation3 For example, Amgen’s Bispecific T-cell Engager (BiTE®) binds both a CD3 epitope on the surface of T cells and a tumor-associated antigen,Citation4,Citation5 effectively acting as a bridge to link immunologically active T cells and target tumor cells. To date, over 100 bispecific formats have been reported, with over 85 in development and three receiving US Food and Drug Administration approval.Citation1,Citation6,Citation7 Generally, bispecifics can be classified in three categories: 1) fragment fusion (e.g., tandem single-chain variable fragment (scFv), dual-affinity re-targeting antibodies); 2) IgG fusion (e.g., IgG-scFv, dual-variable domain-Ig); and 3) IgG-like molecule (e.g., hetero-IgG).Citation7,Citation8 While fragment fusions and IgG-fusions show a simple engineered configuration (only one or two polypeptide chains) that favors purification and stable cell line generation, these formats often display low yields and undesirable stability profiles. In contrast, IgG-like bispecifics that mimic the native structure of IgG molecules (e.g., hetero-IgG) show higher stability and superior cell production. Moreover, they are among the most represented bispecific formats in clinical trials, possibly due to the good half-life profile in serum and low potential for immunogenicity.Citation1 However, due to the high number of chains with differing sequences (3–4) in these formats, multiple purification steps are needed to remove undesired mispaired species, which can lead to substantial reduction of the final purification yield.
In an engineered IgG-like bispecific antibody, multiple heavy chains (HCs) and light chains (LCs) are assembled into a single molecule to enable the recognition of two distinct epitopes. Therefore, ensuring that the correct chains pair to yield the desired bispecific molecule is challenging. In the case of 4-chain hetero-IgGs, the co-expression of these chains in the same cell can result in nine possible combinations of mispaired IgG species.Citation9,Citation10 Several strategies, including knobs-into-holes,Citation11 strand-exchange engineered domainCitation12 and charge pair mutations (CPMs),Citation13,Citation14 have been developed to address the HC/HC and HC/LC pairing problems. In most cases of HC/LC engineering, the rationale is to engineer the chain interface in such a way that favors cognate HC/LC pairing over non-cognate. However, despite the best engineering efforts, sequence diversity in the complementarity-determining regions (CDRs), framework, and LC isotype often limits the success of these engineering tools when applied as a rigid platform to HC/LC pairing.
To overcome these difficulties, the use of a common light chain (cLC) is appealing because it avoids the need to drive pairing between specific HCs and LCs. However, the identification of a cLC that maintains the desired binding profile to distinct epitopes when paired with different HCs is difficult and often requires substantial investment early in the drug development process.Citation15 In general, two methods are most commonly used to discover antibodies that carry a cLC. The first involves the screening of display libraries that consists of diverse HC sequences, but only one or few LCs. Alternatively, mice expressing a universal LC can be immunized for each of the desired targets.Citation16,Citation17 Since both approaches restrict the available LCs, these cLC antibodies often show suboptimal binding affinities requiring extensive engineering, mostly in the HC, to optimize target affinity. In contrast, both HC and LC can be targeted for optimization efforts in monospecific mAbs. Moreover, structures of antibody-antigen complexes reveal that much of antibody/epitope interactions are HC driven and in some rare cases the LC does not make any productive interactions.Citation18 This led us to hypothesize that some LCs may be suitable for pairing with non-cognate HCs while retaining target binding affinity. The cLC hetero-IgGs assembled with such LCs, together with the cognate and non-cognate HCs, will require less optimization because they retain the native affinity in their cognate HC/LC arms.
The development of therapeutic antibodies usually starts with the immunization of humanized animal models with selected antigens, leading to the identification and isolation of lead mAbs.Citation19,Citation20 These mAbs are selected to meet design goals such as target specificity, binding affinity, cross-species reactivity, yield, stability and lack of immunogenicity, among others, but little is known of the properties required for those that will become building blocks for bispecifics. Frequently, this demands the empirical testing of hundreds of bispecifics to evaluate every parental mAb combination, resulting in extended timelines and increased need for resources.
In this study, we conceptualized, developed, and validated two high-throughput screening methods to facilitate the rational selection of lead mAbs to make bispecifics: competition and non-competition Chain Selectivity Assessment (CSA). Using competition CSA (cCSA), we selected for mAbs whose HCs and LCs assemble effectively into hetero-IgG molecules with no or minimal cross-pairing between two antigen-binding fragment (Fab) regions. From two distinct panels of lead mAbs, each recognizing a different target, we identified mAb combinations showing a native preference (low cross-paring) toward their cognate HC/LC pairing. The 4-chain hetero-IgGs assembled from these selected mAb combinations showed high expression levels and suitable purification profiles in the absence of HC/LC interface engineering. Through non-competition CSA (ncCSA), we were able to identify LCs that efficiently pair with non-cognate HCs. Besides the optimal manufacturing profile, binding analysis of these cLC hetero-IgGs also revealed molecules that retain binding to both targets, suggesting that ncCSA is a powerful tool to identify cLCs in a cost-effective manner.
Results
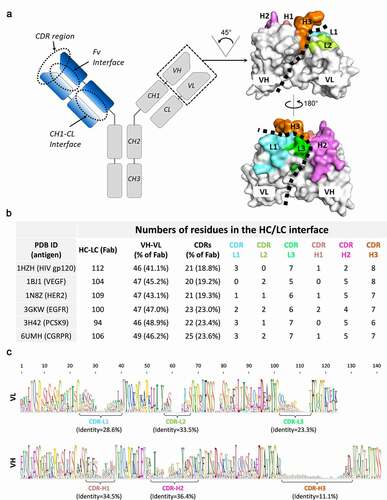
Diversity of antibody variable regions provides rationale for the chain selectivity assessment
The IgG molecule is composed of two Fabs that recognize the epitope and a crystallizable fragment (Fc) region that interacts with receptors on the cell surface (). In the Fab region, two main points of contact mediate the HC/LC interaction: the VH/VL and CH1/CL domains. Although antibodies mostly conserve the CH1/CL interface (with only the LC kappa/lambda diversity to account for), the VH-VL interface is highly variable and unique to each antibody. Indeed, here both framework and CDRs directly engage across the interface (). To further evaluate the native properties of the VH/VL interface, we sought to analyze six x-ray structures of mAbs generated against a variety of relevant therapeutic targets. Across these six structures, we calculated that ~100 (94 to 112) residues mediate the HC/LC interface (buried surface area of 1,620–1,880 Å2), with over 40% of those located in the VH/VL interface, with the CDRs contributing ~20% of the total (). Surprisingly, among all six CDRs, the CDR-H2 and CDR-H3 in the HC and CDR-L3 in the LC have the most interfacing residues and are responsible for over 15% of the overall HC/LC interaction. Since CDR-H3 and CDR-L3 are major determinants for antibody target specificity, they are highly variable, displaying low sequence identity (11.1 and 23.3%, respectively ()). Due to the fact that highly diverse CDRs can contribute to HC/LC interaction, we hypothesized that some HC/LC pairs and interfaces may be more favorable than others. Moreover, as the VH/VL interaction is reportedly the first step during the assembly of the quaternary IgG structure,Citation22,Citation23 a favorable VH/VL pair may be key to prime the cognate HC/LC pairing. Therefore, as correct HC/LC pairing is critical to the assembly of 4-chain hetero-IgGs, we rationalized that parental mAbs showing a high preference for cognate HC/LC pairing over non-cognate can serve as ideal building blocks. Simultaneously, those chains shown to be nonspecific (i.e., promiscuous LCs) present an opportunity to explore cLCs. Consequently, the deep understanding of the native properties of the antibody’s sequences may facilitate the production of bispecifics more amenable to its design goals. Since we recognize that the molecular details driving chain pairing selectivity are not yet fully understood, we envisioned a method to experimentally identify selective and nonselective HC/LC pairs that can be used to assemble hetero-IgGs and other formats requiring HC/LC pairing.
Figure 1. Structural analysis of HC-LC interface
Description of chain selectivity assessment screening methods
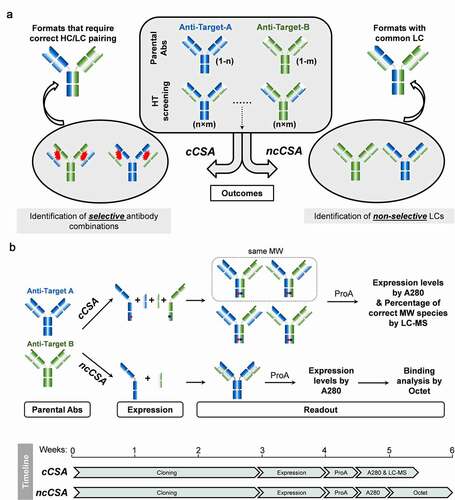
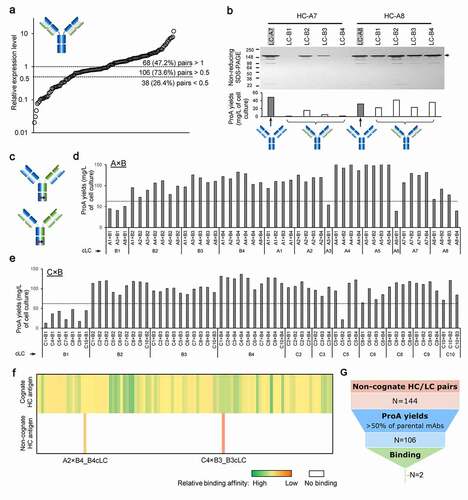
To facilitate the development of IgG-like bispecifics, we envisioned a high-throughput screening process, CSA, to evaluate HC/LC selectivity (). Since the antibody HC is only secreted when bound to the LC,Citation22,Citation23 the measure of expression level of a given HC/LC pair may correlate to the HC/LC pairing efficiency. Starting from two panels of parental antibodies (anti-Target-A and anti-Target-B), we deployed high-throughput CSA in two different scenarios (competition and non-competition) to evaluate the specificity in the assembly of hetero-IgGs (4 chains) and to identify promiscuous LCs for cLC hetero-IgGs (3 chains), respectively ( and 2b). The cCSA experiment mimics a co-expression scenario of four different chains (2×HCs and 2×LCs), resulting in all possible combinations between anti-Target-A and anti-Target-B parental antibodies. The expressed hetero-IgGs were purified from conditioned medium with ProA beads and the percentage of correct HC/LC pairing was relatively quantitated through liquid chromatography-mass spectrometry (LC-MS) ( and S1). It is worth noting that 2 of the 4 hetero-IgG species may show the same molecular weight (MW), with one of them representing a scenario where both LCs (LCA and LCB) have a perfect cross pair with the opposite non-cognate HCs (). Although LC-MS cannot distinguish such species, this scramble of LCs shows low frequency even after limited proteolysis (unpublished data), strongly suggesting that the correct MW detected by LC-MS indicates correct HC/LC pairing. The high percentage of correct species is an indication of the preference for cognate HC/LC pairing over the non-cognate. The antibody combinations with high levels of correct species will be selected as the optimal building blocks for bispecifics that require correct/specific HC/LC pairing (). As for the ncCSA experiment, every single HC from anti-Target-A pairs with every LC from anti-Target-B at a 1:1 ratio, and vice versa. High expression levels (comparable to HC/LC cognate pair or higher) measured by ProA capture indicate that LCs pair well and allow the non-cognate HCs to fold and to be secreted (). In contrast, low expression levels suggest that the VH/VL interface of these chain(s) are specific to their cognate interfaces, and as a result cannot accommodate others. Next, using the same ProA purified material, we integrated a second high-throughput step to characterize the binding affinity using ForteBio Octet. This method enables a quick (less than 6 weeks) and efficient tool to screen and identify rare and valuable LCs that can be used as building blocks for cLC bispecific Abs.
Figure 2. Schematic representation of the design of high-throughput Chain Selective Assessment methods (CSA)
Identification of low cross-pairing antibody combinations with competition csa method
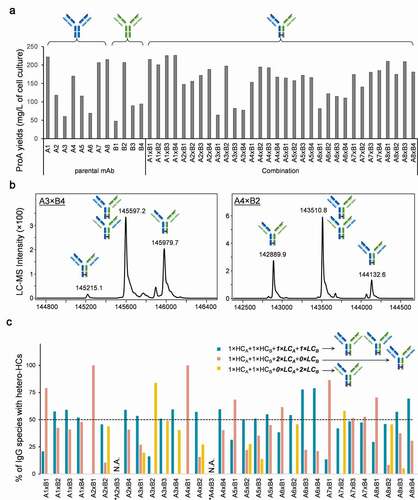
To validate the cCSA method, we selected two panels of parental antibodies, eight anti-Target-A Abs (A1–A8) and four anti-Target-B (B1–B4), representing a diversity in framework composition (Table S1). We then introduced mutations to drive heavy-heavy chain pairing and combined antibodies from each target, resulting in 32 combinations for evaluation. Following the purification of the secreted IgGs from conditioned media, we measured the expression levels of the combinations and parental mAbs by A280 ( and S2). Notably, parental mAbs demonstrated a high variation in expression levels ranging from 50 to 250 mg/L. Surprisingly, most of the antibody combinations expressed at levels higher than 100 mg/L with few exceptions (A3×B1, A3×B3, A3×B4 and A6×B1), which may be related to the low expressing parents A3 and B1. To identify whether the species assembled and secreted have correct HC/LC pairing, we used non-reducing LC-MS to analyze the ProA purified proteins (exemplified in ). By using the Fc region of the molecules for purification with ProA beads, we selected for HC-containing molecules only, and all other species (e.g., LC dimers) are discarded. Although the LC-MS analysis cannot confirm whether the two pairs of cognate HC/LC are correctly paired, it can determine whether each species has a copy of each of the four chains. While not definitive, this is a required condition toward the assembly and production of hetero-IgGs. Therefore, we sought to calculate the percentage for each of these three possible HC/LC scenarios: 1×HCA+1×HCB+1× LCA+1×LCB, 1×HCA+1×HCB+2×LCA+0×LCB and 1× HCA+1× HCB+0×LCA+2×LCB, all containing HC heterodimers (). Although species of two combinations (A2×B3 and A4×B3) were not able to be evaluated in LC-MS analysis due to the small MW difference between two LCs (<60 Da), we were able to successfully quantify the IgG species in all other 30 combinations. Furthermore, although we deployed Fc CPMs in the mAb combinations to enhance HC heterodimerization, there were still small amounts of homodimers and/or half mAbs (Table S2). However, since the focus of this study is on HC/LC pairing, we excluded those species from further analysis. Interestingly, our data showed that in about half of the molecules tested (17/32), the percentage of the desired species (1×HCA+1×LCA+1×HCB+1×LCB) detected was ≥50% of the total species present in the ProA-purified samples (). Surprisingly, in two combinations (A6×B3 and A6×B4) the percentage of the correct product was >75%. In contrast, the ProA purified samples of the remaining 13 combinations contained <50% of the species of interest, with 5 of those combinations (A1×B1, A2×B1, A3×B2, A4×B1 and A7×B1) containing less than 25% of the desired species (). Interestingly, in most cases (23/32) the most undesired products contained only one of the 2 LCs (1×HCA+1×HCB+2×LCA+0×LCB and 1× HCA+1× HCB+0×LCA+2×LCB), suggesting that either one of the LCs is overexpressing or LCA and LCB may compete during expression and molecule assembly, leading to a complete or partial suppression of the other (). Thus, with this information, the cCSA method can efficiently screen and identify suitable combinations of parental mAbs with native properties that enable the correct assembly of the hetero-IgGs upon expression in a single cell. This allows for the deselection of candidates while directing efforts toward those with more promising characteristics, resulting in time and resource savings.
Figure 3. High-throughput screening for low cross-pairing antibody combinations in cCSA experiment
Low cross-pairing antibody combinations are good candidates for making 4-chain hetero-IgGs
In the cCSA method described above, more than half (17/32) of the combinations showed high levels (≥50%) of desired species (1×HCA+1×LCA+1×HCB+1×LCB) (). However, to assess whether this high-throughput method was truly predictive of good hetero-IgG candidates, we decided to scale-up the 32 hetero-IgGs and perform a 2-step purification with ProA followed by a cation exchange chromatography (CIEX). The goal was not only to quantify the final yields of the desired species, but also to study the separation profile in the CIEX step. From the 32 hetero-IgGs, we successfully purified 11 with a final purity >90% as independently determined by size exclusion chromatography (SEC), micro capillary electrophoresis (MCE), and LC-MS ( and Table S2). Interestingly, the final yields for the parental mAbs, the building blocks for these hetero-IgGs, appear to correlate with the yields of the resulting bispecific molecules. Indeed, hetero-IgGs A1×B3, A1×B4, A4×B3, and A8×B3 displaying the highest protein yields all contain at least one of the high-expressing parental mAbs A1, A4, and A8 (). For further verification, the final pools with purity >90% were evaluated for binding against Target-A and -B, critical to evaluate the cognate HC/LC pairing. The data shows that the affinity of each arm in the bispecific molecule to its respective target was comparable to that of the parent mAbs (Figure S3), confirming that all 11 hetero-IgGs have correct HC/LC in both arms.
Figure 4. Predictability of HC/LC pairing in 4-chain hetero-IgGs
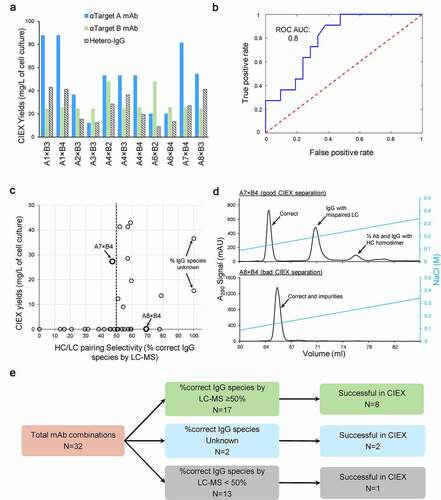
Overall, this data aligns well with the cCSA experiment (). From the 11 hetero-IgGs successfully purified, the percentage of correct HC/LC pairing predicted by LC-MS was high for 8 molecules (≥50% correct HC/LC species, A1×B3, A1×B4, A3×B3, A4×B2, A4×B4, A6×B2, A6×B4, and A8×B3) and unknown for A2×B3 and A4×B3 (indeterminate due to the small MW difference between chains). Most importantly, only one molecule (A7×B4 with 47.4% correct species) that showed low HC/LC pairing by cCSA could be purified by CIEX (). Indeed, the percentage of correct species determined by cCSA was predictive of which Ab combinations resulted in successful hetero-IgGs (receiver operating characteristic curve (ROC) area under the curve 0.80; ). Furthermore, we also attempted to build a correlation between the percentage of HC/LC pairing predicted by cCSA and the final yields. As shown in , most of the combinations (12/13) with <50% correct HC/LC pairing by cCSA failed later during the CIEX purification, highlighting that effectiveness of chain pairing can facilitate purification by decreasing the amount of impurities present. One exception was the A7× B4 molecule that, while displaying 47.4% pairing, showed a CIEX profile with suitable separation (). Interestingly, CIEX could not purify 9 of 17 molecules with ≥50% correct HC/LC pairing predicted by cCSA (). Indeed, to illustrate this phenomenon we selected the example of the A8× B4 hetero-IgG that displayed 69.4% correct pairing in the cCSA experiments. This molecule exhibits a well-shaped peak that conceals the mispaired species in such a way that no resolution between the two different species (1×HCA+1×HCB+1×LCA+1×LCB and 1×HCA+1×HCB+2×LCA+0×LCB) is observed (). Thus, while the cCSA method can predict high-performing hetero-IgG molecules based on the native VH/VL interface, properties not screened by cCSA (such as those influencing separation profiles on ion-exchange chromatography columns) also play a role in selecting hetero-IgGs.
Screening for cLCs with the ncCSA method
As shown above, while some LCs pair preferably with their cognate HCs, other LCs can also bind to non-cognate HCs efficiently. If the resulting non-cognate HC/LC pair retains binding, such an LC could serve as a cLC. We envisioned the ncCSA method as an opportunistic approach to evaluate whether the parental mAbs offer such LCs (). To demonstrate the efficacy of this method, we selected two bispecific programs (A×B and C×B) with 8 anti-Target-A, 4 anti-Target-B and 10 anti-Target-C parental mAbs (Table S1). As shown in Table S3, we paired every LC of anti-Target-B with individual HCs from anti-Target-A or -C mAbs. Meanwhile, we also combined each HC of anti-Target-B with individual LCs from different anti-Target-A or -C mAbs. We then expressed the resulting 144 non-cognate HC/LC pairs together with 22 parental mAbs (control) in 293-6E and purified with ProA beads, followed by analysis with non-reducing SDS-PAGE gel and A280 quantitation (Figure S4). Further analysis of the ProA yields for these 144 non-cognate HC/LC pairs ( and Table S3) showed that only 38 of the 144 combinations (26.4%) displayed a significant reduction in expression levels (50% or lower relative to the controls), suggesting an overall widespread promiscuous behavior within HC/LC pairing. Of the remaining 106 combinations, 68 (47.2%) HC/LC non-cognate pairing molecules displayed higher expression levels than the corresponding parental mAb controls (). However, many LCs were not broadly promiscuous. A closer look into two examples is useful to illustrate this phenomenon. When LCs-B1–4 were paired with HC-A7, protein expression was significantly lower when compared to the cognate LC-A7 (control) (). In contrast, the expression levels of the same anti-Target-B LCs while paired with HC-A8 are comparable or higher than cognate LC-A8 (control). These results suggest that the anti-Target-B LCs are promiscuous with respect to HC-A8 but not toward HC-A7, highlighting the role that HCs also play a role in determining LC cross-pairing.
Figure 5. High-throughput screening for cLC hetero-IgGs by ncCSA
Binding analysis identified cLC hetero-IgG candidates
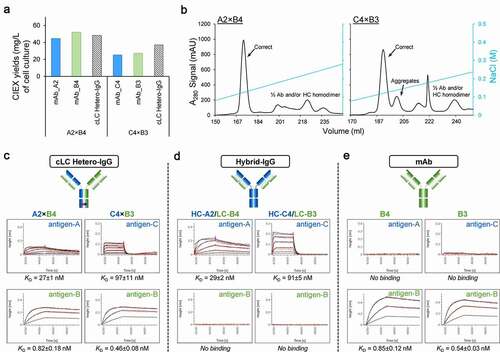
Although well-expressing non-cognate HC/LC pairs are promising candidates to assemble cLC bispecifics, expression levels alone provide no insight into function. To select for functional bispecifics, we assembled selected promiscuous LCs (demonstrating 50% or higher expression levels relative to cognate controls during the ncCSA assay () into a cLC hetero-IgG format with both the cognate and non-cognate HCs, containing Fc CPMs to promote HC dimerization (). We then evaluated these cLC hetero-IgGs with a high-throughput binding assay to identify candidates that allow binding to both targets. After high-throughput expression in 4 mL deep-well blocks, with 293-6E cells, the cLC hetero-IgGs were purified by ProA. The yields for 92 of 106 of the molecules (86.8%), was ~100 mg/L, which is comparable to the parental mAbs ( and S5), with only 14 cLC hetero-IgGs (13.2%) showing ProA yields lower than 60 mg/L ( and 5e). Interestingly, all molecules with cLC B1 showed a remarkably lower expression, suggesting that this LC may act as a limiting factor in the overall expression of these bispecifics (). Indeed, the ProA yield for B1 mAb with 41.6 mg/L was the lowest among all parental mAbs used as building blocks for the generation of these bispecifics (). Although A3 and A6 parental mAbs also showed a relatively low ProA recovery (60.3 and 69.3 mg/L, respectively), the cLC hetero-IgGs containing these 2 building blocks showed acceptable protein yields when combined with any B parental but B1, suggesting that in this case the non-cognate HCs may have rescued the expression levels (). Another important observation is that the cLC hetero-IgGs (A×B and C×B) also showed a ~2-fold increase overall in correct pairing over the 4-chain hetero-IgGs just after ProA purification, highlighting the impact of HC/LC pairing in the production levels of these molecules (Figure S6).
For rapid binding screening, these single-step purified samples were then assessed by ForteBio Octet. To minimize interference by residual impurities, the cLC hetero-IgG molecules were first captured onto Streptavidin fiber optic biosensors with a biotinylated anti-human IgG Fc polyclonal antibody via the Fc region, and then soluble antigen-A, -B, or -C were loaded for incubation. As expected, all cLC hetero-IgGs displayed binding to their respective targets via the cognate HC/LC arm, with comparable affinity to the parental mAbs (). Interestingly, two cLC hetero-IgGs (A2×B4 and C4×B3) also showed detectable binding via the non-cognate HC/LC arm recognizing Target-A or -C (). In the case of A2×B4, the B4 LC was paired with both HCs (A2 and B4), whereas for C4×B3 the HCs (C4 and B3) were both paired with the B3 LC. Of note, these cLCs were both generated against Target-B. Although this non-canonical binding is lower than the single-digit nM binding typically observed for parental mAbs, it demonstrates how the ncCSA method provides a new opportunity to identify LCs with unique structural features allowing for highly efficient pairing with non-cognate HCs (). Furthermore, rapid binding analysis can reveal those rare cLCs that also support binding to epitopes recognized by the non-cognate HCs. Since the manufacturability of IgG-like bispecifics is often challenging, and production levels are typically below that of monospecific mAbs,Citation24 we sought to explore the expression and purification properties of these cLC hetero-IgGs. To better mimic the scale and purification process required for therapeutic candidates, these 2 molecules were expressed in 250 mL 293-6E cells and subjected to a 2-step purification with ProA followed by CIEX to meet the purity target of >95%. Notably, the levels of protein secretion, by ProA, were about 2-fold higher for these 2 cLC hetero-IgGs when compared to the parental mAbs (Table S4). More importantly, these cLC hetero-IgGs showed a final yield comparable to or higher than the parental mAbs (), all with over 97% purity of the desired species (Table S4). Moreover, these bispecifics showed favorable CIEX profiles, with the correct species easily separated from the impurities (). We then repeated the binding assay using the fully purified cLC hetero-IgGs to confirm their affinity for the respective antigens. As observed initially (), these two molecules showed binding affinity via their non-cognate HC/LC arms to antigen-A or -C while retaining the binding properties in the cognate arms to antigen-B ( and S7). To validate the affinity measured for these cLC hetero-IgGs, we also expressed and purified two hybrid IgGs composed of a non-cognate HC and LC each (HC-A2/LC-B4 and HC-C4/LC-B3). The comparable affinities of the hybrid molecules to antigen-A and -C via their non-cognate arms () further confirmed the cLC hetero-IgGs binding. Interestingly, the binding signal for the hybrid IgGs was ~2-fold higher than the signal observed for the non-cognate arm in the cLC hetero-IgGs, which agrees with the number of binding sites present in these molecules (2 vs 1, respectively). Moreover, the fact that neither of them seems to retain binding to antigen-B suggests that the binding capability of hybrid IgGs is mostly driven by HC CDRs, but not LC. Inversely, to also exclude the possibility of nonspecific binding to antigen-A or -C by the cognate arms in the cLC hetero-IgGs, we tested the binding for B4 and B3 parental mAbs. As shown in , B4 and B3 mAbs did not bind to these antigens, further demonstrating that the binding detected for the non-cognate arm is neither derived from a nonspecific interaction between cognate arm and antigen-A or -C nor the result of cLC alone.
Figure 6. Expression, purification and binding properties of two selected cLC hetero-IgG molecules
In summary, the ncCSA method successfully identified two rare LCs from a pool of mAbs that paired with non-cognate HCs showing mAb like productivity while allowing the HC in this non-cognate HC/LC arm to retain binding activity.
Discussion
Despite breakthroughs in protein engineering, the development of bispecifics, particularly those with non-native quaternary structures, has been hampered by manufacturing challenges, including low yields, inefficient separation from impurities, and molecule instability.Citation1,Citation7 Bispecific formats like hetero-IgGs and cLC hetero-IgGs with 4 and 3 chains, respectively, maintain their overall IgG-like native quaternary structure, which may in turn enable the desired therapeutic properties. However, these formats require extensive engineering efforts to drive chain pairing.Citation11,Citation13,Citation14,Citation25 cLC hetero-IgGs avoid alteration of the HC/LC interface altogether, but this approach demands a substantial investment of resources and time.Citation15 Another aspect to consider is the lower affinity that Fabs carrying a cLC may display, which may not fit specific therapeutic needs.Citation15,Citation26
The design goals and selection process to identify best-in-class mAb therapeutics are well understood. However, little is currently known about how the native properties of these same mAbs may impact the assembly of bispecifics when used as building blocks, and determining the molecular basis of folding, activity, and stability of antibodies is a time-consuming process.Citation27 Although the structural features of mAbs are largely understood,Citation18,Citation28,Citation29 the highly diverse Fv regions, necessary to confer epitope specificity, make each IgG molecule distinctly unique. This sequence diversity across panels of mAbs often undercuts the successful application of protein engineering platforms.
Here, we wanted to illuminate the HC/LC pairing preference mediated by the VH/VL interface. Therefore, we developed and validated the CSA method to effectively select the best building blocks for hetero-IgGs. While cCSA examines the HC/LC paring specificity by recreating the competing environment of four antibody chains expressed in a single cell, the ncCSA simply evaluates whether a single LC has the potential to pair with a given non-cognate0 HC.
In the cCSA experiment, it was paramount to set a benchmark to separate combinations of two parental mAbs that were likely to form a specific HC/LC cognate pair. While high-throughput LC-MS readout cannot confirm correct pairing, it accurately determines whether the four chains of interest are present. Thus, assuming an HC/HC heterodimer, there are three possible events detected by LC-MS: 2×LC1, 2×LC2 and 1×LC1 + 1×LC2, resulting in a random probability of 25%, 25%, and 50%, respectively. Our analysis showed that about half (8/17) of the molecules with a 50% or higher correct chain pairing expressed well and could be fully purified at a large scale. Strikingly, most candidates (12/13) with less than 50% in cCSA experiment failed in the purification. This is extremely important, as we can confidently use these metrics to deselect candidates during the workflow. As ion-exchange columns are used as a standard method for purifying bispecifics, surface properties will have a direct effect on how molecules interact with a charged resin, leading to good/poor separation. Future work will be necessary to evaluate how to integrate electrostatic charges (i.e., isoelectric point) with the cCSA method to aid in the rational selection for mAb combinations.
The ncCSA experiment provided an opportunity for the identification of cLCs. From our screening, 73.6% of these non-cognate combinations show a high level of promiscuity (>50% of expression levels of their parental mAbs), further highlighting the challenges associated with generating 4-chain hetero-IgG molecules with minimal mispairing. However, non-cognate LCs can often impair epitope binding by either lacking favorable binding interactions with the new epitope or by more globally perturbing the CDR structure in the HCs and further disrupting the overall paratope surface. Therefore, the positive binding detected for two molecules (1.39% of the panel) is a remarkable result, suggesting that nature can produce such molecules and that it is up to us to find them. Moreover, with this approach one of the arms retains the cognate HC/LC pair, leaving the binding affinity toward one target intact. This can provide a valuable feature enabling the successful and broader application of cLC bispecifics in programs where one Fv is required to exhibit high affinity, but not the other (i.e., agonistic, cis-binding and/or bivalent formats where avidity can compensate for the lower affinity of the cLC arm). This is in striking contrast with cLC mice and display-focused technologies that often force both HCs to accept a non-cognate LC.Citation16,Citation17
For multispecific therapeutics to truly deliver on their promise, the manufacturability of these custom-made large molecules must improve. We thus sought to reveal the molecular basis that enables the production of these molecules. Here, we have shown that the number of chains used in a single cell transfection do not seem to impair the secretion levels of these 3 and 4-chain molecules. Although it seems to correlate with an increase in chain mispairing, the number of polypeptide chains may be easier for the current mammalian cell machinery to deal with than non-IgG-like formats (containing scFv fragments and other fusions) that show suboptimal folding, assembly and/or secretion.Citation7 Moreover, we have demonstrated that the sequence diversity in mAbs plays a decisive role in the success of making multispecifics. With the CSA methods presented here, we can rationally triage these building blocks and ensure successful development of the molecules.
Materials and methods
Plasmid construction
The antibody HC and LC genes were synthesized by Twist Bioscience and then individually cloned into a mammalian transient expression vector using a Golden Gate assembly method.Citation30 To reduce protein heterogenicity, all HCs were constructed with human IgG1 scaffold (IgG1-SEFL2) carrying an aglycosylation mutation and a novel engineered disulfide bond.Citation31 For hetero-IgGs that require HC heterodimerization, CPMs were introduced into the Fc regions. After sequencing confirmation by Sanger, transfection-grade DNA was prepared using Maxi plasmid purification kits (Qiagen, catalog # 12165), and then mixed at a ratio of 1:1 (HC:LC) for mAbs and hybrid IgGs (non-cognate HC/LC pairs), 1:1:1:1 (HCA:LCA:HCB:LCB) for 4-chain hetero-IgGs and 2:1:1 (cLC:HCA:HCB) for cLC hetero-IgGs (3-chain hetero-IgGs).
Cell culture and protein expression
All proteins were transiently expressed in suspension human embryonic kidney 293–6E cells (NRC-BRI) using an in-house improved protocol. Briefly, cells were maintained in FreeStyle F-17 medium (Gibco, catalog # A1383502) with 0.1% Kolliphor P188 (Sigma, catalog # K4894), 25 μg/ml G418 (Gibco, catalog # 10131027) and 6 mM L-glutamine (Gibco, catalog # 25030149). To achieve a density of 2 × 106 viable cells per ml for optimal transfection, cells were passaged 26 hours in advance of transfection. For each ml of cells, 0.5 μg DNA was complexed with 1.5 μl PEImax reagent (Polysciences, catalog # 24765–2) in 100 μl FreeStyle F-17 medium for 10 min, and then added to cell culture. One day after transfection, cell cultures were fed with Tryptone N1 solution (Organotechnie, catalog # 19553) and glucose (Thermo Fisher, catalog # A2494001) to a final concentration of 2.5 g/L and 4.5 g/L, respectively. Three days later, 3.75 mM sodium valproate (MP Biomedicals, catalog # 0215206480) was added to enhance protein expression. At day 6 post transfection, conditioned medium was harvested for purification.
High-throughput protein purification with ProA magnetic beads
The KingFisher® Flex system (Thermo Fisher) was used for high-throughput protein purification with magnetic ProA beads (GE Life Sciences). Briefly, 4 ml 293–6E cells in 24-well deep blocks were transfected for protein expression, followed by the addition of 100 μl magnetic ProA beads were added one day before harvest. Then, beads were collected and subjected to KingFisher purification with a 24 deep-well magnetic head. After washing 3 times with phosphate-buffered saline and twice with Milli-Q water, proteins were eluted with 500 μl of 100 mM sodium acetate at pH 3.6 for 10 min, and then immediately neutralized by adding 10 μl of 3 M Tris, pH 11.0.
Two-step purification with ProA and cation ion exchange chromatography
Proteins expressed in 40 ml 293–6E cells were purified using ProA affinity capture (1 ml HiTrap MabSelect SuRe, GE Life Sciences, catalog # GE11-0034-93), eluted with 100 mM sodium acetate, pH 3.6 followed immediately by buffer exchange into 10 mM sodium acetate, 150 mM NaCl, pH 5.2 using a 5 ml HiTrap Desalting column (GE Life Sciences, catalog # GE17-1408-01) as described previously.Citation32
For ion-exchange chromatography, ProA eluates (1.5–1.8 ml) were diluted with 20 ml of 20 mM MES, pH 6.2 and loaded onto 1 ml cation ion-exchange column (SP-HP HiTrap, GE Life Sciences, catalog # GE29-0513-24) at 1 ml/min. The column was washed with 8 column volumes of the same buffer at 1 ml/min and the proteins were eluted with a linear 0–400 mM NaCl gradient over 40 column volumes at 0.4 ml/min. Fractions of 90% or higher purity (as determined by SEC and MS) were pooled and their concentration was determined using Multiscan GO microplate reader (Thermo Fisher).Citation33 The final pooled samples were independently analyzed by SEC, non-reducing MCE and LC-MS to once again confirm the >90% purity.
Non-reducing SDS-PAGE
To analyze ProA purified samples, 1–2 μg of proteins were loaded onto Novex 4–20% Tris-Glycine gels (Invitrogen, catalog # WXP42026BOXA) in the absence of reducing agent. A PageRuler prestained protein ladder 10‒250kD (Thermo Fisher, catalog # 26620) was included on each gel. After running at 150 V for 1 hour, the gel was stained using InstantBlue Coomassie Protein Stain (Expedeon, catalog # ISB01L), briefly washed with Milli-Q water, and then imaged with a Gel Doc System (Bio-Rad).
Non-reducing micro capillary electrophoresis and analytical size exclusion chromatography
Purity of purified samples was analyzed with non-reducing MCE and analytical SEC. For non-reducing MCE, 6 μl of proteins were mixed with 21 μl of sample buffer (8.4 mM Tris-HCl pH 7.0, 7.98% Glycerol, 2.38 mM EDTA, 2.8% SDS, and 2.4 mM Iodoacetamide), heated at 85°C for 10 min, and then analyzed using a Caliper LabChip GXII Touch instrument (PerkinElmer). For analytical SEC, protein samples were analyzed on an Acquity HPLC instrument (Waters) using a BEH column (200 Å, 1.7 micron, 4.7 × 300 mm) with100 mM sodium phosphate pH 6.9, 50 mM NaCl, 7.5% ethanol as the running buffer at 0.45 ml/min flow rate.
Protein mass spectrometry by liquid chromatography-mass spectrometry
Relative quantitation of protein species was performed on an LC-MS system as previously describedCitation34–36 with some modification. Briefly, about 15 µg of each purified sample was analyzed by non-reduced LC-MS to maintain sample integrity and retain chain pairing information. The LC-MS system consisted of an Agilent 1290 Infinity II UPLC connected to an Agilent 6224 ESI-TOF mass spectrometer. Chromatographic separation utilized a Zorbax RRHD 300SB-C8 2.1 × 50 mm, 1.8 μm UPLC column heated to 70°C and run at a flow rate of 0.5 ml/min. Chromatographic solvents of A: 0.1% trifluoroacetic acid (TFA) in H2O and B: 0.1% TFA in 90% n-propanol were used. The gradient was isocratic at 80% A/20% B for 4 min, then to 28% A/72% B over 2 min, then 10% A/90% B over 0.5 min, and then finally 5% A/95% B over 0.5 min. The MS method scans m/z [1000–7000] acquiring 0.7 spectra/sec. The resulting spectra were summed, then deconvoluted using either the Agilent Mass Hunter Qualitative Analysis software (Version B.07.00) or Intact Program module from Protein Metrics, followed by relative quantification of each deconvoluted species using the MS signal intensity.36 An Excel-based tool was used to calculate the intact mass and intensity of various correct and mispaired species. In the case of a 4-chain hetero-IgG, the following species are calculated: IgG species with four unique chains, IgG species with HC heterodimer but 2× of one LC, IgG species with HC homodimer, and half-mAb species.
Characterization of binding affinity
To measure the binding affinities (KD equilibrium dissociation constant) of the purified antibodies (bispecific or monoclonal) to soluble antigens, the antibodies were first captured onto streptavidin SAX biosensor tips with a biotinylated (1 biotin/molecule), polyclonal capture antibody (Jackson ImmunoResearch, catalog# 109–005-098) and then incubated with a dilution series of each soluble antigen. This assay format was chosen so the bivalent antibodies would be immobilized and on the biosensor tips and tested versus the same serial dilution of each soluble antigen. The measured quantitative KD affinities thus represent monovalent 1:1 binding interaction and can be directly compared. Experiments were run in a ForteBio Octet HTX instrument using the 96-tip mode with standard 5 Hz data acquisition rate at 27°C and 1000 RPM. Using Genedata Screener V16 software, raw Octet binding data was processed with installed SPR kinetic curve fitting package and globally fit to a 1:1 binding model to determine the association rate constant (ka) and the dissociation rate constant (kd). The equilibrium dissociation constant (KD) was then calculated as a ratio of kd/ka. The KD values are average ± standard error from at least three independent experiments. The representative sensorgrams shown in are in each case from one of the experiments used in the analysis.
Abbreviations
Author contributions
F.G. and D.G. designed the project; F.G. and Z.W. supervised research; D.G. designed and generated all DNA constructs; D.L. performed mammalian expression and high-throughput KingFisher purification; D.G. and D.L. analyzed protein samples using SDS-PAGE gels; K.P.B. and A.R.C. performed large-scale purification with ProA and CIEX columns; C.S. and J.H.R. analyzed purified proteins by LC-MS; R.B.C. performed binding analysis; D.G., T.P.R. and F.G. interpreted the results; D.G. and F.G. wrote the paper. All authors reviewed the paper.
Declaration of interests
The authors declare no competing interests.
Supplemental Material
Download Zip (11.2 MB)Acknowledgments
We are very grateful to Fuyi Chen, Kathryn Douglas and Elisa Ayala for assistance with DNA cloning; Michelle Wu for assistance with expression; Dwight Winters, Cai Guo, Evyn Mirasol and Hannah Catterall for the assistance with protein analytics; Iain Campuzano, Eric Bryant and Alex Partin for revision of the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585–13.
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020; 27:1.
- Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol. 2015;8(1):130. doi:10.1186/s13045-015-0227-0.
- Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005;10(18):1237–44. doi:10.1016/S1359-6446(05)03554-3.
- Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, Wei A, Dombret H, Foa R, Bassan R, et al.Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47. doi:10.1056/NEJMoa1609783.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Wang Q, Chen Y, Park J, Liu X, Hu Y, Wang T, McFarland K, Betenbaugh MJ. Design and production of bispecific antibodies. Antibodies (Basel). 2019;8(3):43. doi:10.3390/antib8030043.
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67(2):95–106. doi:10.1016/j.molimm.2015.01.003.
- Suresh MR, Cuello AC, Milstein C. Bispecific monoclonal antibodies from hybrid hybridomas. Methods Enzymol. 1986;121:210–28.
- Carter P. Bispecific human IgG by design. J Immunol Methods. 2001;248(1–2):7–15. doi:10.1016/S0022-1759(00)00339-2.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9(7):617–21. doi:10.1093/protein/9.7.617.
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23(4):195–202. doi:10.1093/protein/gzp094.
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al.x Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–46. doi:10.1074/jbc.M110.117382.
- Dillon M, Yin Y, Zhou J, McCarty L, Ellerman D, Slaga D, Junttila TT, Han G, Sandoval W, Ovacik MA, et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs. 2017;9(2):213–30. doi:10.1080/19420862.2016.1267089.
- Shiraiwa H, Narita A, Kamata-Sakurai M, Ishiguro T, Sano Y, Hironiwa N, Tsushima T, Segawa H, Tsunenari T, Ikeda Y, et al. Engineering a bispecific antibody with a common light chain: identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods. 2019;154:10–20. doi:10.1016/j.ymeth.2018.10.005.
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16(7):677–81. doi:10.1038/nbt0798-677.
- Krah S, Schroter C, Eller C, Rhiel L, Rasche N, Beck J, Sellmann C, Gunther R, Toleikis L, Hock B, et al. Generation of human bispecific common light chain antibodies by combining animal immunization and yeast display. Protein Eng Des Sel. 2017;30(4):291–301. doi:10.1093/protein/gzw077.
- Garces F, Mohr C, Zhang L, Huang CS, Chen Q, King C, et al. Molecular insight into recognition of the cgrpr complex by migraine prevention therapy aimovig (erenumab). Cell Rep. 2020;30(6):1714–23 e6. doi:10.1016/j.celrep.2020.01.029.
- Bruggemann M, Osborn MJ, Ma B, Hayre J, Avis S, Lundstrom B, Buelow R. Human antibody production in transgenic animals. Arch Immunol Ther Exp (Warsz). 2015;63(2):101–08. doi:10.1007/s00005-014-0322-x.
- Foltz IN, Gunasekaran K, King CT. Discovery and bio-optimization of human antibody therapeutics using the XenoMouse® transgenic mouse platform. Immunol Rev. 2016;270(1):51–64. doi:10.1111/imr.12409.
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–97. doi:10.1016/j.jmb.2007.05.022.
- Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, Buchner J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34(5):569–79. doi:10.1016/j.molcel.2009.04.028.
- Feige MJ, Buchner J. Principles and engineering of antibody folding and assembly. Biochim Biophys Acta. 2014;1844(11):2024–31. doi:10.1016/j.bbapap.2014.06.004.
- Qiong Wang YC, Park J, Liu X, Yifeng H, Wang T, Betenbaugh K. Design and production of bispecific antibodies. Antibodies. 2019.
- Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108(27):11187–92. doi:10.1073/pnas.1019002108.
- Van Blarcom T, Lindquist K, Melton Z, Cheung WL, Wagstrom C, McDonough D, Valle Oseguera C, Ding S, Rossi A, Potluri S, et al. Productive common light chain libraries yield diverse panels of high affinity bispecific antibodies. MAbs. 2018;10(2):256–68. doi:10.1080/19420862.2017.1406570.
- Feige MJ, Hendershot LM, Buchner J. How antibodies fold. Trends Biochem Sci. 2010;35(4):189–98. doi:10.1016/j.tibs.2009.11.005.
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293(5532):1155–59. doi:10.1126/science.1061692.
- Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Strickland C. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. 2015;22(12):953–58. doi:10.1038/nsmb.3129.
- Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3(11):e3647. doi:10.1371/journal.pone.0003647.
- Jacobsen FW, Stevenson R, Li C, Salimi-Moosavi H, Liu L, Wen J, Luo Q, Daris K, Buck L, Miller S, et al. Engineering an igG scaffold lacking effector function with optimized developability. J Biol Chem. 2017;292(5):1865–75. doi:10.1074/jbc.M116.748525.
- Yoo D, Provchy J, Park C, Schulz C, Walker K. Automated high-throughput protein purification using an AKTApurifier and a CETAC autosampler. J Chromatogr A. 2014;1344:23–30. doi:10.1016/j.chroma.2014.04.014.
- Winters D, Chu C, Walker K. Automated two-step chromatography using an AKTA equipped with in-line dilution capability. J Chromatogr A. 2015;1424:51–58. doi:10.1016/j.chroma.2015.10.092.
- Spahr CS, Daris ME, Graham KC, Soriano BD, Stevens JL, Shi SD. Discovery, characterization, and remediation of a C-terminal Fc-extension in proteins expressed in CHO cells. MAbs. 2018;10(8):1291–300. doi:10.1080/19420862.2018.1511197.
- Sawyer WS, Srikumar N, Carver J, Chu PY, Shen A, Xu A, Williams AJ, Spiess C, Wu C, Liu Y, et al. High-throughput antibody screening from complex matrices using intact protein electrospray mass spectrometry. Proc Natl Acad Sci U S A. 2020;117(18):9851–56. doi:10.1073/pnas.1917383117.
- Campuzano IDG, Robinson JH, Hui JO, Shi SD, Netirojjanakul C, Nshanian M, Egea PF, Lippens JL, Bagal D, Loo JA, et al. Native and denaturing ms protein deconvolution for biopharma: monoclonal antibodies and antibody-drug conjugates to polydisperse membrane proteins and beyond. Anal Chem. 2019;91(15):9472–80. doi:10.1021/acs.analchem.9b00062.